Introduction
The progression of aggressive breast cancer is
characterized by genomic instability leading to multiple genetic
defects, phenotypic heterogeneity, chemoresistance and poor outcome
(1,2). An imbalance between oncogene and tumor
suppressor activities plays an important role in the onset of
breast cancer through the inactivation of G1/S and/or
G2/M cell cycle checkpoints, which normally ensure the
orderly progression of cell cycle events (3). In normal cells, checkpoint activation
in response to DNA damage is mediated by p53 activation and
inhibition of Cdk2 activity leading to cell cycle arrest (4–6). The
centrosome has been implicated in the pathogenesis of cancer
through the development of multipolar mitotic spindles leading to
chromosomal instability and tumor cell heterogeneity (7,8). The
centrosome is the major microtubule-organizing center of the cell
and is duplicated once during a normal cell cycle to give rise to
two centrosomes that function as the spindle poles during mitosis
(3). Therefore, tight coordination
between centrosome duplication and DNA replication cycles is
essential to ensure equal segregation of sister chromatids during
cell division. In cancer, loss of coordination between the
centrosome and DNA cycles leads to centrosome amplification,
increased frequency of multipolar mitoses, and consequent
chromosomal instability (5,6,9,10). p53
and cyclin-A/Cdk2 play an important role in coordinating centrosome
duplication with cell cycle events. In mouse model studies, loss of
p53 by gene targeting and gain-of-function p53 mutations resulted
in the development of centrosome amplification and aberrant mitoses
(5,6,11).
Furthermore, cyclin-A has been demonstrated to be a key regulator
of the centrosome cycle (12,13),
and p53 mutations associated with cyclin-A overexpression
synergistically increase the frequency of centrosome defects
(14). Aurora-A mitotic kinase is a
critical Ser/Thr protein kinase that also controls centrosome
maturation and duplication and regulates spindle formation for
appropriate chromosomal segregation during normal mitosis (15). In cancer cells, overexpression of
Aurora-A kinase promotes centrosome amplification and chromosomal
instability (CIN), thus conferring tumor cell heterogeneity
associated with acquired drug resistance and poor outcome (16). Aurora-A is overexpressed in human
breast tumors and is associated with an invasive basal-like
phenotype and poor prognosis (17).
Aurora-A exerts a direct effect on oncogenic transformation in
vitro and in vivo through increased p53 degradation and
inhibition of apoptosis through activation of the PI3K/AKT pathway
leading to chemoresistance (18).
In human breast cancer, the mechanistic relationship between
deregulated activity of the cyclin-A/Cdk2 complex and Aurora-A
kinase in the induction of centrosome amplification has not been
investigated. To establish the molecular mechanisms linking
genotoxic stress, G1/S checkpoint and Aurora-A kinase
activity to the centrosome duplication cycle, we studied the effect
of drugs inducing genotoxic stress in breast tumor-derived cell
lines with abrogated p53 function as previously described (5,6). Our
results demonstrated that induction of genotoxic stress induces
centrosome amplification through stabilization and activation of
Aurora-A kinase mediated by Cdk2 oncogenic signaling in breast
cancer cells.
Materials and methods
Human breast cancer cell lines
The human breast cancer cell line MCF-7 was obtained
from ATCC (Manassas, VA, USA). The MCF-7 cells carrying a
dominant-negative p53 mutant (vMCF-7DNp53) or
overexpressing a constitutive active Raf-1 oncoprotein
(vMCF-7DRaf-1) were generated as previously described
(5,6,19,20).
Induction of genotoxic stress
To investigate the relationship between centrosome
amplification and G1/S checkpoint activation, cell lines
were plated at a density of 3×105. After 48 h, cells
were treated with 2 mM hydroxyurea (HU) or 1 μM methotrexate for 48
h to induce genotoxic stress and centrosome amplification.
Treatment of cancer cells with
small-molecule inhibitors of Cdk2 and Aurora-A
To inhibit Cdk2 or Aurora-A kinase activity, cancer
cells were treated with 1 μM SU9516 or 1 μM Alisertib, and the
resulting cellular phenotype was analyzed by immunofluorescence and
immunoblotting.
Indirect immunofluorescence and
immunoblotting
For indirect immunofluorescence and protein
expression analyses, breast cancer cells were treated as previously
described (5,6,19,20).
Antibodies employed in this study were the following: Aurora-A
(Cell Signaling Technology, Inc., Beverly, MA, USA); cyclin A
(Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and β-actin
(Sigma, St. Louis, MO, USA). Centrin antibody (20H5) was kindly
provided by Dr Salisbury’s Laboratory (Mayo Clinic, Rochester, MN,
USA).
Construction of the shRNA Aurora-A
vector
The PSSH1 shRNA suppression plasmid contains the H1
RNA polymerase III-dependent promoter for the generation of shRNA
molecules. shRNA oligos directed against the 39 UTR of Aurora A
(TAGGGATTTGCTTGG-GATA) were annealed and cloned into the
BglII/HindIII cloning site at the 3′ end of the RNA
polymerase III-dependent H1 RNA promoter driven vector. Clones
containing the insert were identified and sequenced to ensure
fidelity.
Results
To establish the mechanistic linkage between Cdk2
and Aurora-A kinase oncogenic signalings in the induction of
centrosome amplification, we employed MCF-7 breast cancer cells
harboring abrogated p53 function (MCF-7DNp53). The
centrosome phenotype was characterized employing antibodies
directed against the centrosomal protein centrin and the mitotic
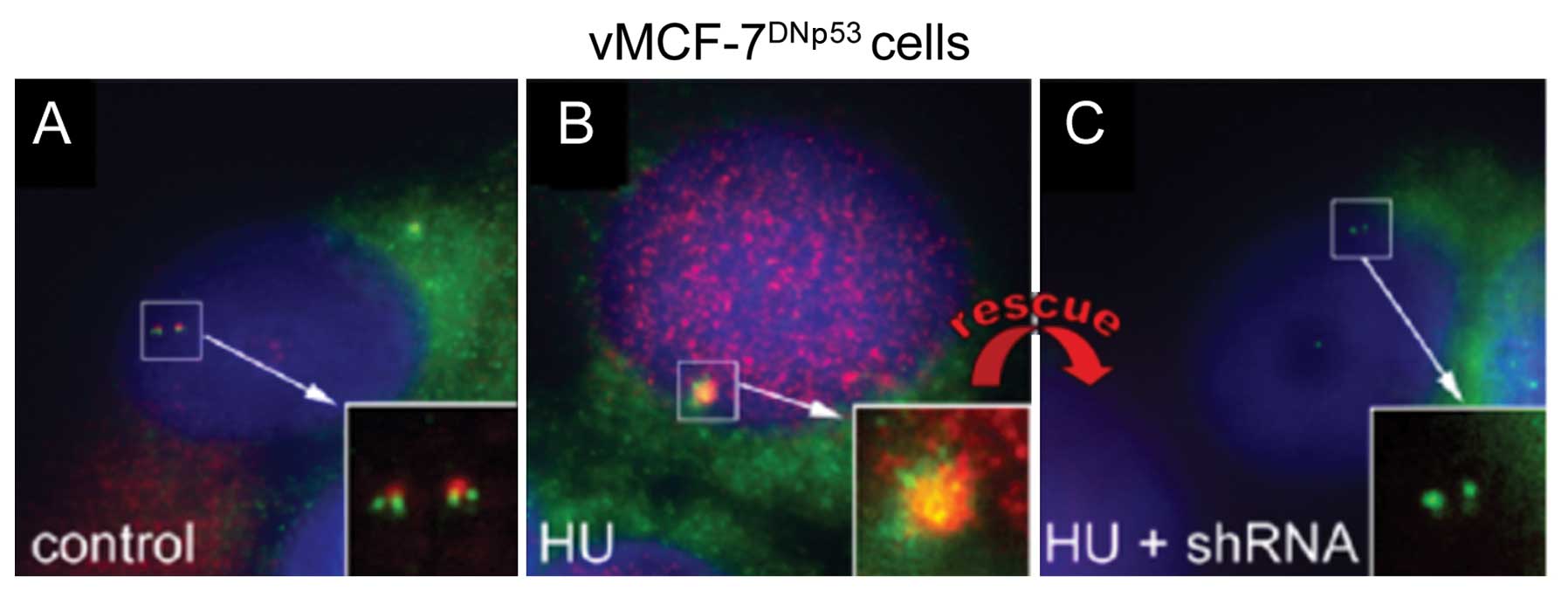
kinase Aurora-A. Treatment of MCF-7DNp53 cells with HU
for 48 h resulted in centrosome amplification characterized by
centriole overduplication (Fig. 1A)
as previously described (5).
Importantly, the amplified centrosomes displayed robust Aurora-A
co-localization suggesting that Aurora-A kinase activity may induce
centrosome amplification after genotoxic stress (Fig. 1B). To investigate whether Aurora-A
kinase activity plays a direct role in the induction of centrosome
amplification, we treated MCF-7DNp53 cells with HU and
shRNA targeting Aurora-A for 48 h. Following knockdown of Aurora-A,
centrosome amplification failed to occur after genotoxic stress,
and remarkably a normal centrosome phenotype was rescued in
MCF-7DNp53 cells (Fig.
1C). These results demonstrate the causal role of Aurora-A
kinase activity in the induction of centrosome amplification
following genotoxic stress.
Next, we investigated the mechanistic linkage
between Cdk2 and Aurora-A kinase activity in the induction of
centrosome amplification following genotoxic stress in
MCF-7DNp53 and parental MCF-7 cells. We previously
demonstrated that abrogation of p53 function leads to deregulated
activity of the cyclin-A/Cdk2 complex resulting in induction of
centrosome amplification (5,6). In
this study, we established an in vitro functional assay
where MCF-7DNp53 and parental MCF-7 cells were treated
with methotrexate, a genotoxic agent commonly employed in the
adjuvant setting of breast cancer. In order to determine the
concentration of methotrexate that will inhibit DNA replication and
induce genotoxic stress, we performed dose response and time course
experiments with MCF-7 and MCF-7DNp53 cells. Our
experiments established that incubation for 48 h with 1 μM
methotrexate induced a G1/S arrest of the cell cycle by
FACS analysis (data not shown). To determine the effect of
methotrexate on the development of centrosome amplification and
Aurora-A centrosomal localization, we incubated MCF-7 and
MCF-7DNp53 cells with 1 μM methotrexate for 48 h and
analyzed the centrosome phenotype using antibodies directed against
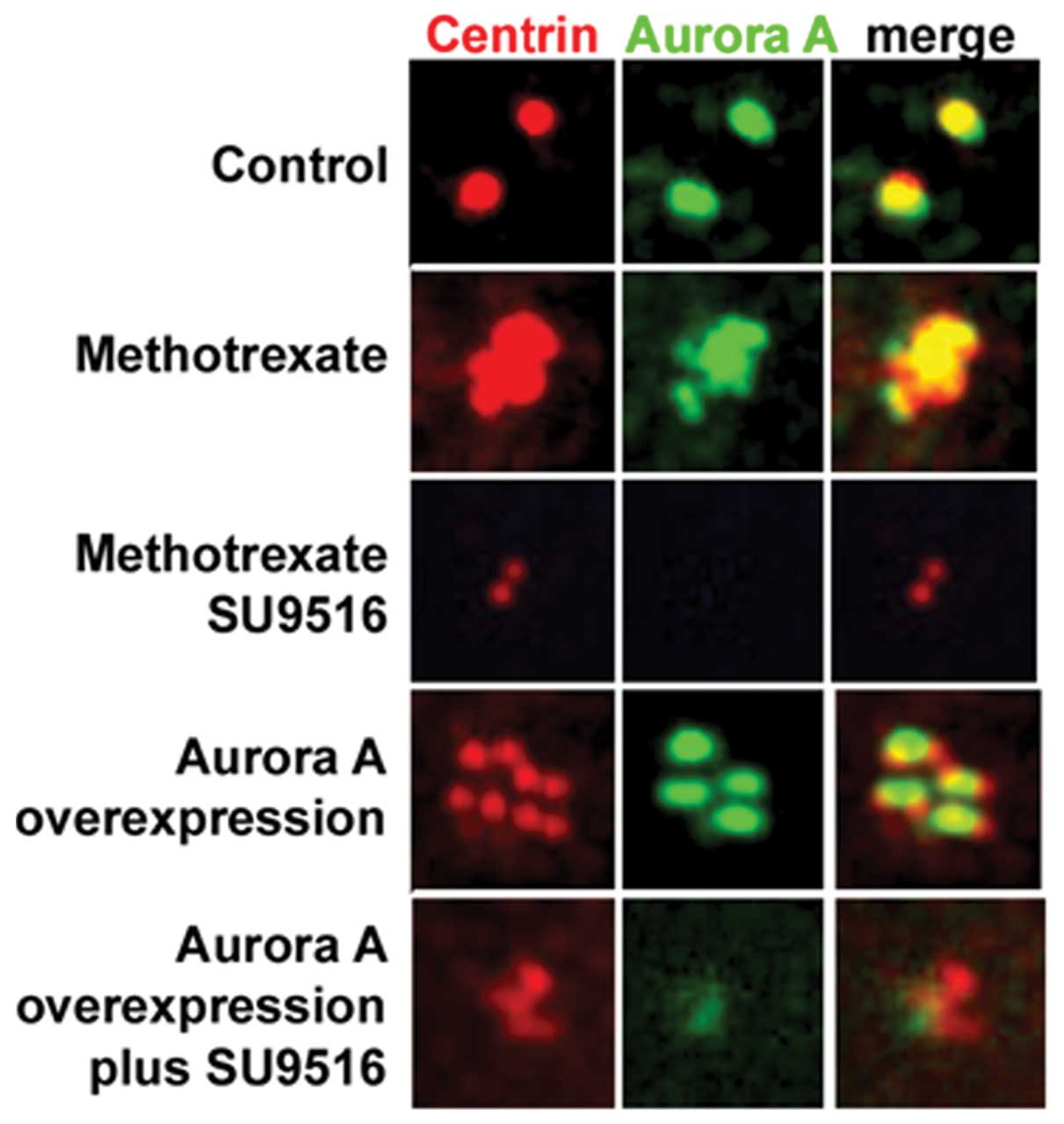
the proteins centrin and Aurora-A. As previously demonstrated,
MCF-7 cells retained a normal centrosome phenotype while
vMCF-7DNp53 cells developed centrosome amplification
(only centrosomes from the vMCF-7DNp53 cells are shown
in Fig. 2). Genotoxic
stress-induced centrosome amplification was associated with an
increase in Aurora-A centrosomal localization as we already
demonstrated in Fig. 1B. This
effect of methotrexate on centrosome amplification and Aurora-A
centrosomal localization was blocked by incubation for 48 h with
the small-molecule inhibitor of Cdk2 activity, SU9516 (Fig. 2). These results indicated that
Aurora-A kinase activity is downstream to Cdk2 signaling in the
induction of centrosome amplification. To investigate at the
mechanistic level the role of Cdk2 kinase in the activation of
Aurora-A-induced centrosome amplification, we engineered
vMCF-7DNp53 cells overexpressing an Aurora-A lentivector
(vMCF-7DNp53/Aurora-A). vMCF-7DNp53/Aurora-A
cells displayed centrosome amplification without induction of
genotoxic stress, confirming the causal role of aberrant Aurora-A
kinase activity in inducing centriole over-duplication (Fig. 2). Importantly, treatment of
vMCF-7DNp53/Aurora-A cells with 1 μM SU9516 for 48 h
reduced Aurora-A centrosomal localization that was linked to
suppression of centrosome amplification (Fig. 2). Since we previously demonstrated
that Cdk2 controls the centrosome duplication cycle through
interaction with cyclin-A (5), we
employed a variant MCF-7 cell line overexpressing a constitutive
active Raf-1 oncoprotein (vMCF-7DRaf-1) that displays
high levels of endogenous cyclin-A (20). To investigate the presence of a
positive feed-back loop between cyclin-A/Cdk2 and Aurora-A
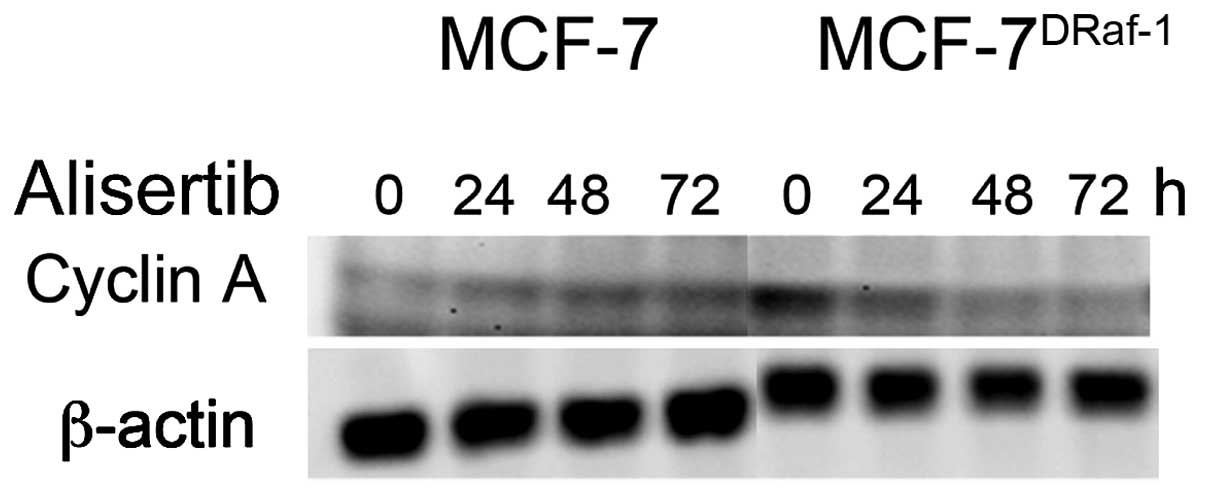
oncogenic signalings, we treated MCF-7 and vMCF-7DRaf-1
cells with a novel Aurora-A inhibitor (Alisertib) to assess the
expression of cyclin-A (Fig. 3).
Importantly, treatment with Alisertib induced the reduction in
cyclin-A expression in vMCF-7DRaf-1 cells demonstrating
that Aurora-A kinase may modulate cyclin-A/Cdk2 activity during
cell cycle progression. Taken together, these findings demonstrate
at the mechanistic level a novel interplay between cyclin-A/Cdk2
and Aurora-A oncogenic signalings in the development of centrosome
amplification in breast cancer cells.
Discussion
Deregulation of cell cycle checkpoints, centrosome
amplification and induction of CIN are hallmarks of breast cancer
(10). CIN represents a major
problem in the management of breast cancer patients due to the
generation of tumor clonal heterogeneity, which in turn may
facilitate the development of chemoresistance and tumor progression
(21). However, molecular
mechanisms associated with the development of CIN are poorly
understood in breast cancer. Several studies suggest that
deregulation of the centrosome cycle leading to centrosome
amplification may lead to the development of CIN through the
formation of multipolar mitotic spindles and unequal chromosome
segregation (3,10,22).
The tumor-suppressor gene p53, mutated in >50% of human cancers
(23), plays an important role in
the maintenance of centrosome homeostasis since loss of p53
function can lead to centrosome defects (5,6).
Furthermore, the Cdk2/cyclin-A complex has also been implicated in
the control of centrosome duplication (5,6,12), and
changes in their expression correlates with centrosome
amplification (24), suggesting an
interplay between the p53 and cdk2/cyclin-A pathways in
coordinating centrosome duplication with other cell cycle events.
Moreover, the centrosome duplication cycle is also regulated by the
mitotic kinase Aurora-A (25).
Aberrant Aurora-A kinase activation induces centrosome
amplification and CIN in solid tumors (16). However, whether or not deregulated
function of the cyclin-A/Cdk2 complex induces centrosome
amplification through aberrant activation of Aurora-A kinase has
not been established. In the present study, we investigated the
mechanistic linkage between cyclin-A/Cdk2 and Aurora-A oncogenic
signalings in the development of centrosome amplification in human
breast cancer cell models. We employed vMCF-7DNp53 cells
that display a deregulated activity of the cyclin-A/Cdk2 complex
and develop centrosome amplification after genotoxic stress
(5,6). Following induction of genotoxic
stress, vMCF-7DNp53 cells developed centrosome
amplification that was functionally linked to an increase in
Aurora-A centrosomal localization. Aurora-A played a causal role in
the induction of centrosome amplification, since suppression of
Aurora-A expression restored a normal centrosome phenotype.
Importantly, we demonstrated that Aurora-A kinase-induced
centrosome amplification was mediated by Cdk2 activity since
molecular targeting of Cdk2 suppressed Aurora-A centrosomal
localization and the consequent centrosome amplification. Moreover,
we employed vMCF-7DRaf-1 cells that display high levels
of endogenous cyclin-A, and we demonstrated that molecular
targeting of Aurora-A reduces cyclin-A expression. In conclusion,
these findings highlight a novel positive feedback loop between
cyclin-A/Cdk2 and Aurora-A pathways in the development of
centrosome amplification in breast cancer cells. They also provide
the operational rationale for targeting ‘druggable cell cycle
regulators’ as an innovative therapeutic strategy to inhibit
centrosome amplification and CIN in breast tumors resistant to
conventional chemotherapeutic drugs.
Acknowledgements
This study was supported by USAMRMC BC022276 and
Intramural RECDA Award to A.B.D., the Mayo Clinic Breast Cancer
Specialized Program of Research Excellence, NIH, CA116201 to J.I.,
and the Mayo Clinic School of Medicine. We also wish to acknowledge
the TACMA Core Facility of the Mayo Clinic Comprehensive Cancer
Center for assisting us with the immunofluorescence analysis and
interpretation of the results.
References
|
1
|
Lengauer C, Kinzler KW and Vogelstein B:
Genetic instabilities in human cancers. Nature. 6712:643–649. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Loeb LA: A mutator phenotype in cancer.
Cancer Res. 61:3230–3239. 2001.PubMed/NCBI
|
|
3
|
D’Assoro AB, Lingle WL and Salisbury JL:
Centrosome amplification and the development of cancer. Oncogene.
21:6146–6153. 2002.
|
|
4
|
Jin S and Levine AJ: The p53 functional
circuit. J Cell Sci. 114:4139–4140. 2001.PubMed/NCBI
|
|
5
|
D’Assoro AB, Busby R, Suino K, Delva E,
Almodovar-Mercado GJ, Johnson H, Folk C, Farrugia DJ, Vasile V,
Stivala F and Salisbury JL: Genotoxic stress leads to centrosome
amplification in breast cancer cell lines that have an inactive
G1/S cell cycle checkpoint. Oncogene. 23:4068–4075. 2004.PubMed/NCBI
|
|
6
|
D’Assoro AB, Busby R, Acu ID, Quatraro C,
Reinholz MM, Farrugia DJ, Schroeder MA, Allen C, Stivala F, Galanis
E and Salisbury JL: Impaired p53 function leads to centrosome
amplification, acquired ERalpha phenotypic heterogeneity and
distant metastases in breast cancer MCF-7 xenografts. Oncogene.
27:3901–3911. 2008.PubMed/NCBI
|
|
7
|
Brinkley BR: Managing the centrosome
numbers game: from chaos to stability in cancer cell division.
Trends Cell Biol. 11:18–21. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
D’Assoro AB, Barrett SL, Folk C, Negron
VC, Boeneman K, Busby R, Whitehead C, Stivala F, Lingle WL and
Salisbury JL: Amplified centrosomes in breast cancer: a potential
indicator of tumor aggressiveness. Breast Cancer Res Treat.
75:25–34. 2002.PubMed/NCBI
|
|
9
|
Lingle WL, Lutz WH, Ingle JN, Maihle NJ
and Salisbury JL: Centrosome hypertrophy in human breast tumors:
implications for genomic stability and cell polarity. Proc Natl
Acad Sci USA. 95:2950–2955. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lingle WL, Barrett SL, Negron VC, D’Assoro
AB, Boeneman K, Liu W, Whitehead CM, Reynolds C and Salisbury JL:
Centrosome amplification drives chromosomal instability in breast
tumor development. Proc Natl Acad Sci USA. 99:1978–1983. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tarapore P and Fukasawa K: Loss of p53 and
centrosome hyperamplification. Oncogene. 21:6234–6240. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Meraldi P, Lukas J, Fry AM, Bartek J and
Nigg EA: Centrosome duplication in mammalian somatic cells requires
E2F and Cdk2-cyclin A. Nat Cell Biol. 1:88–93. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Okuda M: The role of nucleophosmin in
centrosome duplication. Oncogene. 21:6170–6174. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mussman JG, Horn HF, Carroll PE, Okuda M,
Tarapore P, Donehower LA and Fukasawa K: Synergistic induction of
centrosome hyperamplification by loss of p53 and cyclin E
overexpression. Oncogene. 19:1635–1646. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nikonova AS, Astsaturov I, Serebriiskii
IG, Dunbrack RL Jr and Golemis EA: Aurora A kinase (AURKA) in
normal and pathological cell division. Cell Mol Life Sci. Aug
3–2012.(Epub ahead of print).
|
|
16
|
Li JJ, Weroha SJ, Lingle WL, Papa D,
Salisbury JL and Li SA: Estrogen mediates Aurora-A overexpression,
centrosome amplification, chromosomal instability, and breast
cancer in female ACI rats. Proc Natl Acad Sci USA. 101:18123–18128.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Staff S, Isola J, Jumppanen M and Tanner
M: Aurora-A gene is frequently amplified in basal-like breast
cancer. Oncol Rep. 23:307–312. 2010.PubMed/NCBI
|
|
18
|
Yang H, He L, Kruk P, Nicosia SV and Cheng
JQ: Aurora-A induces cell survival and chemoresistance by
activation of Akt through a p53-dependent manner in ovarian cancer
cells. Int J Cancer. 119:2304–2312. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
D’Assoro AB, Leontovich A, Amato A,
Ayers-Ringler JR, Quatraro C, Hafner K, Jenkins RB, Libra M, Ingle
J, Stivala F, Galanis E and Salisbury JL: Abrogation of p53
function leads to metastatic transcriptome networks that typify
tumor progression in human breast cancer xenografts. Int J Oncol.
37:1167–1176. 2010.PubMed/NCBI
|
|
20
|
Leontovich AA, Zhang S, Quatraro C, Iankov
I, Veroux PF, Gambino MW, Degnim A, McCubrey J, Ingle J, Galanis E
and D’Assoro AB: Raf-1 oncogenic signaling is linked to activation
of mesenchymal to epithelial transition pathway in metastatic
breast cancer cells. Int J Oncol. 40:1858–1864. 2012.PubMed/NCBI
|
|
21
|
Pinto AE, André S, Mendonça E, Silva G and
Soares J: Overall survival in advanced breast cancer: relevance of
progesterone receptor expression and DNA ploidy in fine-needle
aspirates of 392 patients. Int J Biol Markers. 18:7–12.
2003.PubMed/NCBI
|
|
22
|
Duensing S and Münger K: Centrosomes,
genomic instability, and cervical carcinogenesis. Crit Rev Eukaryot
Gene. 13:9–23. 2003. View Article : Google Scholar
|
|
23
|
Levine AJ, Momand J and Finlay CA: The p53
tumour suppressor gene. Nature. 351:453–456. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kronenwett U, Castro J, Roblick UJ,
Fujioka K, Ostring C, Faridmoghaddam F, Laytragoon-Lewin N,
Tribukait B and Auer G: Expression of cyclins A, E and
topoisomerase II alpha correlates with centrosome amplification and
genomic instability and influences the reliability of cytometric
S-phase determination. BMC Cell Biol. 4:82003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lukasiewicz KB, Greenwood TM, Negron VC,
Bruzek AK, Salisbury JL and Lingle WL: Control of centrin stability
by Aurora A. PLoS One. 6:e212912011. View Article : Google Scholar : PubMed/NCBI
|