Introduction
Despite recent advances in the understanding of lung
cancer biology, non-small cell lung cancer (NSCLC) is still the
leading cause of death from cancer in the world (1). Aberrant signaling pathways have been
extensively studied in human NSCLC, and targeted therapy has been
applied in the clinic. The epidermal growth factor receptor (EGFR)
is one of the most successive targets (2,3). The
EGFR tyrosine kinase inhibitors (EGFR-TKIs), gefitinib and
erlotinib, that specifically target the EGFR tyrosine kinase domain
have already been approved by the US FDA as monotherapy for the
treatment of patients with locally advanced or metastatic NSCLC,
and in many other countries gefitinib has also gained approval for
the treatment of advanced NSCLC patients with EGFR mutations in all
lines of therapies (2). The
majority of NSCLC cases overexpress EGFR, but only a limited subset
of patients who harbor EGFR mutations benefit from therapy
(1). Intrinsic resistance in
patients without EGFR mutations, as well as acquired resistance
after the initial response to TKIs, is a serious clinical issue.
Therefore, novel therapeutic strategies are under
investigation.
To date, various studies have been performed to
identify the population of patients that may be responsive to
EGFR-TKIs. The possible mechanisms that underlie the primary
resistance of these drugs have been supposed as follows: i)
activation of other signaling pathways alternative to EGFR, such as
the insulin-like growth factor 1 receptor (IGF-1R) and vascular
endothelial growth factor receptor (VEGFR); and ii) deregulation of
downstream signaling pathways such as AKT/mTOR (2–5).
Recently, much research has focused on combined treatment with
different signaling drugs, representing a novel strategy for cancer
therapy (6–8). Here, mTOR inhibitor emerged as an
attractive drug candidate due to the role of the mTOR pathway in
the development of lung cancer resistance.
The mammalian target of rapamycin (mTOR) is a highly
conversed serine-threonine kinase, located in the PI3K-AKT pathway.
It controls cell growth, proliferation, protein synthesis,
autophagy and metabolism, resulting in its increased attention for
use in cancer-targeted therapies (9–11).
Various mTOR inhibitors have been used in trials for anticancer.
RAD001 (everolimus) is an orally bioavailable mTOR inhibitor
derived from rapamycin, that has gained US FDA approval for the
treatment of metastatic renal carcinoma (11). However, in many other clinical
trials, it has demonstrated limited success, which may be a result
of the feedback activation of the PI3K-AKT targeted pathway
(9,10).
Taken together, we proposed that combined treatment
of an EGFR-TKI, gefitinib, and an mTOR inhibitor, RAD001, may have
more efficacy in treating NSCLC cell lines. Thus, in the present
study, we examine the antitumor effect of an mTOR inhibitor
combined with gefitinib on EGFR-TKI primary resistant
EGFR-wild-type NSCLC cell lines. To elucidate the effects and the
underlying mechanisms, we used gefitinib and RAD001 to treat three
NSCLC cell lines A549, NCI-H661, NCI-H460 that have wild-type EGFR
but have a different mutation status of PIK3CA. Oncogenic mutation
of PIK3CA was considered here to evaluate whether there is a
genetically distinct response to the combined treatment.
Materials and methods
Cell culture and reagents
The human NSCLC cell lines, NCI-H460, A549,
NCI-H661, were purchased from the Type Culture Collection of the
Chinese Academy of Sciences, Shanghai, China. NCI-H460 and NCI-H661
were cultured in RPMI-1640 medium (Gibco) supplemented with 10%
fetal bovine serum. A549 was cultured in F12K medium as
recommended. RAD001 (everolimus) was provided by Novartis
Pharmaceuticals, and gefitinib was supplied by AstraZeneca. Drugs
were dissolved in DMSO (Sigma-Aldrich, St. Louis, MO, USA),
respectively, and stored at −20°C. Solutions were finally diluted
in the culture medium to achieve the required concentrations.
Cell proliferation assay
The effects of the drugs on cell proliferation were
determined by a colorimetric assay using
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
Sigma-Aldrich). Cells were plated in 96-well plates at a density of
2,000–4,000 cells and allowed to attach overnight. The next day,
cells were treated with a range of drug concentrations of
gefitinib, RAD001 or the combination as recommended. Control cells
were treated with the same concentration of the vehicle DMSO.
After a 72-h incubation, 10 μl MTT solution was
added to each well. Four hours later, formazan crystals were
solubilized in 100 μl DMSO, and the absorbance was measured at 490
nm.
Combined effect analysis
The combined effect of gefitinib and RAD001 was
determined by using the Bliss additivism model. The Bliss value is
expressed as an equation Ebliss = EA + EB - EA × EB, where EA and
EB are the fractional inhibition effects of drug A and B alone at
an indicated concentration, respectively. Ebliss is the fractional
inhibition that would be expected if the combination of the two
drugs was exactly additive. If the experimentally measured
fractional inhibition of combined treatment is more than the
expected Ebliss value, then the combination is considered to be
synergistic, while if the experimentally combined effect is less
than the expected Ebliss value, then the interaction is
antagonistic. Otherwise, the combined effect is additive (12,13).
Apoptosis and cell cycle analysis
All cells were seeded at 1×105/well in
6-well plates and allowed to attach. Then RAD001, gefitinib or
their combination was added at the indicated concentrations. After
a 48-h incubation, the cells were harvested. For apoptosis
analysis, harvested cells were stained with Annexin V-fluorescein
isothiocyanate (FITC) and propidium iodide (PI) using a commercial
kit (FITC apoptosis kit, Nanjing KeyGen Biotech., Co., Ltd.,
Nanjing, China). The fluorescence levels of apoptotic cells were
determined by flow cytometry on a FACSort, (BD Biosciences,
Franklin Lakes, NJ, USA) and analyzed using the CellQuest software.
To analyze the cell cycle distribution, cell nuclear DNA was
stained with PI and detected with FACSort flow cytometry. Cell
cycle profile was performed using ModFit LT software (BD
Biosciences).
Western blot analysis
Cells were cultured for 24 h and stimulated with
inhibitors at the indicated concentrations. Protein extracts were
normalized according to the protein content, resolved by 10%
SDS-PAGE, transferred to nitrocellulose membranes, probed overnight
at 4°C with antibodies and finally revealed using the ECL western
blot analysis system. The following antibodies were used:
anti-phospho-AKT (S473) and anti-phospho-p70S6K (Thr389) were
obtained from Cell Signaling Technology. Anti-AKT and anti-p70S6K
was purchased from Epitomics. Anti-β-actin and horseradish
peroxidase-conjugated anti-rabbit or anti-mouse IgG were purchased
from Boster Biological Technology (Wuhan, China). Each sample was
assayed in triplicate.
Statistics
The statistical significance between two different
groups was evaluated by the Student's t-test. SPSS 13.0 was
performed for all the statistical tests. All P-values were
two-sided; P<0.05 was considered to indicate a statistically
significant result. All data are presented as means ± SD.
Results
Effects of single-drug treatment with
gefitinib or RAD001 on cell proliferation
We determined the effects of gefitinib treatment
alone on three NSCLC cell lines with wild-type EGFR (NCI-H460, A549
and NCI-H661). First, we compared the proliferation rate of these
cell lines following treatment with gefitinib. Cells were treated
with gefitinib at a range of concentrations for 72 h, and the
median inhibitory concentration (IC50) values were
determined by MTT proliferation assay. IC50 values were
8, 18 and >20 μM for NCI-H460, A549 and NCI-H661, respectively.
According to previous studies (1,14),
cells having an IC50 >1 μM are considered as being
resistant to gefitinib. Therefore, the three NSCLC cell lines had
primary resistance to gefitinib. The cell lines exhibited wild-type
EGFR but had a different mutation status of PIK3CA (Table I), suggesting that the EGFR mutation
status plays an important role in the determination of gefitinib
resistance.
 | Table IEGFR and PIK3CA mutation status of the
selected cell lines. |
Table I
EGFR and PIK3CA mutation status of the
selected cell lines.
| Cell lines | PIK3CA | PTEN | EGFR |
|---|
| A549 | Wild-type | Wild-type | Wild-type |
| NCI-H661 | Wild-type | Wild-type | Wild-type |
| NCI-H460 | Mutant | Wild-type | Wild-type |
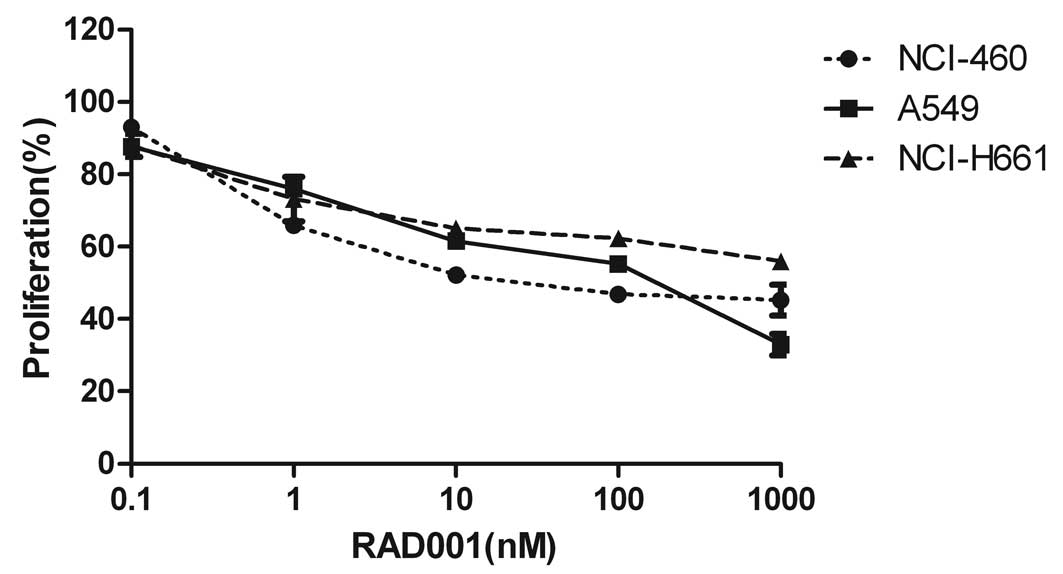
To determine the effects of the mTOR inhibitor
RAD001 on these gefitinib-resistant cells, we also performed a
proliferation assay. We demonstrated that RAD001 treatment caused a
mild but significant growth inhibition in all cell lines studied
(Fig. 1). Specially, following a
72-h treatment, 100 nM of RAD001 achieved a mild reduction in the
percentage of viable A549 (~51%), NCI-H460 (~55%) and NCI-H661
cells (~38%), and the inhibitory effects were dose-dependent.
However, a cell killing effect of >75% was not observed even at
concentrations up to 1,000 nM, which support the need for
alternative regimens to improve monotherapeutic efficacy. Notably,
expression of wild-type EGFR did not affect the responsiveness of
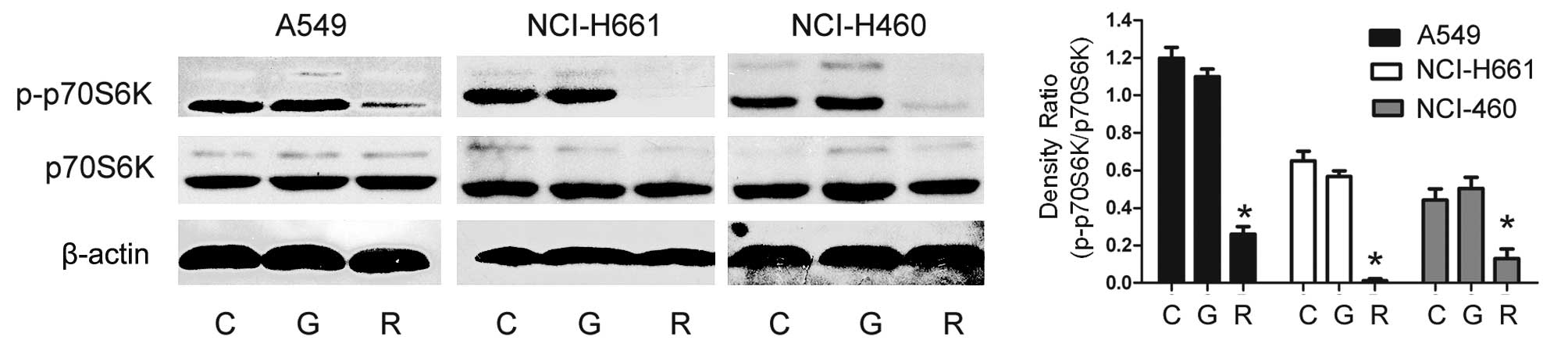
these cells to RAD001. To further explore the effect of RAD001 on
mTOR signaling, we performed a western blot assay. P70S6K is an
important downstream effector of mTOR, and its phosphorylation has
been reported to be effectively inhibited by rapamycin analog
treatment (15). In the present
study, western blot assay was performed after exposure to RAD001 at
a concentration of 10 nM for 24 h, showing a significant decrease
in the phosphorylation of p70S6K at Thr389 in all cell lines
(Fig. 2). Therefore, these results
indicate the importance of the mTOR signaling pathway in the
proliferation of NSCLC cells, suggesting the need for combined
treatment to enhance the antitumor effect.
Gefitinib cooperates with RAD001 to
enhance the inhibitory effect of either drug alone in NCI-H460
cells with PIK3CA mutation
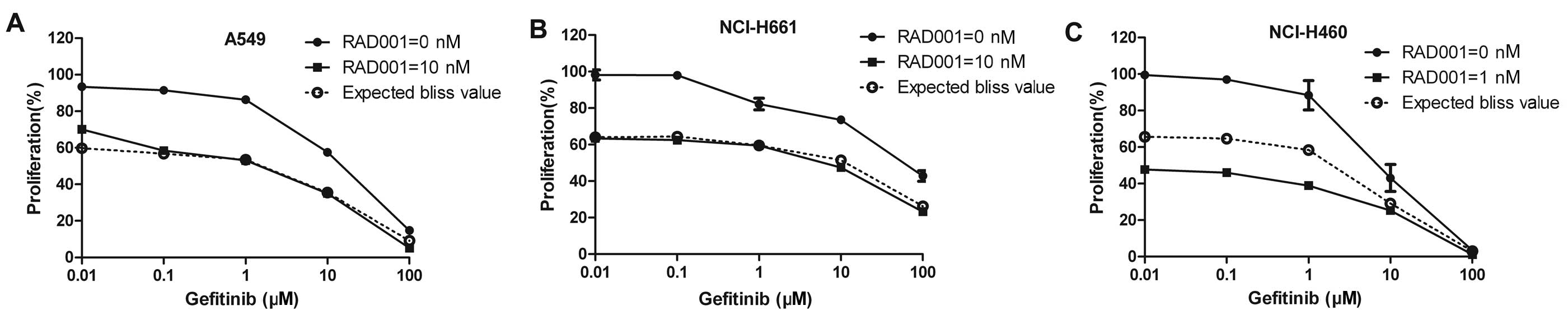
To examine the inhibitory effect following
suppression of both EGFR and mTOR pathways, we evaluated the
combined effect of gefitinib and RAD001 on growth inhibition,
apoptosis and cell cycle arrest. MTT assay was performed on cells
treated with gefitinib at concentrations ranging from 0.01 to 100
μM, in the presence of RAD001 at fixed concentrations from 1 to 10
nM, depending on the cell line. In an attempt to evaluate the
combined efficacy, we used the Bliss additivism model to clarify
whether the effect was additive, synergistic or antagonistic as
described in Materials and methods. The proliferation curve of the
cells following treatment with varying concentrations of gefitinib
in the presence or absence of a fixed concentration of RAD001 are
shown in Fig. 3. To note, the Bliss
theoretical curves that would be expected if the combination was
additive was calculated using the Bliss equation and are shown in
Fig. 3. Comparing the experimental
and the theoretical curves, only in the NCI-H460 cells was the
experimentally measured fractional inhibition of the combined
treatment more than the expected Ebliss value. Therefore, the
effect of the combination treatment on the NCI-H460 cells was
synergistic. The effects on the other two cell lines, A549 and
NCI-H661, showed pure additivity. Of note, NCI-H460 cells harbor a
PI3K mutation, whereas A549 and NCI-H661 harbor no mutation in this
gene.
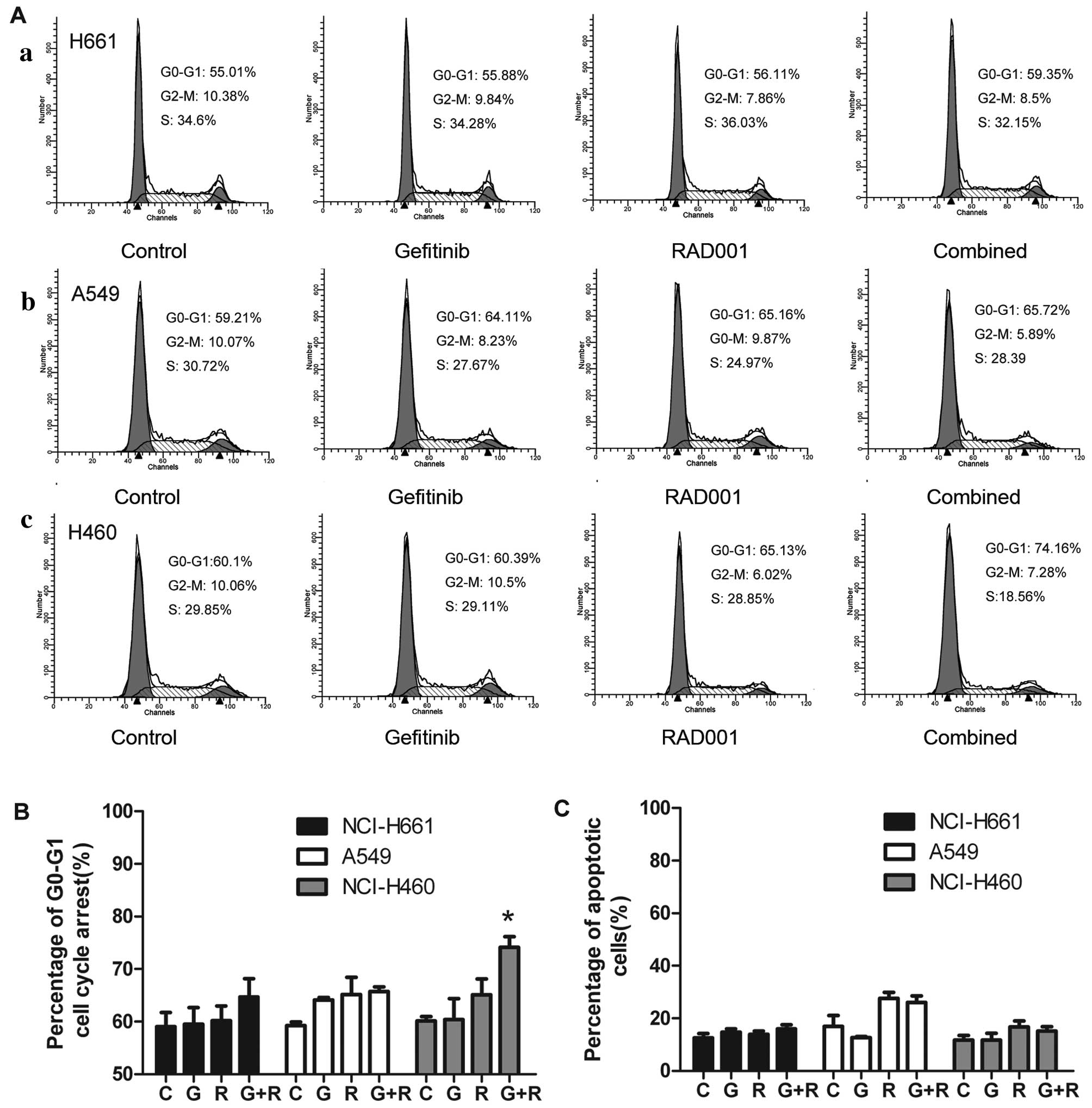
For further investigation, we explored the effects
of the combined treatment on the percentage of cell apoptosis and
cell cycle distribution following treatment with two single drugs
and their combination using flow cytometric analyses of Annexin V
binding. The combination treatment did not enhance the rate of
apoptosis when compared with this rate following treatment with
either drug alone in all of the cell lines studied (Fig. 4C). In addition, we tested the cell
cycle profiles of the three cell lines after treatment with each of
the drugs alone or their combination (Fig. 4A and B). We found that the treatment
with the combination of gefitinib and RAD001 had a great impact on
the cell cycle distribution in the NCI-H460 cells. The combination
caused accumulated of cell cycle arrest in the G0/G1 phase, and the
proportion of cells in the S and G2/M phase decreased following
treatment with 1 μM gefitinib and 10 nM RAD001 in combination for
48 h; the proportion of cells in the G0/G1 phase increased from
60.10 to 74.16% (P<0.05) (Fig.
4A), indicating that RAD001 enhanced a cytostatic effect,
rather than a cytotoxic effect. However, no significant changes
were detected in the NCI-H661 and A549 cells (Fig. 4A and B).
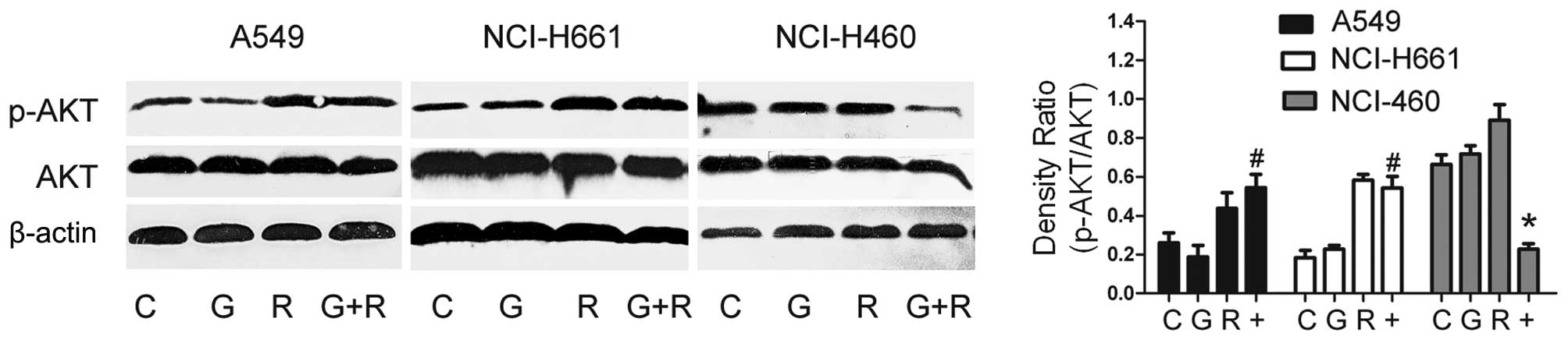
Combination treatment diminishes AKT
signaling in the NCI-H460 cells
To further explore the mechanisms of
genotype-dependent proliferation after the indicated treatments, we
evaluated therapy-associated changes in cell signaling pathway.
Since AKT has been proven to play an important role in cell
survival and is highly linked with mTOR, we aimed to ascertain how
AKT phosphorylation and the mTOR downstream effector are influenced
following treatment with gefitinib and RAD001 alone or in
combination (Fig. 5). As we
previously reported (Fig. 2),
western blot analysis demonstrated that exposure to RAD001 at a
concentration of 10 nM induced a significant decrease in the
phosphorylation level of p70S6K, an important downstream effector
of mTOR, in all the cell lines studied. Yet, no changes were
observed following treatment with gifitinib alone, which partially
supports the idea of combined inhibition with mTOR. Although AKT
phosphorylation is highly suppressed by gefitinib in sensitive cell
lines (8), it persistently remained
the same after treatment with gefitinib alone at a concentration of
1 μM in the gefitinib primary resistant NSCLC cell lines (Fig. 5). RAD001 and other rapamycin analogs
have been shown to indirectly activate mTORC2-dependent AKT
phosphorylation at Ser473 (15). We
found that the p-AKT level was increased after treatment with
RAD001 alone at a concentration of 10 nM, consistent with various
studies due to a negative feedback loop after inhibition of s6k
phosphorylation (10,15).
Therefore, we investigated whether the
phosphorylation level of AKT is influenced by the combination
treatment. Exposure to the combination of RAD001 (10 nM) and
gefitinib (1 μM) led to a significant reduction in p-AKT level in
the NCI-H460 cells (PIK3CA mutant), when compared with the
respective gefitinib or RAD001 treatment. However, no reduction in
AKT phosphorylation was noted after combination treatment in the
549 and NCI-H661 cells, which is consistent with their additive
combined effects.
Discussion
Non-small cell lung cancer represents 80% of all
lung cancer cases, and the majority of patients present with
locally advanced or metastatic disease. Treating advanced NSCLC
patients with platinum-based combination chemotherapy has achieved
more success than best supportive care. However, it yields a median
survival <12–13 months (6).
Novel therapeutic strategies including molecular targeted therapy
such as EGFR-TKIs have been proven to be effective, but only
limited to a subset of patients (1,2).
Signaling-based combination therapies have recently emerged as
novel attractive strategies (6,8,16).
Evidence supports the deregulation of the PI3K/mTOR pathway as a
possible mechanism for primary resistance of NSCLC to EGFR-TKIs
(2,17). Therefore, this study was performed
using three EGFR-TKI-resistant cell lines to evaluate the
therapeutic potential of the concomitant inhibition of EGFR and
mTOR signaling. The studied cell lines are characterized by
expression of wild-type EGFR, but with a different genetic status
of PIK3CA.
Our present study demonstrated that these three cell
lines all exhibit a poor response to gefitinib, and the mTOR
pathway is required for sustained growth in EGFR-TKI-resistant
cells. All of the cells harbored wild-type EGFR, which is
consistent with the role of the EGFR mutational status in
predicting EGFR-TKI efficacy. Phosphorylation of p70S6K, an
important downstream factor of mTOR, was not inhibited by gefitinib
alone in the three resistant cells, which suggests that maintained
activation of this signaling pathway was associated with gefitinib
intrinsic resistance. Indeed, the role of mTOR in tumorigenesis has
already been intensely investigated, and research has previously
revealed that constructive activation of the AKT/mTOR pathway is
present in a defined subset of NSCLC cases (18). Furthermore, we found that the mTOR
inhibitor RAD001 induced growth inhibition in gefitinib
primary-resistant cells, and p70S6K was effectively reduced.
However, it is known that RAD001 is a cytostatic factor and cannot
fully inhibit cell proliferation when a certain percentage of
inhibition is achieved. Inhibition with mTOR inhibitor rapamycin or
its derivates can indirectly activate AKT phosphorylation through a
feedback pathway by IRS or Grb (19–21),
which may partially compensate for the blockage of p70S6K. Here,
p-AKT was shown to be increased in RAD001-treated cells, which may
be a possible explanation for its cytostatic effect and limited
therapeutic efficacy following treatment with RAD001 alone.
However, rapamycin or its analogs were found to be
able to synergize drug sensitivity in many cases using combined
treatment (22,23). Thus, combined blockage of both EGFR
and mTOR is proposed to exhibit a more effective antitumor effect.
This hypothesis has been validated in many human cancer cell lines.
It has already been reported that inhibition of the mTOR pathway by
RAD001 produced a cooperative effect with EGFR inhibitors and
overcame resistance to anti-EGFR drugs according to Bianco et
al(8). Moreover, combined
inhibition using an mTOR inhibitor and anti-EGFR drugs was also
found to be synergistic in selected NSCLC cells or patients in many
other studies. Higher proliferation index or KRAS mutated status
were included (24,25). In our study, the genetic status of
PIK3CA was considered to illustrate the role of genetically
distinct combined therapeutic interventions in EFGR-wild-type
cells. Thus, we performed this study in three NSCLC cell lines A549
(PIK3CA wild-type), NCI-H661 (PIK3CA wild-type) and NCI-H460
(PIK3CA mutant), which all express wild-type EGFR. We aimed to
ascertain whether RAD001 enhances the antitumor effect of gefitinib
in these cells, and whether cells with a genetically distinct
status of PIK3CA showed a different efficacy. Here, we demonstrated
that concomitant therapy with gefitinib and RAD001 had a
significant synergistic inhibitory effect on the NSCLC cell line
NCI-H460 with PIK3CA mutation, but was not effective on cells with
wild-type PIK3CA, according to the Bliss additivism model.
Moreover, consistent with this, cell cycle distribution was
significantly altered in the NCI-H460 cells. We showed that the
drug combination promoted extensive accumulation of cells in the
G0-G1 phase of the cell cycle, while the percentages of cells in
the S and G0/M phase was decreased. The same phenomenon was not
noted in the A549 and NCI-H661 cells. Notably, we did not observe
significant apoptosis following any of the treatments, which may be
consistent with the cytostatic effect of RAD001.
The mechanism responsible for the different efficacy
in NCI-H460 cells compared to the other two cell lines may be
associated with their PIK3CA mutation status. Oncogenic mutations
of the PI3K catalytic a subunit (PIK3CA) are among the most
frequently reported genetic aberrations in many human epithelial
cancers (15), and it seems to be
an important indicator of resistance and poor outcome in patients
with NSCLC treated with EGFR-TKIs (26). Furthermore, oncogenic mutation of
PIK3CA may lead to constitutively activity of downstream signaling
such as the PI3K/AKT/mTOR pathway, which is partially EGFR
independent (27). One recent study
reported that the combination of EGFR monoclonal antibodies with a
PI3K inhibitor was a good therapeutic option in PIK3CA mutated head
and neck squamous cell cancer (28). Therefore, combined treatment with
EFGR-TKI and RAD001 is proposed to effectively inhibit cell
proliferation in cells with PIK3CA mutation. Our research did not
show synergistic inhibition of the combination treatment in the
PIK3CA wild-type A549 and NCI-H661 cell lines, suggesting that
another receptor, tyrosine kinase pathway may be activated in these
cells resulting in AKT activation and cannot be reduced by combined
treatment. Tamburini et al(29) strongly indicated an mTOR-independent
deregulation of oncogenic protein synthesis in human myeloid
leukemogenesis. In addition, consistent with this, it has been
shown that an EGFR-TKI and RAD001 can effectively inhibit AKT
phosphorylation in resistant cells with a PI3KCA mutation. In the
present study, combination treatment disturbed the PI3K/mTOR
pathway and enhanced the antitumor effect of monotherapy on
NCI-H460 cells.
It is critically important to develop specific
biomarkers for predicting a certain response of targeted cancer
therapies (30). Here, our results
demonstrated that the combination therapy was effective only in
selected cells, which may harbor mutations of the PIK3CA gene.
Recently, combined inhibition of EGFR and mTOR has
been previously investigated in patients with advanced NSCLC, and
demonstrated a clinical response in KRAS- mutated cancer patients,
although the study was not concerned with PI3KCA mutations
(24). PIK3CA mutations can be
widely noted in many types of human cancers. Due to its
contribution in cancer pathogenesis, therapy targeting this pathway
has been proven to be effective (15). In the present study, we further
confirmed that combined inhibition with an EGFR-TKI and an mTOR
inhibitor effectively induced cell proliferation inhibition and
cell cycle arrest in EGFR-wild-type NSCLC cells with a PIK3CA
mutation, disturbing AKT signaling. Thus, further study of the
different PI3KCA mutation status in predicting the efficacy of the
concomitant inhibition of the EGFR and mTOR pathways in NSCLC is
warranted. Combined inhibition of the EGFR and mTOR pathways may
provide an excellent therapeutic option for treatment of
PIK3CA-mutated EFGR-wild-type NSCLC.
Acknowledgements
This study was supported by a grant from the
Wujieping Foundation, China.
References
|
1
|
Sharma SV, Bell DW, Settleman J and Haber
DA: Epidermal growth factor receptor mutations in lung cancer. Nat
Rev Cancer. 7:169–181. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wheeler DL, Dunn EF and Harari PM:
Understanding resistance to EGFR inhibitors - impact on future
treatment strategies. Nat Rev Clin Oncol. 7:493–507. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Han SW, Kim TY, Hwang PG, et al:
Predictive and prognostic impact of epidermal growth factor
receptor mutation in non-small cell lung cancer patients treated
with gefitinib. J Clin Oncol. 23:2493–2501. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chakravarti A, Loeffler JS and Dyson NJ:
Insulin-like growth factor receptor I mediates resistance to
anti-epidermal growth factor receptor therapy in primary human
glioblastoma cells through continued activation of phosphoinositide
3-kinase signaling. Cancer Res. 62:200–207. 2002.
|
|
5
|
Engelman JA, Janne PA and Mermel C: ErbB-3
mediates phosphoinositide 3-kinase activity in gefitinib-sensitive
non-small cell lung cancer cell lines. Proc Natl Acad Sci USA.
102:3788–3793. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gorzalczany Y, Gilad Y, Amihai D, Hammel
I, Sagi-Eisenberg R and Merimsky O: Combining an EGFR directed
tyrosine kinase inhibitor with autophagy-inducing drugs: a
beneficial strategy to combat non-small cell lung cancer. Cancer
Lett. 310:207–215. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Morrow PK, Wulf GM, Ensor J, et al: Phase
I/II study of trastuzumab in combination with everolimus (RAD001)
in patients with HER2-overexpressing metastatic breast cancer who
progressed on trastuzumab-based therapy. J Clin Oncol.
29:3126–3132. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bianco R, Garofalo S, Rosa R, Damiano V,
Gelardi T, Daniele G, Marciano R, Ciardiello F and Tortora G:
Inhibition of mTOR pathway by everolimus cooperates with EGFR
inhibitors in human tumours sensitive and resistant to anti-EGFR
drugs. Br J Cancer. 98:923–930. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zoncu R, Efeyan A and Sabatini DM: mTOR:
from growth signal integration to cancer, diabetes and ageing. Nat
Rev Mol Cell Biol. 12:21–35. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yea SS and Fruman DA: Cell signaling. New
mTOR targets Grb attention. Science. 332:1270–1271. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Atkins MB, Yasothan U and Kirkpatrick P:
Everolimus. Nat Rev Drug Discov. 8:535–536. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mordant P, Loriot Y, Leteur C, et al:
Dependence on phosphoinositide 3-kinase and RAS-RAF pathways drive
the activity of RAF265, a novel RAF/VEGFR2 inhibitor, and RAD001
(Everolimus) in combination. Mol Cancer Ther. 9:358–368. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Goldoni M and Johansson C: A mathematical
approach to study combined effects of toxicants in vitro:
evaluation of the Bliss independence criterion and the Loewe
additivity model. Toxicol In Vitro. 21:759–769. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Noro R, Gemma A, Kosaihira S, et al:
Gefitinib (Iressa) sensitive lung cancer cell lines show
phosphorylation of Akt without ligand stimulation. BMC Cancer.
6:2772006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Di Nicolantonio F, Arena S, Tabernero J,
et al: Deregulation of the PI3K and KRAS signaling pathways in
human cancer cells determines their response to everolimus. J Clin
Invest. 120:2858–2866. 2010.PubMed/NCBI
|
|
16
|
Balko JM, Jones BR, Coakley VL and Black
EP: Combined MEK and EGFR inhibition demonstrates synergistic
activity in EGFR-dependent NSCLC. Cancer Biol Ther. 8:522–530.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Janmaat ML, Kruyt FA, Rodriguez JA and
Giaccone G: Response to epidermal growth factor receptor inhibitors
in non-small cell lung cancer cells: limited antiproliferative
effects and absence of apoptosis associated with persistent
activity of extracellular signal-regulated kinase or Akt kinase
pathways. Clin Cancer Res. 9:2316–2326. 2003.
|
|
18
|
Dobashi Y, Suzuki S, Kimura M, et al:
Paradigm of kinase-driven pathway downstream of epidermal growth
factor receptor/Akt in human lung carcinomas. Human Pathol.
42:214–226. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yu Y, Yoon SO, Poulogiannis G, et al:
Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate
that negatively regulates insulin signaling. Science.
332:1322–1326. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hsu PP, Kang SA, Rameseder J, et al: The
mTOR-regulated phosphoproteome reveals a mechanism of
mTORC1-mediated inhibition of growth factor signaling. Science.
332:1317–1322. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shi Y, Yan H, Frost P, Gera J and
Lichtenstein A: Mammalian target of rapamycin inhibitors activate
the AKT kinase in multiple myeloma cells by up-regulating the
insulin-like growth factor receptor/insulin receptor
substrate-1/phosphatidylinositol 3-kinase cascade. Mol Cancer Ther.
4:1533–1540. 2005. View Article : Google Scholar
|
|
22
|
Goudar RK, Shi Q, Hjelmeland MD, et al:
Combination therapy of inhibitors of epidermal growth factor
receptor/vascular endothelial growth factor receptor 2 (AEE788) and
the mammalian target of rapamycin (RAD001) offers improved
glioblastoma tumor growth inhibition. Mol Cancer Ther. 4:101–112.
2005.
|
|
23
|
Gemmill RM, Zhou M, Costa L, Korch C,
Bukowski RM and Drabkin HA: Synergistic growth inhibition by Iressa
and rapamycin is modulated by VHL mutations in renal cell
carcinoma. Br J Cancer. 92:2266–2277. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Price KA, Azzoli CG, Krug LM, et al: Phase
II trial of gefitinib and everolimus in advanced non-small cell
lung cancer. J Thorac Oncol. 5:1623–1629. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
La Monica S, Galetti M, Alfieri RR, et al:
Everolimus restores gefitinib sensitivity in resistant non-small
cell lung cancer cell lines. Biochem Pharmacol. 78:460–468.
2009.PubMed/NCBI
|
|
26
|
Vienna L, Fortunato B, Lorenza P, et al:
Phosphoinositide-3-kinase catalytic alpha and KRAS mutations are
important predictors of resistance to therapy with epidermal growth
factor receptor tyrosine kinase inhibitors in patients with
advanced non-small cell lung cancer. J Thorac Oncol. 6:707–715.
2011. View Article : Google Scholar
|
|
27
|
Elizabeth B, Alexandra E, Eric B, et al:
Rapamycin synergizes with the epidermal growth factor receptor
inhibitor erlotinib in non-small cell lung, pancreatic, colon, and
breast tumors. Mol Cancer Ther. 5:2627–2684. 2006.PubMed/NCBI
|
|
28
|
Magali R, Paul P, Amelie D, et al:
Mechanisms underlying resistance to cetuximab in the HNSCC cell
line: role of AKT inhibition in by passing this resistance. Int J
Oncol. 38:189–200. 2011.PubMed/NCBI
|
|
29
|
Tamburini J, Green AS and Bardet V:
Protein synthesis is resistant to rapamycin and constitutes a
promising therapeutic target in acute myeloid leukemia. Blood.
114:1618–1627. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mohseni M and Park BH: PIK3CA and KRAS
mutations predict for response to everolimus therapy: now that's
RAD001. J Clin Invest. 120:2655–2658. 2010. View Article : Google Scholar : PubMed/NCBI
|