Introduction
Metastasis of cancer cells is the most common reason
for therapy failure. Although researchers have proposed a broad
spectrum of mechanisms for cell migration and invasion, cancer
therapeutics designed to block tumor progression by modulating
these mechanisms have not yet proven effective in clinical trials.
This may reflect the fact that cancer cells can operate different
migration programs under different environmental conditions
(1). Therefore, comprehensive
understanding of the molecular and cellular underpinnings of cancer
cell migration/invasion to better understand cancer metastasis and
support the development of new treatment strategies is needed.
Circadian clocks, which are the body’s molecular
time-keeping systems, form the basis for the daily rhythms of
multiple biochemical, physiological and behavioral processes in
most organisms (2,3). Importantly, substantial evidence
suggests that dysfunctions of the circadian system are associated
with pathological conditions, such as the formation and progression
of cancer. For example, an increased risk of breast cancer was
reportedly associated with female workers who were exposed to
chronic disruptions of the sleep-wake cycle, such as flight
attendants and rotating or permanent night-shift workers (4–6).
Numerous other epidemiological studies have shown that perturbation
of the normal circadian rhythm increases the risk of not only
breast cancer, but also prostate, colorectal and endometrial
cancers (7).
In mammals, the circadian system is regulated by a
set of core clock factors, including Bmal1, Clock, casein kinase
Iɛ, the cryptochromes (Cry1 and 2) and the periods (Per1-3), as
well as supplementary regulators such as RORα and REV-ERBα
(8–10). Per1 and Per2 are relatively well
characterized in terms of their roles in cancer. They are
reportedly downregulated in various types of human cancer (11–14),
and Per2 gene-deficient mice exhibit an increased rate of lymphoma
formation in response to ionizing radiation (15). At the molecular level, Per1 and Per2
are involved in the DNA damage response (16), and overexpression of either protein
inhibits cancer cell growth and increases the apoptotic rate
(16–18), supporting the notion that they
participate in tumor suppression. Aside from these findings,
however, there is little information regarding the molecular
linkage between circadian rhythms and tumor
formation/progression.
Bmal1 [brain and muscle aryl hydrocarbon receptor
nuclear translocator (ARNT)-like] is a central clock factor that
regulates the expression levels of the Cry and Per genes (19). Based on a recent report that
downregulation of Bmal1 promotes tumor growth in cell culture and
mice (20), we herein investigated
whether Bmal1 also influences the invasiveness of cancer cells. The
obtained data are presented in this study and the importance of our
findings is discussed.
Materials and methods
Antibodies and inhibitors
Antibodies were purchased from the following
institutions: anti-Bmal1 and anti-Akt from Santa Cruz Biotechnology
(Santa Cruz, CA, USA); anti-phosphoinositide 3-kinase (PI3K) from
Upstate Biotechnology (Lake Placid, NY, USA); anti-Bcl-w,
anti-PTEN, and anti-phospho-Akt from Cell Signaling Technology
(Danvers, MA, USA); anti-β-actin from Sigma-Aldrich (St. Louis, MO,
USA); and anti-MMP-2 from Calbiochem (La Jolla, CA, USA). The
synthetic inhibitors were obtained from Calbiochem.
Cell culture, transfection and
treatment
Human lung cancer cells (A549 and H1299) and glioma
cells (U251) were cultured in RPMI-1640 and DMEM, respectively,
supplemented with 10% heat-inactivated FBS. The Bmal1-expressing
pCMV-SPORT6 vector (Thermo Fisher Scientific, Rockford, IL, USA),
Bcl-w-expressing pcDNA3 vector (21), and siRNAs against Bmal1, Per3 and
RORα (Ambion, Austin, TX, USA) were introduced into cells using
Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the
manufacturer’s protocol. All transfections were performed
transiently, and transfectants were used for the indicated
experiments following 40–48 h of the recovery.
Western blot analysis
Cells were lysed on ice for 30 min in a buffer
containing 20 mM Tris-HCl (pH 7.4), 100 mM NaCl, 0.5% NP-40, 0.1 mM
Na3VO4, 50 mM NaF, 30 mM
Na4O7P2 · 10 H2O and a
protease inhibitor cocktail (GenDepot, Barker, TX, USA). To compare
the levels of secreted MMP-2, cells were cultured for 24 h in
serum-free medium and conditioned media were obtained. Proteins in
the lysates or conditioned media were resolved by SDS-PAGE, and
transferred to nitrocellulose filters (Millipore, Bedford, MA, USA)
using an ECL Semi-Dry Transfer unit (Amersham Life Sciences,
Uppsala, Sweden). The nitrocellulose filters were incubated with a
blocking buffer (10% non-fat dry milk and 0.05% Tween in PBS) for 1
h and then incubated overnight with primary antibodies at 4°C.
After incubation with secondary antibodies, peroxidase activity was
assessed using a chemiluminescence-based detection system (Thermo
Fisher Scientific).
Invasion assay
The invasion assay was performed as previously
described (21). Briefly, 0.2 ml of
transfected cells (1–1.75×105 cells/ml) in serum-free
medium were seeded onto the upper surfaces of Matrigel-coated
polycarbonate filters (BD Biosciences, Bedford, MA, USA) in a
modified Boyden chamber (Corning Inc., Corning, NY, USA). The lower
compartments of the chambers were filled with 0.6 ml of medium
supplemented with 10% heat-inactivated FBS. After 24 h of
incubation at 37°C and 5% CO2, the cells that had
migrated to the lower surface of the filter were fixed, stained
using a Diff-Quick kit (Fisher Scientific, Pittsburgh, PA, USA),
and counted under a microscope.
PI3K activity assay
The PI3K assay was conducted using a PI3K ELISA kit
(Echelon Biosciences, Salt Lake City, UT, USA) according to the
manufacturer’s protocol. Briefly, cells were lysed with ice-cold
lysis buffer (20 mM Tris-HCl, pH 7.4, 137 mM NaCl, 1 mM
CaCl2, 1 mM MgCl2, 1% NP-40, 1 mM PMSF and
0.1 mM sodium orthovanadate) for 20 min, and the cell lysates were
subjected to immunoprecipitation with an anti-PI3K antibody. The
immunoprecipitated PI3K was incubated with the PI(4,5)P2
substrate, and the generated PI(3,4,5)P3 was
assayed by competitive ELISA.
Statistical analysis
All experiments were carried out at least three
times to obtain means and standard deviations as shown in the
graphs in the figures. Results were analyzed for statistical
significance using the Student’s t-test. Differences were
considered significant at P<0.05.
Results
Bmal1 suppresses cancer cell
invasion
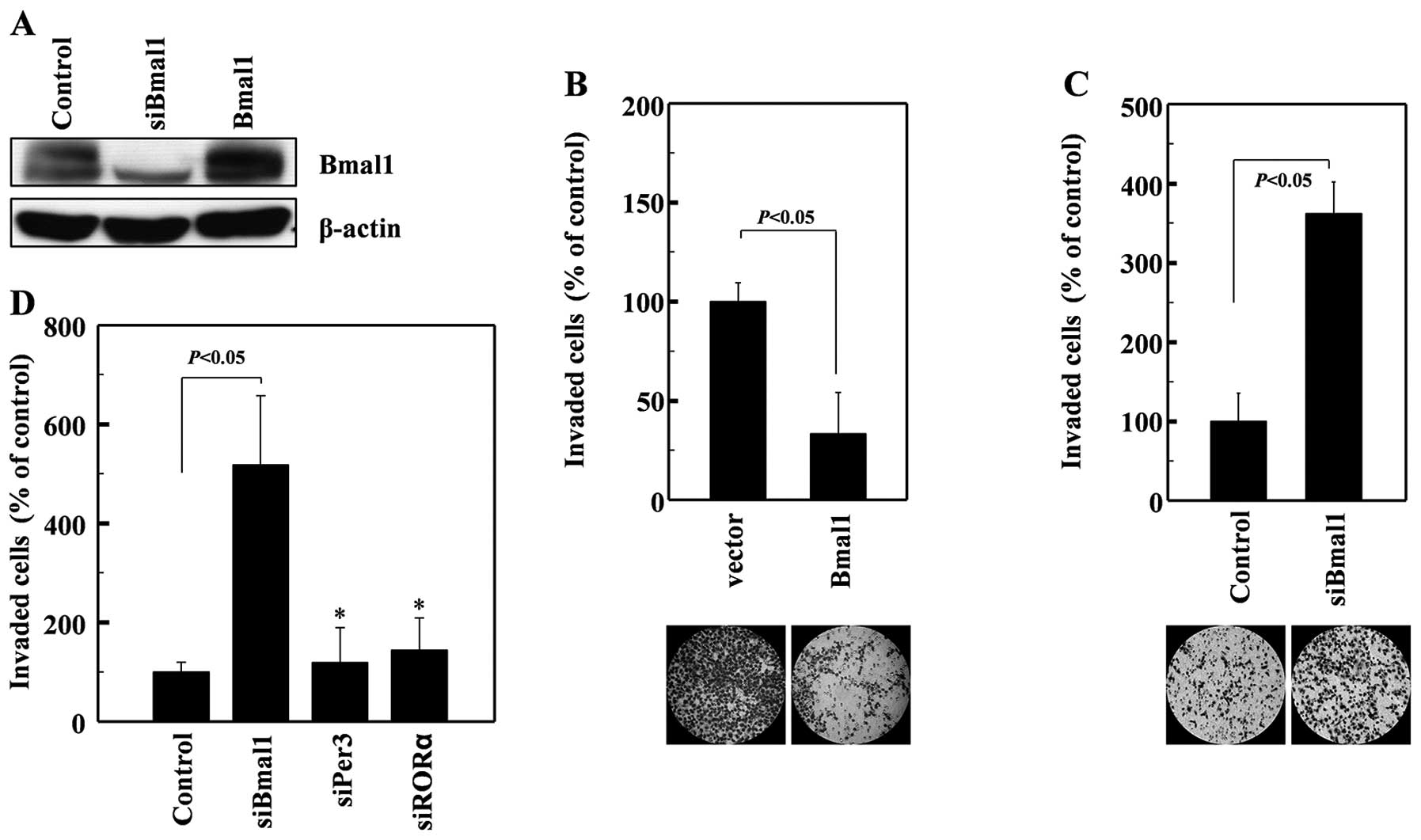
To determine whether Bmal1 influences cellular
invasiveness, we overexpressed Bmal1 in human A549 lung cancer
cells (Fig. 1A). This resulted in a
dramatic reduction in cellular invasiveness, as analyzed on
Matrigel-coated polycarbonate filters (Fig. 1B). Conversely, the siRNA-mediated
reduction in endogenous Bmal1 levels (Fig. 1A) promoted cell invasion (Fig. 1C). These findings suggest that Bmal1
suppresses cancer invasion. These effects were not mimicked by
siRNAs against Per3 or RORα (Fig.
1D), suggesting that this invasion-suppressing activity is not
common to all circadian factors.
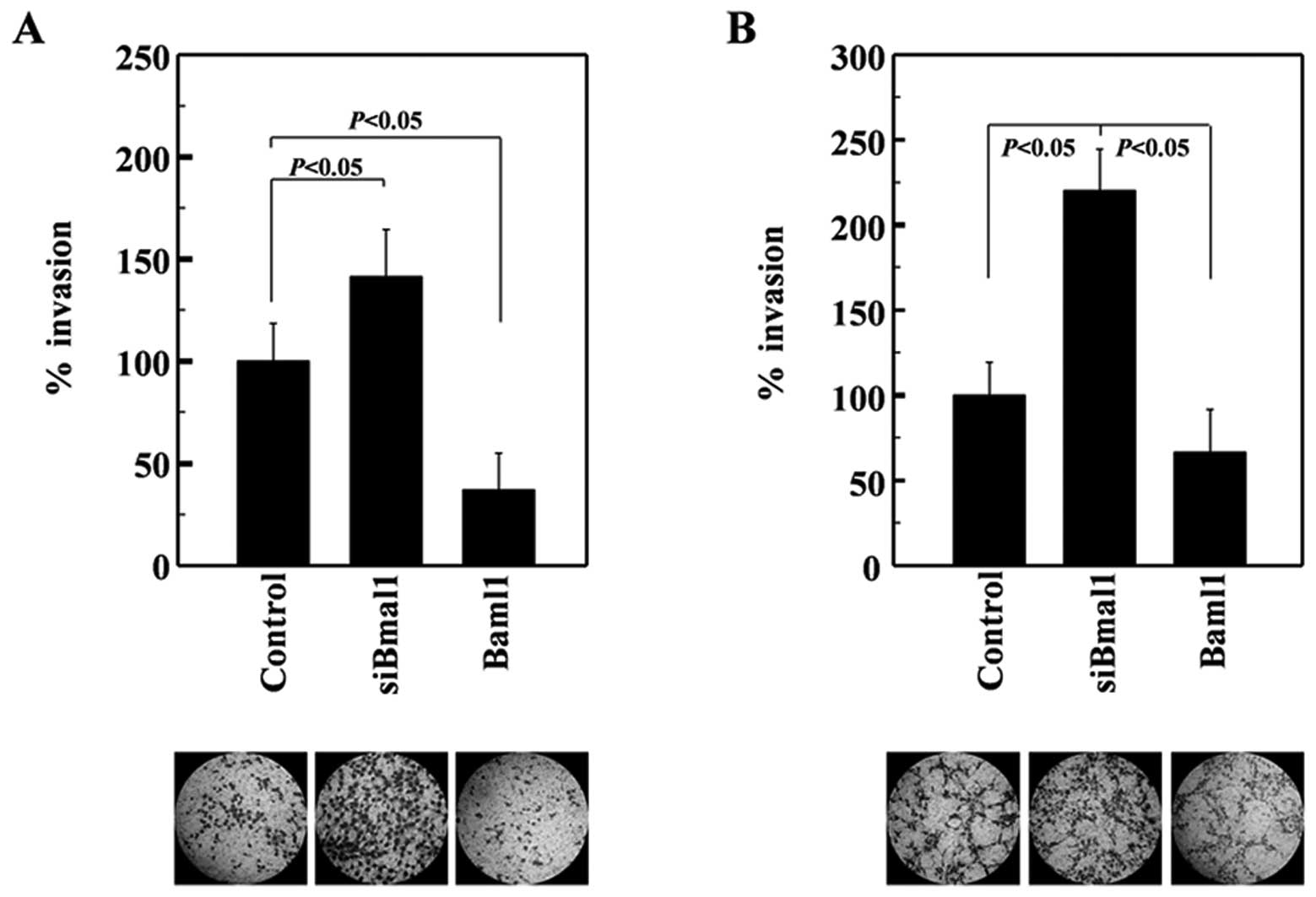
A549 cells express wild-type p53 (22). Given that this tumor suppressor is
mutated in >50% of human tumors (23), we next investigated whether Baml1
suppresses the invasiveness of H1299 lung cancer cells, which were
used as representative p53-null cells (24). The invasiveness of H1299 cells was
also enhanced and suppressed by siRNA-mediated Baml1 knockdown and
its overexpression, respectively (Fig.
2A). Therefore, Baml1 appears to suppress lung cancer cell
invasion regardless of p53 expression. To determine whether Bmal1
exerts this function in cancer cells from other organs, we examined
U251 glioma cells. The invasiveness of these cells was again
increased and decreased by Bmal1 knockdown and overexpression,
respectively (Fig. 2B), suggesting
that Bmal1 suppresses the invasion of glioma cells. Together, these
results suggest that Bmal1 reduces the invasiveness of multiple
cancer types in a p53-independent manner.
Bmal1 suppresses the PI3K-Akt-MMP-2
pathway
The PI3K-Akt-MMP-2 pathway is involved in promoting
cell invasion under many experimental conditions (25–27).
Here, we found that PI3K activity was elevated in Baml1-knockdown
cells (Fig. 3A), while no change
was evident in the levels of the p85 subunit of PI3K or PTEN, an
endogenous inhibitor of PI3K (28)
(Fig. 3B). This suggests that Baml1
knockdown induces PI3K activation. Bmal1 knockdown also elevated
Akt phosphorylation and MMP-2 protein levels, suggesting that Baml1
may suppress the PI3K-Akt-MMP-2 invasion pathway. To confirm this,
cells were treated with the Baml1-targeting siRNAs in the presence
or absence of inhibitors for PI3K (LY294002), Akt (Akt inhibitor)
and MMP-2 (OA-Hy). As expected, these inhibitors attenuated the
Bmal1 siRNA-induced invasion of A549 cells (Fig. 3C), suggesting that Bmal1 reduces
cellular invasiveness by suppressing the PI3K-Akt-MMP-2
pathway.
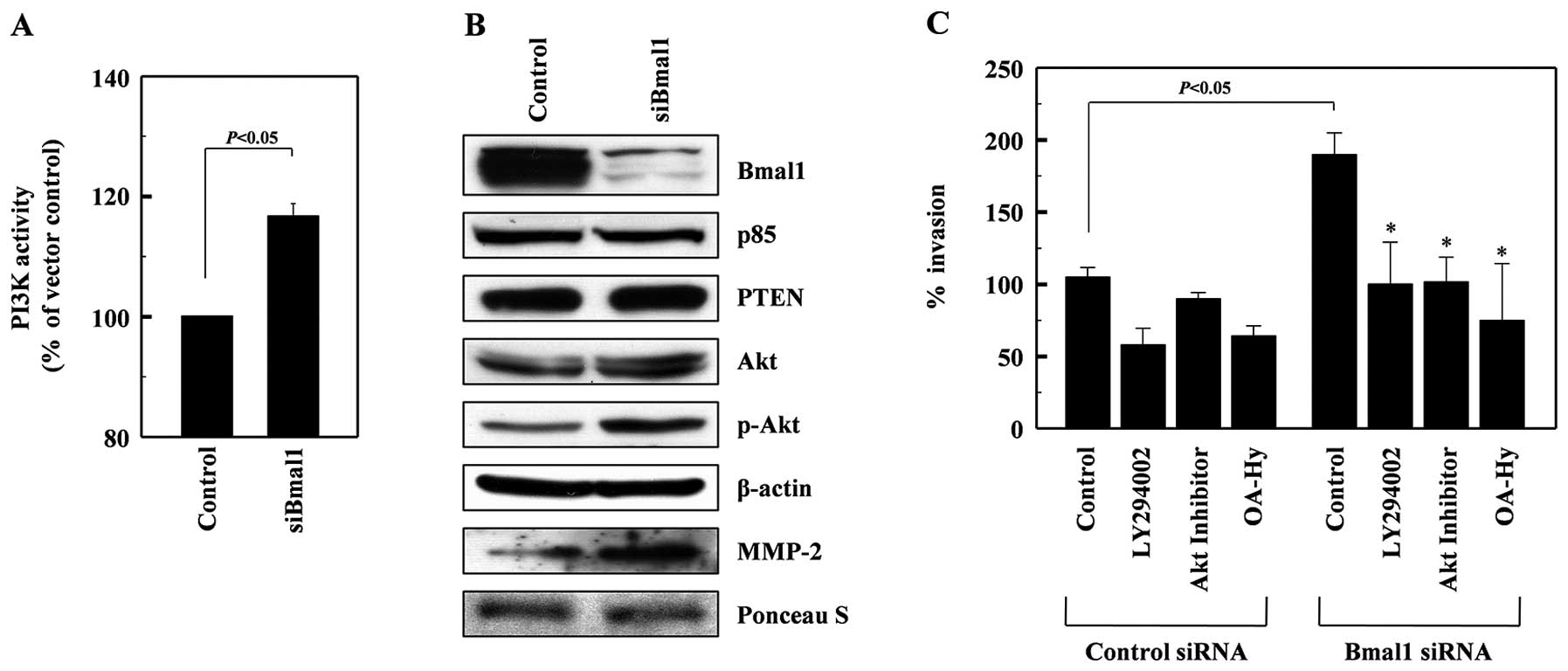 | Figure 3Bmal1 suppresses the PI3K-Akt-MMP-2
pathway. (A) Cell lysates from control or Bmal1-knockdown A549
cells were analyzed for PI3K activity by competitive ELISA. (B) The
levels of Bmal1, the p85 subunits of PI3K, PTEN, Akt, phospho-Akt,
and β-actin in the lysates were analyzed by western blotting.
Alternatively, conditioned media were prepared using
Bmal1-knockdown cells, and MMP-2 levels were analyzed by western
blotting. Protein loading was verified by Ponceau S staining. (C)
Control and Bmal1-knockdown cells were incubated in the presence or
absence of LY294002 (5 μM), Akt inhibitor (5 μM), or OA-Hy (10 μM)
for 24 h, and cellular invasiveness was compared.
*P<0.05 vs. Bmal1 siRNA control. |
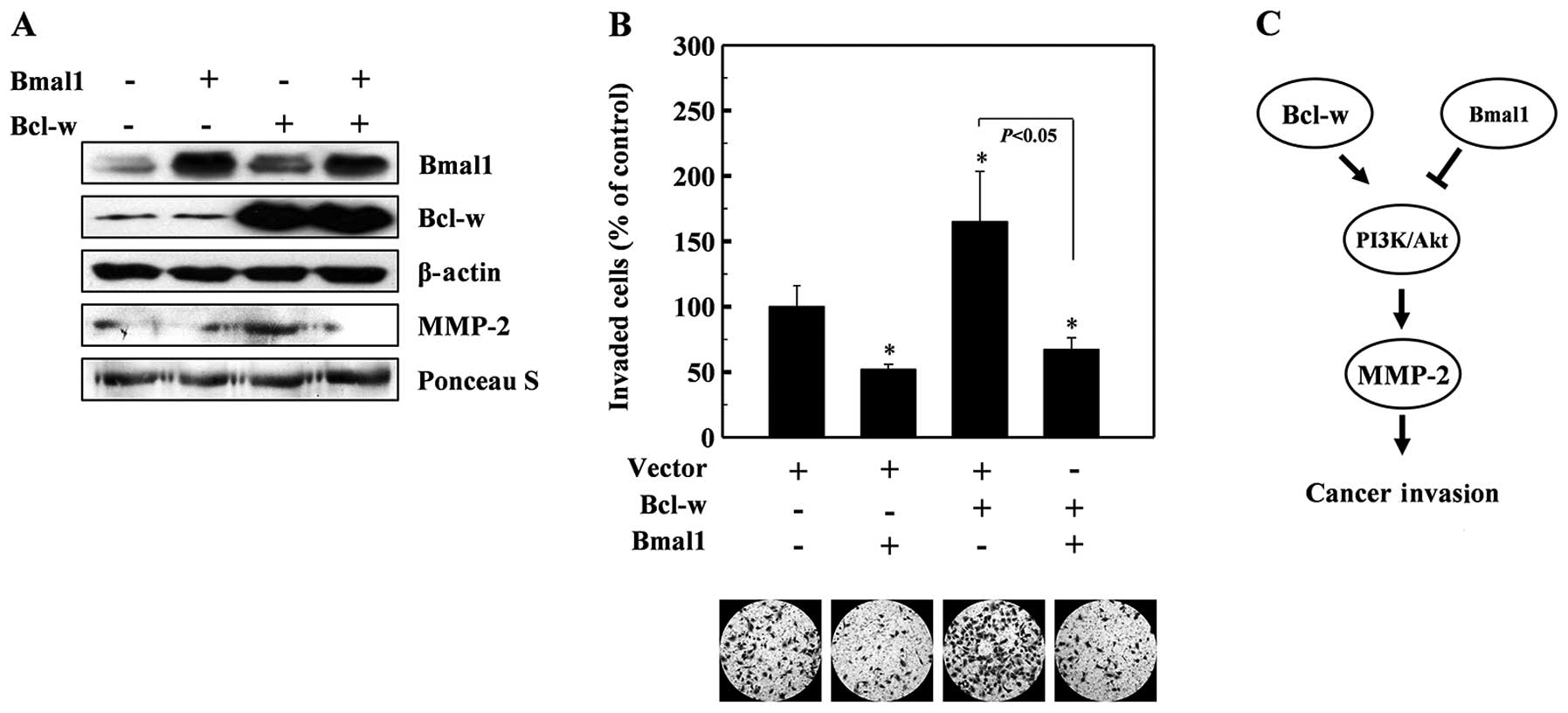
Bmal1 antagonizes the invasion-promoting
action of Bcl-w
Bcl-w is a pro-survival member of the Bcl-2 family
of proteins (29), which are
upregulated in various types of cancer cells (30,31).
Bcl-w protects cells from apoptotic stimuli and also promotes the
invasion of cancer cells by activating the PI3K-Akt-MMP-2 pathway
(26,27). Therefore, we aimed to ascertain
whether Bmal1 antagonizes the invasion-promoting action of Bcl-w.
To accomplish this, we co-expressed Bmal1 and Bcl-w in lung cancer
cells, and examined MMP-2 levels and cellular invasiveness. As
reported previously (26,27), Bcl-w overexpression enhanced the
levels of MMP-2 (Fig. 4A) and
cellular invasiveness (Fig. 4B).
However, both of these events were abolished by the co-expression
of Bmal1 (Fig. 4A and B),
suggesting that Bmal1 acts against Bcl-w to reduce cellular
invasiveness.
Discussion
In the present study, we demonstrated that Bmal1
suppresses cancer cell invasion. This was demonstrated by both
Bmal1 overexpression and RNA interference experiments. This effect
of Bmal1 was observed in lung cancer and glioma cells, indicating
that Bmal1 reduces the invasiveness of multiple types of cancer. We
believe that Bmal1 exerts this function by suppressing the invasion
pathway that involves PI3K, Akt and MMP-2. This was initially
suggested by the observation that Bmal1 knockdown increased the
levels of PI3K activity, Akt phosphorylation, and MMP-2 protein,
and was further confirmed by our observation that Bmal1
knockdown-induced cell invasion was blocked by inhibitors of PI3K,
Akt, and MMP-2. Although certain circadian factors have been
reported to regulate tumor growth and resistance (32), this is the first report that such a
factor can also regulate cancer cell invasiveness.
Previous studies have shown that Bmal1 is
downregulated in certain types of cancer (33) and suppresses tumor growth in cell
culture and animal models (20).
Together, these reports and our present results suggest that Bmal1
functions as a tumor suppressor. This view is further supported by
our finding that Baml1 acts against the oncogene, Bcl-w, to prevent
MMP-2 accumulation and cell invasion. As Bcl-w activates the
PI3K-Akt-MMP-2 pathway, we propose that Bmal1 and Bcl-w may
reciprocally regulate this invasion pathway (Fig. 4C).
The theory that Bmal1 acts as a tumor suppressor led
us to examine the possible relationship between Bmal1 and p53, a
well-characterized tumor suppressor that also suppresses cancer
cell invasion (34). However, we
found that knockdown of Bmal1 elevates the invasiveness of
p53-expressing and p53-null lung cancer cells to almost equal
extents. Therefore, Bmal1 appears to act as a tumor suppressor via
a p53-independent mechanism, at least for the regulation of
cellular invasiveness.
In conclusion, we showed that Bmal1 attenuates
cancer cell invasion by suppressing the PI3K-Akt-MMP-2 pathway.
This result supports the notion that there is a tight molecular
link between circadian rhythms and tumor formation/progression.
Acknowledgements
This study was supported by a grant from the Nuclear
Research and Development Program of the National Research
Foundation of Korea (NRF) funded by the Korean Government (MEST)
(2012M2A2A7010459), and in part by the Basic Science Research
Program through the NRF (2012-0000482).
References
|
1
|
Friedl P and Alexander S: Cancer invasion
and the microenvironment: plasticity and reciprocity. Cell.
147:992–1009. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mohawk JA, Green CB and Takahashi JS:
Central and peripheral circadian clocks in mammals. Annu Rev
Neurosci. 35:445–462. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Takahashi JS, Hong HK, Ko CH and McDearmon
EL: The genetics of mammalian circadian order and disorder:
implications for physiology and disease. Nat Rev Genet. 9:764–775.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rafnsson V, Tulinius H, Jónasson JG and
Hrafnkelsson J: Risk of breast cancer in female flight attendants:
a population-based study (Iceland). Cancer Causes Control.
12:95–101. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Viswanathan AN, Hankinson SE and
Schernhammer ES: Night shift work and the risk of endometrial
cancer. Cancer Res. 67:10618–10622. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schernhammer ES, Kroenke CH, Laden F and
Hankinson SE: Night work and risk of breast cancer. Epidemiology.
17:108–111. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fu L and Lee CC: The circadian clock:
pacemaker and tumour suppressor. Nat Rev Cancer. 3:350–361. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dunlap JC: Molecular bases for circadian
clocks. Cell. 96:271–290. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Preitner N, Damiola F, Lopez-Molina L,
Zakany J, Duboule D, Albrecht U and Schibler U: The orphan nuclear
receptor REV-ERBalpha controls circadian transcription within the
positive limb of the mammalian circadian oscillator. Cell.
110:251–260. 2002. View Article : Google Scholar
|
|
10
|
Sato TK, Panda S, Miraglia LJ, Reyes TM,
Rudic RD, McNamara P, Naik KA, FitzGerald GA, Kay SA and Hogenesch
JB: A functional genomics strategy reveals Rora as a component of
the mammalian circadian clock. Neuron. 43:527–537. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gery S, Gombart AF, Yi WS, Koeffler C,
Hofmann WK and Koeffler HP: Transcription profiling of C/EBP
targets identifies Per2 as a gene implicated in myeloid leukemia.
Blood. 106:2827–2836. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Winter SL, Bosnoyan-Collins L, Pinnaduwage
D and Andrulis IL: Expression of the circadian clock genes Per1 and
Per2 in sporadic and familial breast tumors. Neoplasia. 9:797–800.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yeh KT, Yang MY, Liu TC, Chen JC, Chan WL,
Lin SF and Chang JG: Abnormal expression of period 1 (PER1) in
endometrial carcinoma. J Pathol. 206:111–120. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pogue-Geile KL, Lyons-Weiler J and
Whitcomb DC: Molecular overlap of fly circadian rhythms and human
pancreatic cancer. Cancer Lett. 243:55–57. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fu L, Pelicano H, Liu J, Huang P and Lee
C: The circadian gene period2 plays an important role in tumor
suppression and DNA damage response in vivo. Cell. 111:41–50. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gery S, Komatsu N, Baldjyan L, Yu A, Koo D
and Koeffler HP: The circadian gene per1 plays an important role in
cell growth and DNA damage control in human cancer cells. Mol Cell.
22:375–382. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hua H, Wang Y, Wan C, Liu Y, Zhu B, Wang
X, Wang Z and Ding JM: Inhibition of tumorigenesis by intratumoral
delivery of the circadian gene mPer2 in C57BL/6 mice. Cancer Gene
Ther. 14:815–818. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Oda A, Katayose Y, Yabuuchi S, Yamamoto K,
Mizuma M, Shirasou S, Onogawa T, Ohtsuka H, Yoshida H, Hayashi H,
Rikiyama T, Kim H, Choe Y, Kim K, Son H, Motoi F, Egawa S and Unno
M: Clock gene mouse period2 overexpression inhibits growth of human
pancreatic cancer cells and has synergistic effect with cisplatin.
Anticancer Res. 29:1201–1209. 2009.PubMed/NCBI
|
|
19
|
King DP, Zhao Y, Sangoram AM, Wilsbacher
LD, Tanaka M, Antoch MP, Steeves TD, Vitaterna MH, Kornhauser JM,
Lowrey PL, Turek FW and Takahashi JS: Positional cloning of the
mouse circadian clock gene. Cell. 89:641–653. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zeng ZL, Wu MW, Sun J, Sun YL, Cai YC,
Huang YJ and Xian LJ: Effects of the biological clock gene Bmal1 on
tumour growth and anti-cancer drug activity. J Biochem.
148:319–326. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim EM, Kim J, Park JK, Hwang SG, Kim WJ,
Lee WJ, Kang SW and Um HD: Bcl-w promotes cell invasion by blocking
the invasion-suppressing action of Bax. Cell Signal. 24:1163–1172.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lehman TA, Bennett WP, Metcalf RA, Welsh
JA, Ecker J, Modali RV, Ullrich S, Romano JW, Appella E, Testa JR,
Gerwin BI and Harris CC: p53 mutations, ras mutations, and p53-heat
shock 70 protein complexes in human lung carcinoma cell lines.
Cancer Res. 51:4090–4096. 1991.PubMed/NCBI
|
|
23
|
Soussi T, Dehouche K and Béroud C: p53
website and analysis of p53 gene mutations in human cancer: forging
a link between epidemiology and carcinogenesis. Hum Mutat.
15:105–113. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Roberson RS, Kussick SJ, Vallieres E, Chen
SY and Wu DY: Escape from therapy-induced accelerated cellular
senescence in p53-null lung cancer cells and in human lung cancers.
Cancer Res. 65:2795–2803. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shukla S, Maclennan GT, Hartman DJ, Fu P,
Resnick MI and Gupta S: Activation of PI3K-Akt signaling pathway
promotes prostate cancer cell invasion. Int J Cancer.
121:1424–1432. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bae IH, Park MJ, Yoon SH, Kang SW, Lee SS,
Choi KM and Um HD: Bcl-w promotes gastric cancer cell invasion by
inducing matrix metalloproteinase-2 expression via phosphoinositide
3-kinase, Akt, and Sp1. Cancer Res. 66:4991–4995. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bae IH, Yoon SH, Lee SB, Park JK, Ho JN
and Um HD: Signaling components involved in Bcl-w-induced migration
of gastric cancer cells. Cancer Lett. 277:22–28. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kong D and Yamori T: Advances in
development of phosphatidylinositol 3-kinase inhibitors. Curr Med
Chem. 16:2839–2854. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cory S and Adams JM: The Bcl-2 family:
regulators of the cellular life-or-death switch. Nat Rev Cancer.
2:647–656. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lee HW, Lee SS, Lee SJ and Um HD: Bcl-w is
expressed in a majority of infiltrative gastric adenocarcinomas and
suppresses the cancer cell death by blocking stress-activated
protein kinase/c-Jun NH2-terminal kinase activation. Cancer Res.
63:1093–1100. 2003.
|
|
31
|
Hoelzinger DB, Mariani L, Weis J, Woyke T,
Berens TJ, McDonough WS, Sloan A, Coons SW and Berens ME: Gene
expression profile of glioblastoma multiforme invasive phenotype
points to new therapeutic targets. Neoplasia. 7:7–16. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen-Goodspeed M and Lee CC: Tumor
suppression and circadian function. J Biol Rhythms. 22:291–298.
2007. View Article : Google Scholar
|
|
33
|
Mazzoccoli G, Panza A, Valvano MR, Palumbo
O, Carella M, Pazienza V, Biscaglia G, Tavano F, Di Sebastiano P,
Andriulli A and Piepoli A: Clock gene expression levels and
relationship with clinical and pathological features in colorectal
cancer patients. Chronobiol Int. 28:841–851. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Muller PA, Vousden KH and Norman JC: p53
and its mutants in tumor cell migration and invasion. J Cell Biol.
192:209–218. 2011. View Article : Google Scholar : PubMed/NCBI
|