Introduction
Nasopharyngeal carcinoma (NPC), which has the
highest incidence in Southeast Asia, remains one of the leading
causes of cancer-related mortality in the Cantonese region of
Southern China. Radiotherapy is the major treatment for NPC
(1). However, radioresistance
remains a serious obstacle to successful treatment in many cases
(2,3). Thus, an effective way to improve
radiation sensitization of nasopharyngeal carcinoma is by
identifying the mechanisms involved in NPC radiation resistance.
Autophagy is a catabolic process involved in cell growth,
development and homeostasis, maintaining a balance between the
synthesis, degradation and subsequent recycling of cellular
products. The process starts with the formation of the
autophagosome or autophagic vacuole. The vacuole membrane then
fuses with the lysosomal compartment to deliver the contents into
the organelle lumen, where they are degraded and the resulting
macromolecules are recycled (4,5).
Recent studies have revealed the importance of autophagy in the
immune response, inflammatory response, cardiovascular disease,
cancer and neurodegenerative disease (6–9).
However, the major role that autophagy plays in cancer is
controversial (10). Genetic
knockout of autophagy-related genes enhances the development of
spontaneous malignancies whereas mice deficient in
autophagy-related genes exhibit sensitivity to radiotherapy,
chemotherapy or immunotherapy (10–13).
Poly(ADP-ribose) polymerase-1 (PARP-1), activated by
DNA strand breaks, participates in the DNA repair process
physiologically (14). PARP-1 has
been implicated in the G2/M cell cycle checkpoint and has been
shown to play an important role in the recovery of DNA damage in
in vivo studies. Specifically, PARP-1 has been demonstrated
to be an important mediator of DNA base excision repair, which is
important in the repair of single-stranded breaks. Moreover, PARP-1
is also known to bind the more lethal double-stranded DNA breaks
(15–17). IR induces DNA strand breaks, which
mediate its cytotoxic effects. Excessive activation of PARP-1
mediates ionizing radiation (IR)-induced cell death under the
status of oxidative stress and DNA damage (18,19).
However, it remains elusive whether and how PARP-1 activation is
involved in autophagy and the exact function of PARP-1-mediated
autophagy under oxidative stress and DNA damage in CNE-2 cells.
Recently, a study demonstrated that IR induces autophagy through a
novel autophagy signaling mechanism linking PARP-1 activation to
the mammalian target of rapamycin (mTOR) pathway in prostate cancer
cell (20).
Questions concerning the role that autophagy plays
in response to IR in CNE-2 cells and whether it promotes cell
survival or induces cell death and the methods to promote
radiosensitivity by regulating autophagy are yet unanswered. Thus,
it is not surprising that the bulk of research has focused on the
regulation of autophagy in a variety of models (8,11,20–26).
Our data illustrate the importance of IR-induced autophagy, and we
demonstrated that autophagic inhibition may be used as a new method
to promote radiosensitivity. PARP-1-mediated autophagy plays a
cytoprotective role in IR-induced CNE-2 cell death as suppression
of autophagy greatly sensitizes IR-induced cell death.
Materials and methods
Cell culture and irradiation
conditions
CNE-2, a human NPC cell lines, was purchased from
the Cancer Hospital of Shanghai Fudan University, and was cultured
in RPMI-1640 medium (HyClone, USA) supplemented with 10% fetal calf
serum (Gibco, USA), penicillin (100 U/ml), streptomycin (100 U/ml)
and maintained at 37°C in a humidified incubator with 5%
CO2. All irradiations were delivered using 6-MV X-rays
with a linear accelerator (Elekta, Sweden) with a dose rate of 220
cGy/min; SSD, 100 cm.
Reagents
Chloroquine diphosphate (CDP), an inhibitor of
autophagy, was purchased from MP Biomedicals (Santa Ana, CA, USA).
Rapamycin (RAPA), an inducer of cellular autophagy, was purchased
from LC Laboratories (Woburn, MA, USA). PARP-1 inhibitor, 3-amino
benzamide (3AB), was purchased from Sigma-Aldrich Co. (St. Louis,
MO, USA), and rabbit anti-human microtubule-associated protein 1
light chain 3 (MAP1LC3B) was purchased from Sigma. Rabbit
anti-PARP-1 primary antibody was purchased from Cell Signaling
Technology (Danvers, MA, USA), and the rabbit anti-PAR primary
antibody was supplied by BD Biosciences (San Jose, CA, USA). The
GAPDH primary antibody was purchased from Boster Co. (Wuhan,
China), and the goat anti-mouse/rabbit IgG secondary antibody was
purchased from the KPL Co.; Annexin V-FITC apoptosis necrosis
detection kit was purchased from Nanjing Kaiji Co. (Nanjing,
China).
Western blotting
CNE-2 cells were washed with ice-cold PBS twice and
lysed at 4°C. The lysates were centrifuged with 12,000 rpm at 4°C
for 30 min, at a centrifugal acceleration of 18,500 × g. Protein
content in the supernatants was determined using the BCA Protein
Assay kit (Beyotime, Nanjing, China). Equal amounts of protein (25
μg) were submitted to a 15% sodium dodecyl sulfate-polyacrylamide
gel and then electrotransferred onto PVDF membranes (Merck
Millipore, Billerica, MA, USA). After blocking for 2 h, the
membranes were incubated with appropriate antibodies overnight:
anti-LC3B (1:3,000), anti-PARP-1 (1:1,000), anti-PAR (1:1,500) and
anti-GAPDH (1:800). After washing and incubating with fluorescently
labeled goat anti-mouse/rabbit IgG secondary antibody (1:5,000),
the fluorescence intensity was detected using the Odyssey Infrared
Imaging System (LI-COR Biosciences, Lincoln, NE, USA).
Autophagosome detection
Transmission electron microscopy (TEM) (H-600 IV;
Hitachi, Tokyo, Japan) was utilized for analyzing the
ultrastructural images of autophagosomes and autolysosomes. CNE-2
cells were harvested by trypsinization, washed twice with PBS, and
fixed with ice-cold glutaraldehyde (3% in 0.1 M cacodylate buffer,
pH 7.4) for 24 h. The cells were post-fixed in OsO4 and
dehydrated in a graded series of 70 to 100% acetone and then
embedded in Epon 812. One micrometer thin sections were cut, double
stained by uranium tetraacetate and lead citrate trihydrate, and
viewed using TEM with a scanning attachment.
Flow cytometry
The samples were washed with phosphate-buffered
saline (PBS) twice and centrifuged at 1,500 rpm for 5 min, at a
centrifugal acceleration of 1,800 × g. The cells were suspended in
500 μl of binding buffer [Annexin V-fluorescein isothiocyanate
(FITC) kit; Kaiji, Nanjing, China], containing 5 μl of Annexin
V-FITC and 5 μl of PI for determination of phosphatidylserine
exposure on the outer plasma membrane. After incubation for 5–15
min at room temperature in a light-protected area, the samples were
quantified by flow cytometry (BD FACSCalibur, San Jose, CA,
USA).
Assessment of cell viability using MTT
assay
Cells were plated into 96-well plates
(1×103 cells/well, 200 μl cell suspension/well) and
cultured overnight to allow for cell attachment. After irradiation
(0, 24, 48 and 72 h) with 6 Gy X-rays, 20 μl of MTT (5 g/l) was
added into each well. The cells were then incubated at 37°C for 4
h, the supernatant was removed and 200 μl DMSO was added. When the
blue crystals were dissolved, a 96-well multiscanner autoreader
(Bio-Rad M550; Bio-Rad, Hercules, CA, USA) was used to measure the
absorbance value at 490 nm for each well. The survival rate was
calculated as follows: (OD values of the experimental samples/OD
values of the control) × 100%.
Clonogenic survival assay
CNE-2 cells were enzymatically dissociated with
trypsin and seeded at 200, 200, 400, 600, 1,000, 5,000 and 10,000
cells/well. The cells were then irradiated (2 ml of the cell
suspension/well) at room temperature with 6 MV X-rays with an
Elekta linear accelerator with a dose rate of 220 cGy/min,
accordingly, with the exposure dose corresponding to 0, 1, 2, 4, 6,
8 and 10 Gy. Fourteen days later, cells were fixed with 75% ethanol
and stained with Giemsa, and colonies containing >50 cells were
counted. The surviving fraction was calculated as: Survival
fraction (SF) = experimental group colony forming
efficiency/control group colony forming efficiency; Colony
formation efficiency (PE) = the number of colonies/plant cell
number. Experiments were conducted in triplicate. Survival curves
were fitted using the linear-quadratic model
(y=exp(-(α*x+β*x2))) using GraphPad Prism 5.0 (GraphPad
Software Inc., La Jolla, CA, USA).
Statistical analysis
Statistical data are presented as means ± standard
deviation (SD). All data were subjected to analysis of variance
(ANOVA) with significant differences among means identified by LSD
multiple range tests using SPSS 16.0 (SPSS, Inc., Chicago, IL,
USA). The criterion for statistical significance was taken as
P<0.05. At least three independent experiments were
performed.
Results
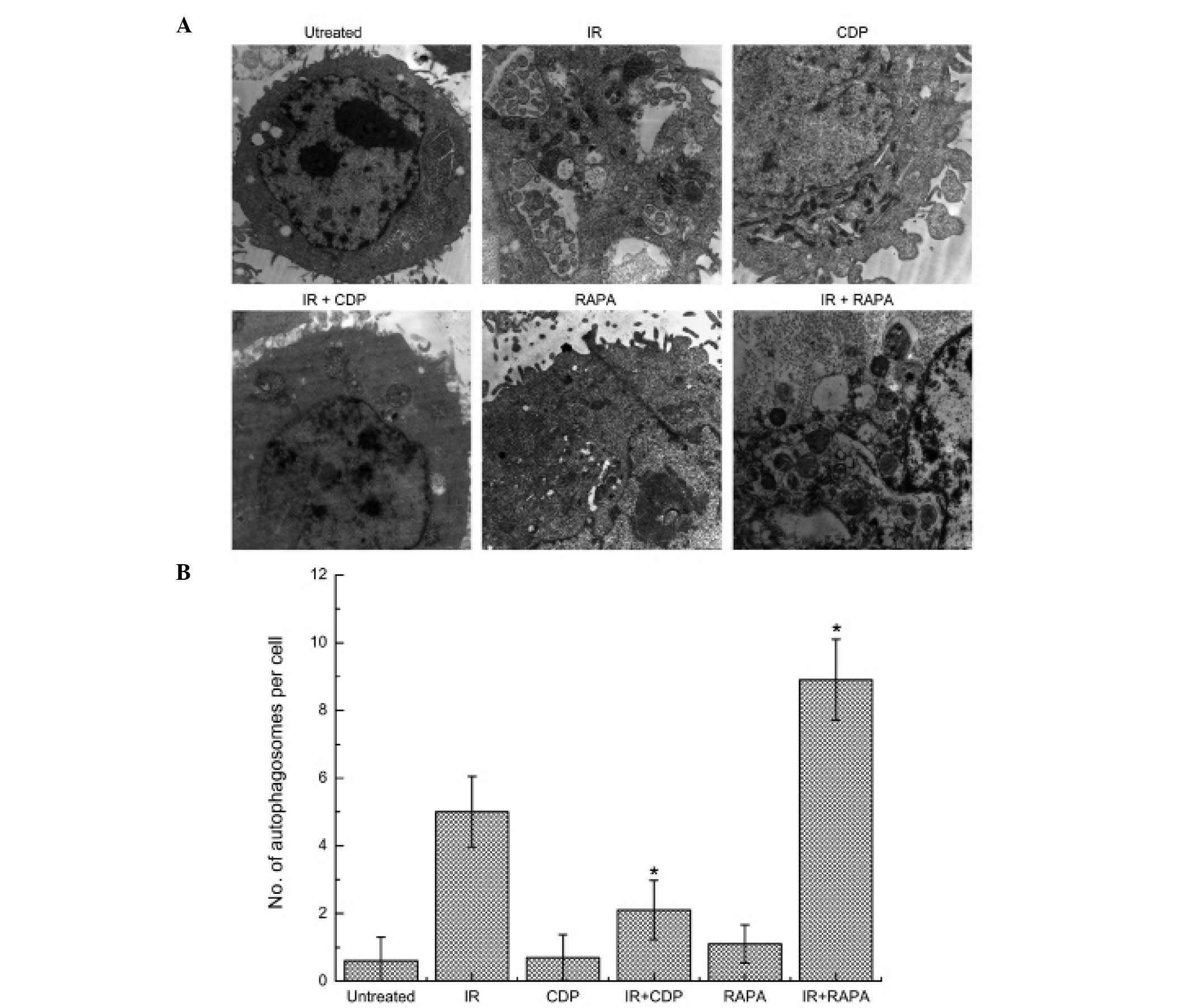
Irradiation-induced accumulation of
autophagosomes
Intracellular autophagosomes were observed by TEM in
CNE-2 cells after IR. CDP is widely used as an inhibitor of
autophagy and RAPA is widely used as an inductor of autophagy. We,
therefore, examined whether CDP or RAPA had effects on the
autophagy in CNE-2 cells. As shown in Fig. 1, subcellular structure analysis
revealed typical morphological features of autophagy in CNE-2 cells
24 h after treated with IR. Autophagosomes decreased in the CNE-2
cells treated with 10-Gy irradiation combined with 40 μM CDP, but
increased in cells treated with 10-Gy irradiation combined with 20
nM RAPA. However, in the untreated cells or cells treated with CDP
or RAPA alone for 24 h, normal nuclei surrounded by cytoplasm with
normal appearing mitochondria were presented, and only occasional
autophagosomes were observed.
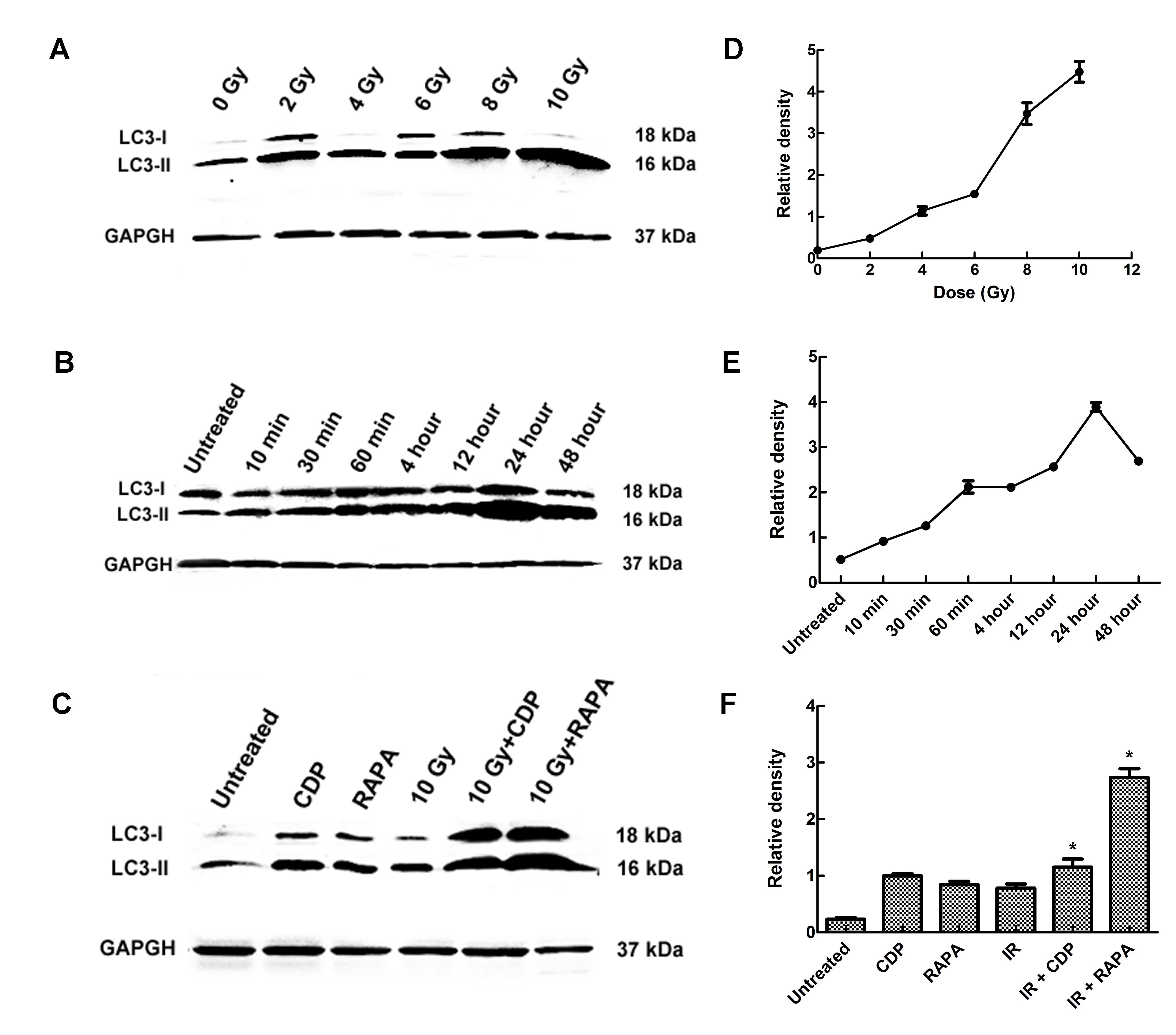
LC3-II expression level in CNE-2 cells
treated with CDP or RAPA combined with IR
To further determine the effect of IR on autophagy,
the conversion of cytosolic LC3-I to LC3-II was examined in the
CNE-2 cells. LC3-II protein level demonstrated a slight dose- and
time-dependent increase induced by IR in the CNE-2 cells (Fig. 2A and B). LC3-II levels were
correlated with the extent of autophagosome formation. Western blot
assays showed that the levels of LC3-II were strongly elevated in
the 10 Gy group and at 48 h following IR; a similar phenomenon was
observed in CNE-2 cells. Significant differences in the expression
level of LC3-II were found between the different experimental
groups (F=231.68, P<0.01). In the RAPA group, the LC3-II level
was increased, and in the CDP group the LC3-II level was also
increased, indicating that RAPA-induced autophagy occurred. CDP
inhibits the phenomenon of autophagy. CDP destroys the structure
and function of the lysosomal to inhibit autophagy, resulting in
autophagic lysosomal aggregation. LC3-II was not effectively
degraded, thus the LC3 expression level was upregulated in the
CDP-treated group, confirming that CDP inhibits autophagy. Compared
with the untreated group, the level of LC3-II was significantly
higher in the RAPA + 10 Gy group (P<0.01) and CDP group
(P<0.01) (Fig. 2C).
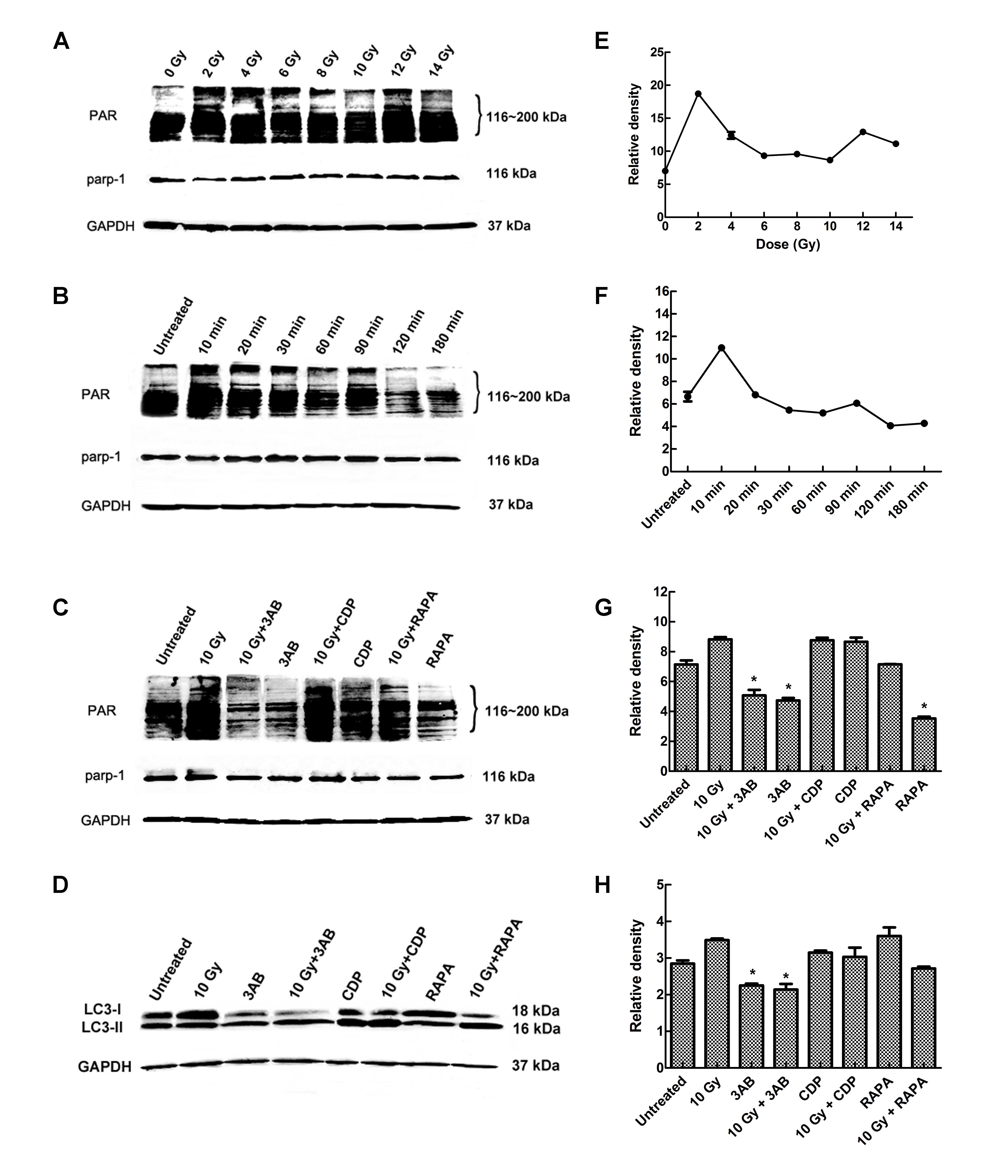
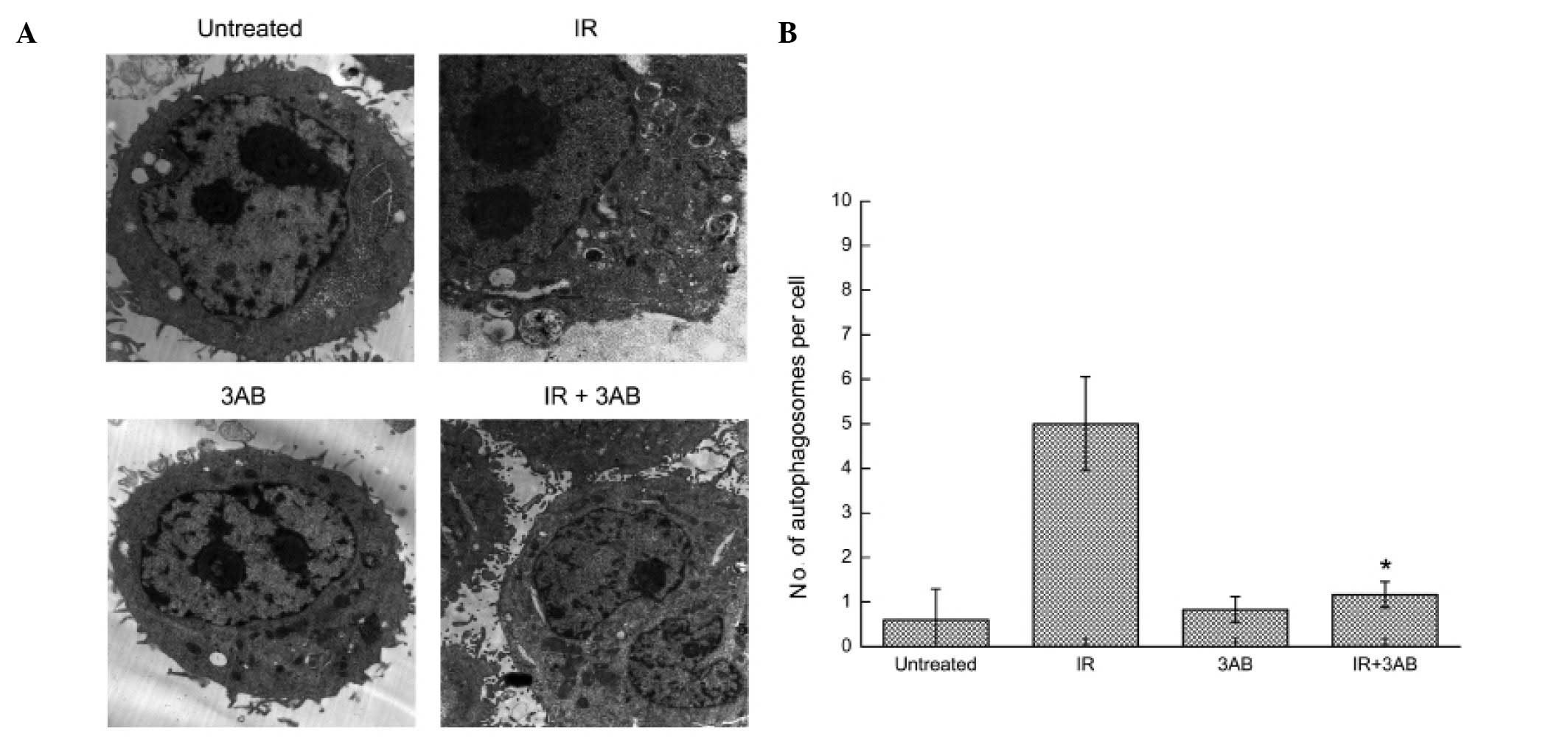
PARP-1 activation contributes to
irradiation-induced cell autophagy
PARP-1 is readily activated in response to DNA
damage and is well associated with cell death. As shown in Fig. 3A and B, increased formation of the
PAR polymer, a direct result of PARP-1 activation, was detected as
early as 10 min after IR exposure. To verify whether PARP-1
activation contributes to the cell autophagy induced by IR, cells
were treated with 10 Gy X-rays in the presence or absence of
3-amino benzamide (3AB), a specific PARP-1 inhibitor. Pretreatment
with 3AB significantly inhibited IR-induced PAR formation. PAR and
LC3-II both showed a certain degree of dose-dependence. Changes in
PAR content were noted obviously earlier than LC3-B changes, and
PAR was soon degraded. This suggests that PAR may be upstream of
the signaling pathways. In order to further verify if the PARP-1
signaling pathway is upstream, we used the PARP-1 chemical
inhibitor 3AB, autophagy inhibitor CDP and autophagy inducer RAPA
to pretreat CNE-2 cells, and 2 h after 10 Gy IR, cells were
collected and the protein was extracted, and changes in the levels
of LC3-I and -II and PAR were determined. (Fig. 3C and D). The results clearly
suggested that PARP-1 activation contributed to IR-induced cell
autophagy. TEM examination further confirmed this finding (Fig. 4).
Autophagy or PARP-1 inhibition increases
the apoptosis rate in CNE-2 cells following irradiation
IR combined with autophagy inhibitor or inducer
caused CNE-2 cell apoptosis; 48 h after IR, the apoptosis rates of
CNE-2 cells in the untreated group, CDP group, RAPA group, 3AB
group, IR group, IR + CDP group, IR + RAPA group and IR + 3AB
group, were 7.71±0.90, 8.29±1.60, 9.20±1.59, 9.91±0.75, 26.63±3.11,
31.28±1.58, 34.19±1.15 and 30.80±3.33%, respectively. Significant
differences in the apoptosis rates were observed between the groups
(F=109.69, P<0.01). The apoptosis rate was significantly higher
in the CDP + IR, RAPA + IR and 3AB + IR group than that in the IR
alone group (P<0.01) (Fig. 5A).
At 24 or 48 h after IR, a similar phenomenon was observed, but at
72 h after IR, RAPA or 3AB combined with IR promoted cell survival
rather than enhanced CNE-2 cell apoptosis (Fig. 5B).
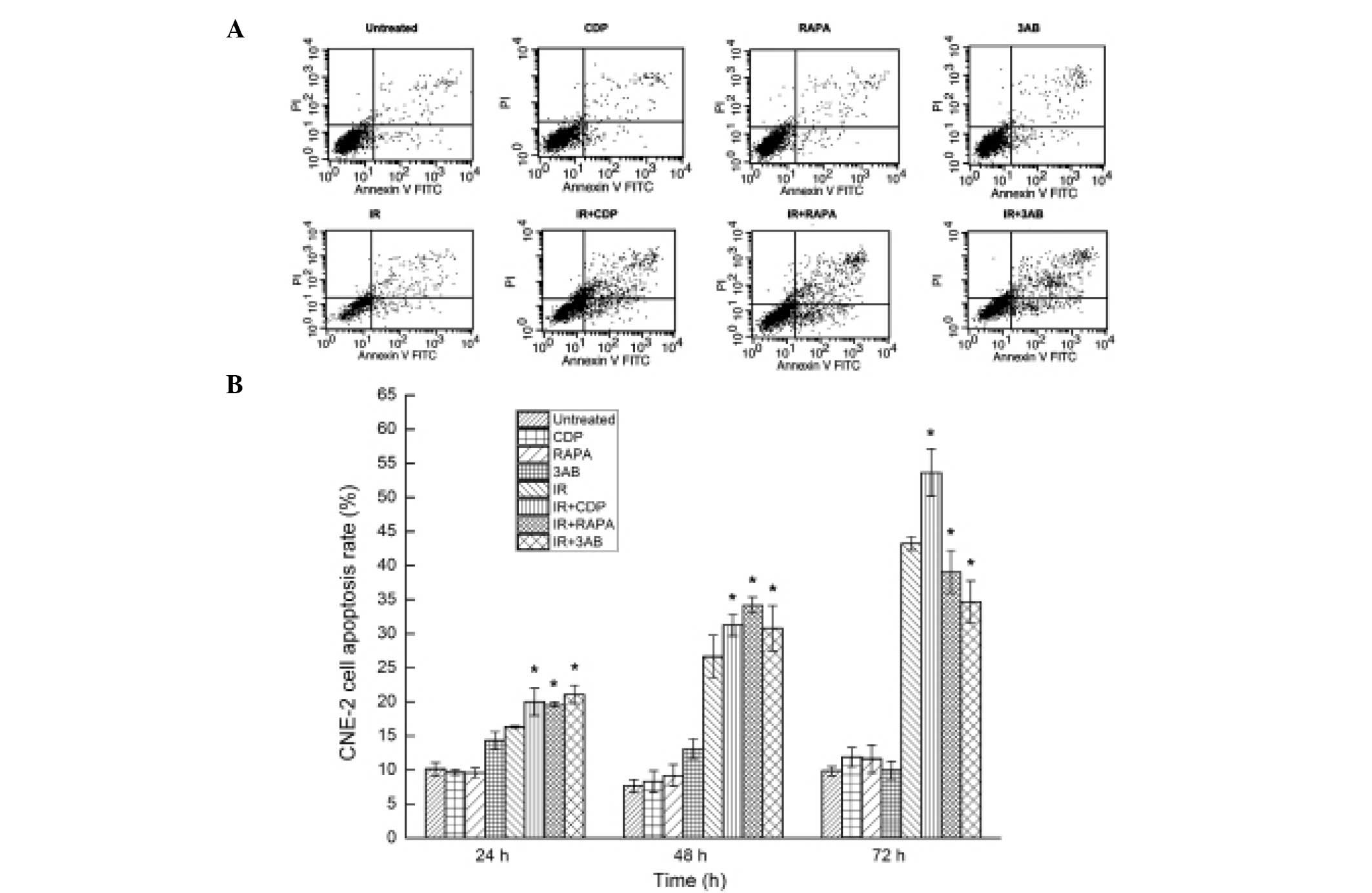 | Figure 5Analysis of CNE-2 cell apoptosis. (A)
Cell apoptosis was detected by flow cytometry 48 h after IR. The
apoptosis rate was evaluated by staining of cells with Annexin
V/PI. In the dual parameter dot plots cells undergoing early
apoptosis are shown in the lower-right quadrant (Annexin
V+/PI-), and cells undergoing late apoptosis
are shown in the upper right quadrant (Annexin
V+/PI+). The apoptosis rate was higher in the
IR + CDP, IR + RAPA and IR + 3AB groups, when compared with that in
the IR alone group; the difference was statistically significant
(P<0.05). (B) Cell apoptosis was detected by flow cytometry at
24, 48 and 72 h after IR, respectively. CDP combined with IR, RAPA
combined with IR, and 3AB combined with IR all enhanced CNE-2 cell
apoptosis at 24 and 48 h after irradiation. However, CDP combined
with IR continued to enhance cell apoptosis at 72 h after IR while
RAPA combined with IR and 3AB combined with IR promoted cell
survival rather than enhanced cell apoptosis. *P<0.05
vs. IR alone. CDP, chloroquine diphosphate; RAPA, rapamycin; 3AB,
3-amino benzamide; IR, ionizing radiation. |
Autophagy or PARP-1 inhibitor contributes
to inhibition of CNE-2 cell proliferation and survival rate after
irradiation
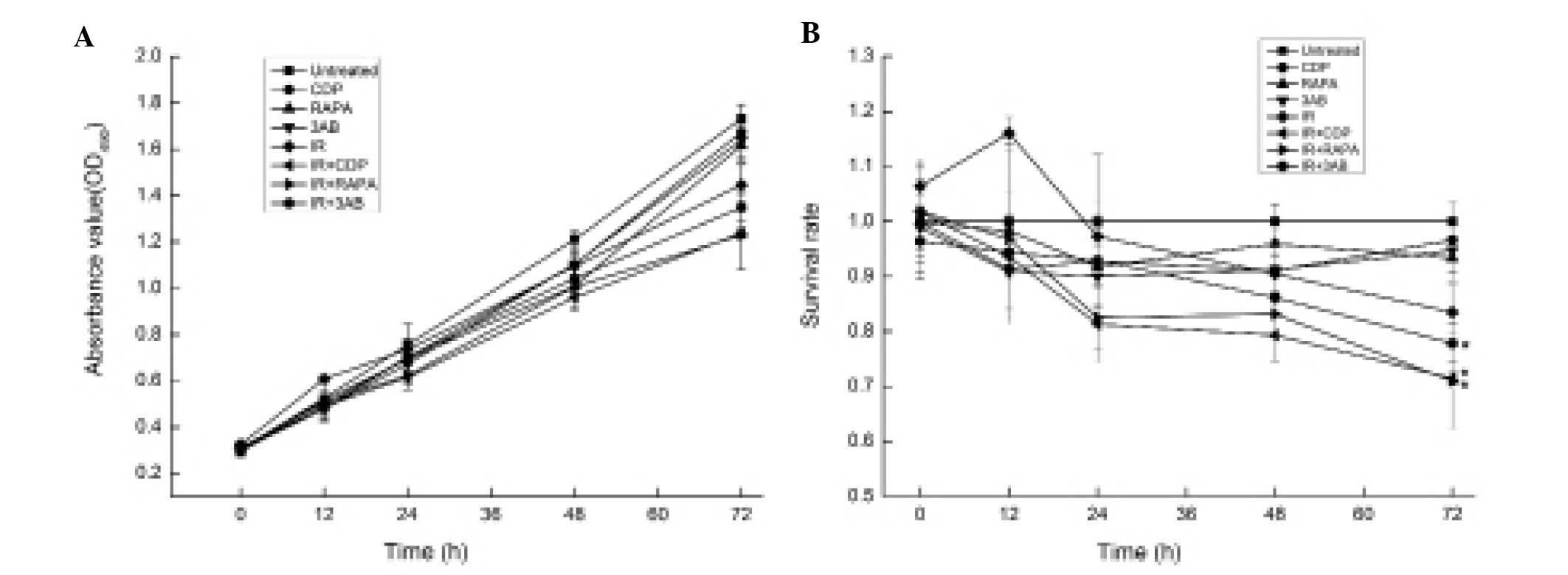
MTT assay was used to further elucidate whether the
sensitivity of CNE-2 cells to IR occurred through autophagy or
PARP-1 inhibition. The absorbance value indicates the proliferation
and survival of cells; the higher the absorbance value, the higher
the number of surviving cells. The proliferation rate of the CDP,
RAPA or 3AB combined with 6 Gy IR groups was lower than that of the
IR alone group (Fig. 6A). There
were significant differences in the number of surviving cells and
the survival rate between the different treatment groups 72 h after
IR (F=50.40, P<0.01) (Fig. 6A).
The survival rates of the CDP, RAPA or 3AB combined with 6 Gy IR
group were significantly lower than that of the IR alone group, at
each time point (P<0.01) (Fig.
6B).
Autophagy or PARP-1 inhibitor contributes
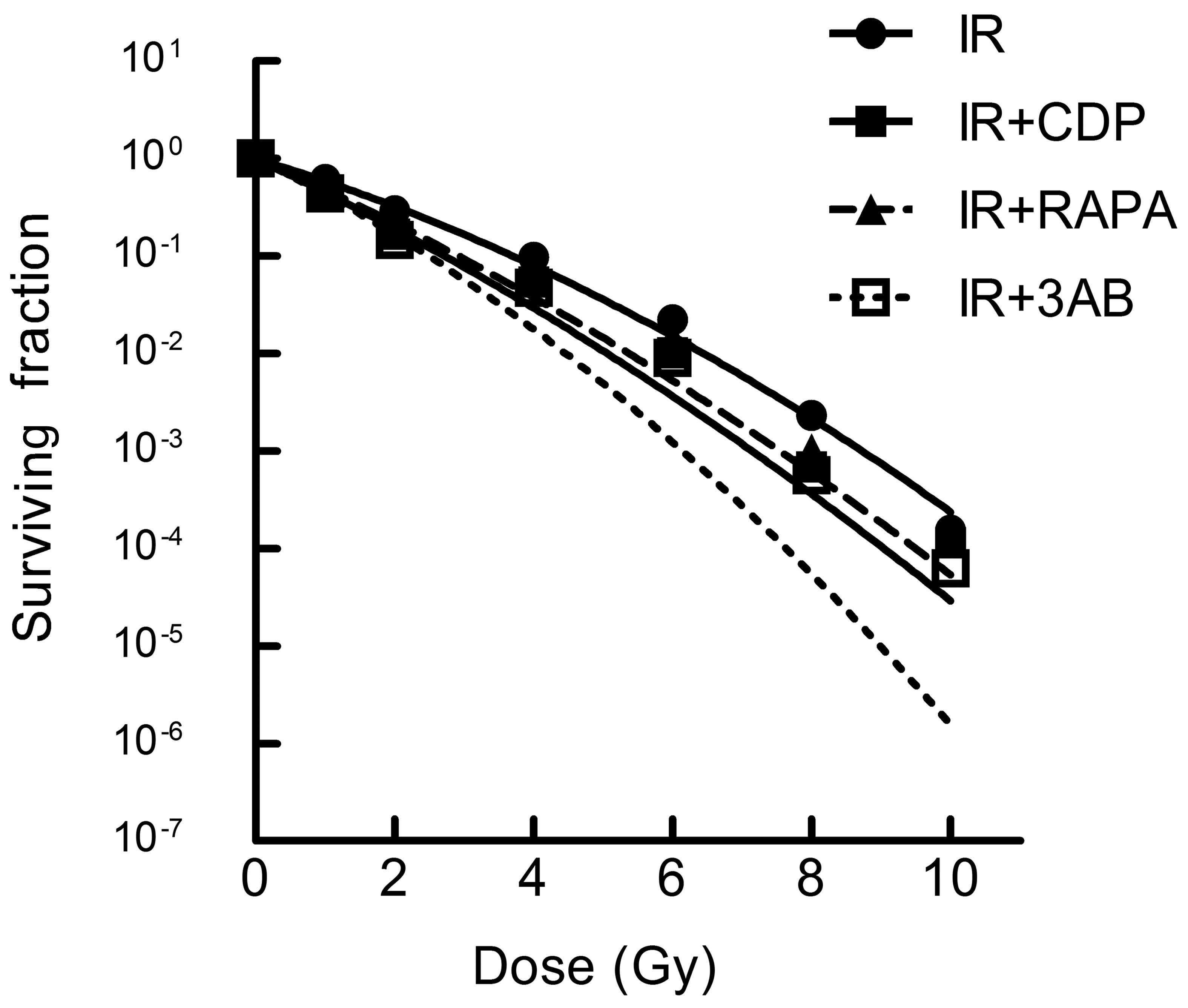
to the radiation sensitization of CNE-2 cells
As expected, the effect of CDP, RAPA and 3AB on the
lethality of IR was ascertained by clonogenic survival assay.
GraphPad Prism 5.0 Software using the linear-quadratic model was
used to calculate the radiobiology parameters and fitting dose
survival curve (Table I and
Fig. 7). Compared with IR alone,
the IR + CDP group caused increased radiosensitivity of CNE-2
cells; RAPA or 3AB combined with IR showed a similar role and
suggests that autophagy inhibition may be an approach to enhance
the lethality of radiation.
 | Table IRadiobiology parameters (the
linear-quadratic model). |
Table I
Radiobiology parameters (the
linear-quadratic model).
| Group | α | β | α/β |
|---|
| IR | 0.496±0.013 | 0.034±0.005 | 14.898±2.932 |
| IR + CDP | 0.770±0.017a | 0.029±0.014 | 34.028±22.440 |
| IR + RAPA | 0.706±0.026a | 0.029±0.015 | 29.673±16.713 |
| IR + 3AB | 0.785±0.033a | 0.057±0.023 | 15.769±7.553 |
| F-value | 97.631 | 2.247 | 1.333 |
| P-value | 0.000 | 0.160 | 0.330 |
Discussion
Apoptosis is a process of programmed cell death,
autophagy is a process of programmed cell survival (27). Yet, too much or too little autophagy
can damage cells. In some cases, autophagy can cause cell death.
Several reports in the early literature also called autophagy ‘type
II programmed cell death’, but now it is a misnomer (28,29).
At present, concerning the relationship between autophagy and
radiation sensitization of tumor cells, several scholars believe
that inhibition of autophagy of tumor cells can improve the effect
of radiotherapy (11,21,23).
Chen et al(21) found that
autophagy inhibitor 3-methyladenine (3-MA) combined with radiation
increased the apoptosis of esophageal squamous carcinoma cells.
Restraining autophagy increased the cytotoxicity of radiotherapy in
esophageal carcinoma cells and arrested the cell cycle in the
G2/M phase, increasing the sensitivity of radiotherapy.
Ito et al(23) found that
autophagy inhibitors, 3-MA and bafilomycin A1, increased the
sensitivity of malignant glioma U373-MG cells. Meanwhile, radiation
caused increased DNA double chain ruptures after inhibition of
autophagy. Thus, autophagy inhibitors may become a type of new
radiotherapy sensitization agents for malignant glioma. Several
researchers also suggest that inducing autophagy improves the
effect of radiotherapy on tumors (20,22).
Cao et al(20) found that
mTOR inhibitors, rapamycin and RAD001, inhibit mTOR function and
induce autophagy, consquently improving the radiotherapy effect in
breast cancer cells.
The present study demonstrated that radiation causes
autophagy, and LC3-II protein levels were increased in a dose- and
time-dependent manner induced by irradiation in CNE-2 cells. LC3-II
is currently the only protein identified which is present in
autophagic bodies and autophagy-lysosome membrane (30). Our research also detected autophagy
body formation and the quantity in nasopharyngeal carcinoma cells
by TEM which is the gold standard of autophagy detection, which
further corroborated the test results of the western blot
analysis.
Autophagy inhibitor CDP damages the structure and
function of lysosomes to inhibit autophagy. It causes
autophagy-lysosome to gather together and the degradation of LC3-II
is reduced. RAPA is an autophagy inducer and causes Atg13
dephosphorylated and Atg1 activation by inhibiting mTOR activity to
induce autophagy (24). In this
study, CDP inhibited CNE-2 cell autophagy and RAPA induced CNE-2
cell autophagy caused by irradiation (31). It is important to note that CDP, by
destroying the lysosome structure and function, inhibits autophagy,
causes autophagy-lysosome together, and reduces LC3-II degradation.
LC3-II failed to undergo effective decomposition, thus in the
CDP-treated group, LC3-II expression level was increased, which
confirmed that CDP inhibits autophagy; the characteristic of CDP is
different from the autophagic inhibitor 3-MA (21).
Flow cytometry detected the apoptosis rate of the
different groups at 24, 48 and 72 h after radiation. Our study
showed that the apoptosis rate of the CDP + IR group significantly
increased at the three time points compared with the IR alone
group, which indicated that autophagy inhibitors can improve the
sensitivity of the early response of nasopharyngeal carcinoma cells
to IR. Although the autophagy inducer RAPA combined with IR
significantly decreased the apoptosis rate at 72 h after radiation
compared with the IR alone group, it significantly increased rather
than decreased the apoptosis rate at 24 or 48 h after radiation.
The results concerning the effect of RAPA were contradictory, and
the reasons may be as follows: i) the characteristics of the RAPA
drug itself, which was used widely as an antifungal drug; ii)
physiological autophagy is a dynamic balance; a dysregulated
balance may cause damage; 10 Gy IR combined with RAPA have synergy,
and excessive autophagy may be also a type of cell death
consequently leading to an increase in the apoptosis rate.
Autophagy is a double-edged sword. Autophagy caused by IR inhibits
the cell death process and plays a protective role. Thus,
inhibiting autophagy can increase the radiation sensitization of
CNE-2 cells. However, excessive autophagy may also cause cell death
(6,7,12,13).
According to the radiation biology, the surviving cells after
radiation require concern. Only those cells that lose their
proliferation capability and are unable to form effective clone
cells are unable to cause cancer recurrence. We used a quadratic
linear model to construct dose survival curves and calculate the
radiation biology parameters. The results suggest that autophagy
inhibitors significantly increased radiation sensitization,
autophagy inducers showed a similar effect. The proliferation of
the cells treated with an autophagy inhibitor combined with IR
decreased.
In the present study, we identifed a novel function
for PARP-1 in mediating IR-induced autophagy and such autophagy
plays a pro-survival function in IR-induced cell death. Although at
the 72-h time point, 3AB promoted CNE-2 cell survival rather than
enhanced cell apoptosis, a clonogenic survival assay was performed
to evaluate long-term cell survival. The result showed that the
PARP-1 inhibitor contributed to radiation sensitization of CNE-2
cells (16). It appears that PARP-1
is able to elicit dual pathways with opposite functions in response
to oxidative stress (31), as
illustrated in Fig. 5B. The
decision of cell life or death is dependent on the balance between
apoptosis and autophagy mediated by these two distinct pathways.
Oxidative DNA damage includes modifications to bases and the sugar
phosphates, as well as single- or double-strand DNA breaks. Such
damage leads to PARP-1 activation, suppression of ATP production
and finally cell death. Furthermore, overactivation of PARP-1
produces large quantities of PAR polymers, leading to autophagy,
and finally cell survival.
In summary, we demonstrated a novel function of
PARP-1 in the regulation of IR-induced autophagy through the mTOR
signaling pathway, and such autophagy serves as a cell survival
mechanism against IR-mediated cell death. Autophagy inhibitors and
PARP-1 inhibitors contributed to the radiation sensitization of
CNE-2 cells. This suggests that CDP or 3AB may be used as adjuvant
treatment for nasopharyngeal carcinoma. Further animal experiments
and clinical tests are warranted to verify our findings.
Acknowledgements
The authors thank Zhangyu Zou from the Department of
Neurology of Peking Union Medical College Hospital, Chinese Academy
of Medical Sciences and Peking Union Medical College, for the
valuable discussions. The study was supported by the NSFC (Natural
Science Foundation of China) (81160285) and the Guangxi Natural
Science Foundation (2010gxnsfa013240).
References
|
1
|
Wei WI and Sham JST: Nasopharyngeal
carcinoma. Lancet. 365:2041–2054. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Feng XP, Yi H, Li MY, et al:
Identification of biomarkers for predicting nasopharyngeal
carcinoma response to radiotherapy by proteomics. Cancer Res.
70:3450–3462. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Guo Y, Zhu XD, Qu S, et al: Identification
of genes involved in radioresistance of nasopharyngeal carcinoma by
integrating gene ontology and protein-protein interaction networks.
Int J Oncol. 40:85–92. 2012.
|
|
4
|
Mizushima N: Autophagy: process and
function. Genes Dev. 21:2861–2873. 2007. View Article : Google Scholar
|
|
5
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Degenhardt K, Mathew R, Beaudoin B, et al:
Autophagy promotes tumor cell survival and restricts necrosis,
inflammation, and tumorigenesis. Cancer Cell. 10:51–64. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shintani T and Klionsky DJ: Autophagy in
health and disease: a double-edged sword. Science. 306:990–995.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Komatsu M, Waguri S, Chiba T, et al: Loss
of autophagy in the central nervous system causes neurodegeneration
in mice. Nature. 441:880–884. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hara T, Nakamura K, Matsui M, et al:
Suppression of basal autophagy in neural cells causes
neurodegenerative disease in mice. Nature. 441:885–889. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dalby KN, Tekedereli I, Lopez-Berestein G
and Ozpolat B: Targeting the prodeath and prosurvival functions of
autophagy as novel therapeutic strategies in cancer. Autophagy.
6:322–329. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Apel A, Herr I, Schwarz H, Rodemann HP and
Mayer A: Blocked autophagy sensitizes resistant carcinoma cells to
radiation therapy. Cancer Res. 68:1485–1494. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Marx J: Autophagy: is it cancer’s friend
or foe? Science. 312:1160–1161. 2006.
|
|
13
|
Wu WK, Coffelt SB, Cho CH, et al: The
autophagic paradox in cancer therapy. Oncogene. 31:939–953. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schreiber V, Dantzer F, Ame JC and de
Murcia G: Poly(ADP-ribose): novel functions for an old molecule.
Nat Rev Mol Cell Biol. 7:517–528. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu SW, Wang H, Poitras MF, et al:
Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by
apoptosis-inducing factor. Science. 297:259–263. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jagtap P and Szabo C: Poly(ADP-ribose)
polymerase and the therapeutic effects of its inhibitors. Nat Rev
Drug Discov. 4:421–440. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ha HC and Snyder SH: Poly(ADP-ribose)
polymerase is a mediator of necrotic cell death by ATP depletion.
Proc Natl Acad Sci USA. 96:13978–13982. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Albert JM, Cao C, Kim KW, et al:
Inhibition of poly (ADP-ribose) polymerase enhances cell death and
improves tumor growth delay in irradiated lung cancer models. Clin
Cancer Res. 13:3033–3042. 2007. View Article : Google Scholar
|
|
19
|
Hagan MP, Yacoub A and Dent P:
Radiation-induced PARP activation is enhanced through EGFR-ERK
signaling. J Cell Biochem. 101:1384–1393. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cao C, Subhawong T, Albert JM, et al:
Inhibition of mammalian target of rapamycin or apoptotic pathway
induces autophagy and radiosensitizes PTEN null prostate cancer
cells. Cancer Res. 66:10040–10047. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen YS, Song HX, Lu Y, et al: Autophagy
inhibition contributes to radiation sensitization of esophageal
squamous carcinoma cells. Dis Esophagus. 24:437–443. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lomonaco SL, Finniss S, Xiang CL, et al:
The induction of autophagy by gamma-radiation contributes to the
radioresistance of glioma stem cells. Int J Cancer. 125:717–722.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ito H, Daido S, Kanzawa T, Kondo S and
Kondo Y: Radiation-induced autophagy is associated with LC3 and its
inhibition sensitizes malignant glioma cells. Int J Oncol.
26:1401–1410. 2005.PubMed/NCBI
|
|
24
|
Paglin S, Lee NY, Nakar C, et al:
Rapamycin-sensitive pathway regulates mitochondrial membrane
potential, autophagy, and survival in irradiated MCF-7 cells.
Cancer Res. 65:11061–11070. 2005. View Article : Google Scholar
|
|
25
|
Rieber M and Rieber MS: Sensitization to
radiation-induced DNA damage accelerates loss of bcl-2 and
increases apoptosis and autophagy. Cancer Biol Ther. 7:1561–1566.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Peng PL, Kuo WH, Tseng HC and Chou FP:
Synergistic tumor-killing effect of radiation and berberine
combined treatment in lung cancer: the contribution of autophagic
cell death. Int J Radiat Oncol Biol Phys. 70:529–542. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kroemer G and Levine B: Autophagic cell
death: the story of a misnomer. Nat Rev Mol Cell Biol. 9:1004–1010.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Levine B and Yuan JY: Autophagy in cell
death: an innocent convict? J Clin Invest. 115:2679–2688. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Galluzzi L, Aaronson SA, Abrams J, et al:
Guidelines for the use and interpretation of assays for monitoring
cell death in higher eukaryotes. Cell Death Differ. 16:1093–1107.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huang Q and Shen HM: To die or to live:
the dual role of poly(ADP-ribose) polymerase-1 in autophagy and
necrosis under oxidative stress and DNA damage. Autophagy.
5:273–276. 2009. View Article : Google Scholar
|