Introduction
Cardiac myxomas (CMs) are mostly sporadic and only a
few are familial myxomas that are mainly referred to as Carney
complex (1,2). Clinical features and long-term outcome
such as recurrence, heart failure, sudden death and embolisms have
been reported (3,4). However, due to the limited
understanding of the molecular mechanisms leading to the
development and progression of CM, no target drug is available.
Surgical excision of the tumor is still the only current treatment
option, however this approach has increased risk of death and may
be followed by recurrences, myocardial infarction and stroke
(5). Therefore, there is an urgent
need to elucidate the molecular mechanisms of CM and to develop new
targeting chemotherapy agents.
To date, dozens of molecules and molecular markers
of sporadic cardiac myxoma have been studied which include
interleukin-6, interleukin-8 and growth-related oncogenes (5–10).
Insulin-like growth factor-1 (IGF-1) is a well-known survival
factor for both normal and malignant cells in many tissues,
including the prostate and the vascular system (11–13).
Elevated IGF-1 levels have been found in certain patients with
Carney complex, and it has been recommended as being suggestive of
or possibly associated with the disorder (14–16).
However, the role of IGF-1 in sporadic myxoma and the related
signaling pathways are largely unknown.
Studies using cancer cell lines revealed that the
signal transduction cascade underlying IGF-1 involved
phosphoinositide 3-kinase/protein kinase-B (PI3K/Akts) (17–20).
The Akts are indirectly downregulated from upstream by the lipid
and protein double phosphatase and tensin homolog deleted on
chromosome ten (PTEN) (21–25) and inactivated by PH domain
leucine-rich repeat phosphatase 1 and 2 (PHLPP1 and 2) through
dephosphorylating its hydrophobic motif directly from downstream.
We previously demonstrated that IGF-1 increased the
over-proliferation of vascular smooth muscle cells (VSMCs) by
increasing phosphorylation of Akt and repressing PTEN activation.
Transgene experiments found that the functional loss of PTEN or
PHLPP induced the activation of Akt, a critical component of the
survival and oncogenic function of IGF-1, and led to the
development of various types of cancer (18,26–29).
IGF-1 has been regarded as a potential target for tumors and
over-proliferating disorders such as atherosclerosis. Statins, as
cholesterol lowering drugs, have also exhibited cancer-retardant
efficacy in several in vitro and in vivo models and
epidemiological studies (30–39).
However, the study of whether the PTEN/PHLPP signaling pathway is
involved in the pleiotropic effect of statins is still obscure.
The aim of the present study was to elucidate
whether PTEN and PHLPPs are expressed in CM cells constitutively or
following stimulation by IGF-1, and to clarify the effect and
molecular mechanism of statins.
Materials and methods
Reagents
The antibodies against PHLPP1 and PHLPP2 were
obtained from Bethyl Laboratories Inc. (Montgomery, TX, USA).
Anti-PTEN was from Cell Signaling (Beverly, MA, USA). Atorvastatin
was from Pfizer (New York, NY, USA).
Culture of CM cells
The culture of CM cells was performed as previously
described (8). Myxoma tissues from
a 46-year-old patient with sporadic cardiac myxoma were obtained
during surgical operation at our hospital. The patient provided
informed consent, and the study protocol was approved by the China
PLA General Hospital Medical Ethics Committee. The cardiac myxoma
cells were extracted ex vivo by enzymatic digestion with
collagenase and were maintained, after differential trypsinization
and morphological confirmation, in Dulbecco’s modified Eagle’s
medium. Before stimulation experiments, the medium was replaced
with serum-free DMEM for 24 h and then replaced with fresh medium
plus the indicated agents for different times. For examination of
the expression of the signaling proteins, cells were treated for 24
h without control or with IGF-1 (100 μg/l) or with atorvastatin (10
μM), or pretreated for 10 min with atorvastatin (10 μM) and then
with IGF-1 (100 μg/l) for 24 h. To assess the effect of IGF-1 and
atorvastatin on the activity of PTEN and PHLPPs, the cells were
treated for 0, 1/12, 1/6, 1, 6, 24 h with atorvastatin (0, 0.1, 1,
5, 10, 100 μM) or IGF-1 (0, 0.1, 1, 5, 10, 100 μg/l), or pretreated
for 10 min with atorvastatin (10 μM) and thereafter with IGF-1 (100
μg/l) for 10 min. CM cells were seeded in triplicate at a density
of 2.5×105 cells/ml on 24-well plates. After incubation
for the indicated time, the medium was removed and the cells were
stored at -80°C until assay.
Proliferation assays
The proliferation assays were performed as
previously described (8). Cells
were seeded into plastic wells and allowed to grow for 48 h in
culture medium with 10% FBS. After 24 h in serum-free medium, the
cells were treated with atorvastatin (0, 0.1, 1, 5, 10, 100 μM) or
IGF-1 (0, 0.1, 1, 5, 10, 100 μg/l) or pretreated for 30 min with
atorvastatin (10 μM) followed by stimulation with IGF-1 (0, 0.1, 1,
5, 10, 100 μg/l) for 48 h under high serum conditions and exposed
to DMEM containing 1 mCi [3H]thymidin for a further 24
h. The cells were washed, harvested and processed for counting in a
liquid scintillation counter.
Western blot analysis
The cellular lysates were prepared as previously
described (26,27). Lysates containing equal protein were
separated by SDS-PAGE and transferred to polyvinyldifluoride
membranes. For anti-PHLPP1, anti-PHLPP2 and anti-PTEN blotting,
membranes were incubated for 2 h with the indicated antibodies.
Blots were then washed and incubated with horseradish
peroxidase-linked anti-rabbit secondary antibody for 3 h.
Densitometric analysis was performed with ImageJ analysis
software.
Immunoprecipitation
Whole cell lysates were incubated for 1 h with 1 μg
of PTEN, PHLPP1 or PHLPP2 antibody and then with protein A/G
Plus-agarose for 24 h at 4°C (28,40).
PTEN lipid phosphatase assay
The immunoprecipitated PTEN was added to reaction
buffer (100 mM Tris pH 8.0, 10 mM DTT, 0.01% Brij 35, 1 g/l BSA, 1
mM EDTA, 25 μM diC16PIP3) for 30 min at 37°C,
then terminated by the addition of Biomol Green reagent (Biomol).
The amount of phosphate in the supernatant was determined by
reading the absorbance of the samples at 620 nm following
incubation for 30 min. Phosphate concentrations were estimated by
comparison to KH2PO4 standards diluted
(0–1000 pM) (11,27).
Assay of PHLPP activity
Briefly, PHLPP1 and PHLPP2 were immunoprecipitated,
and their activities were measured using pNPP as a substrate.
Dephosphorylation of pNPP was measured by continuously monitoring
the change in absorbance at 405 nM (28,40).
Statistical analysis
Values are expressed as means ± SD from at least 3
independent experiments. The significance of differences between
groups was determined using one-way ANOVA statistical analysis. The
threshold for significance was set at a p-value <0.05.
Statistical analysis was performed using SPSS 19.0 statistical
software.
Results
Effect of IGF-1 and atorvastatin on the
proliferation of CM cells
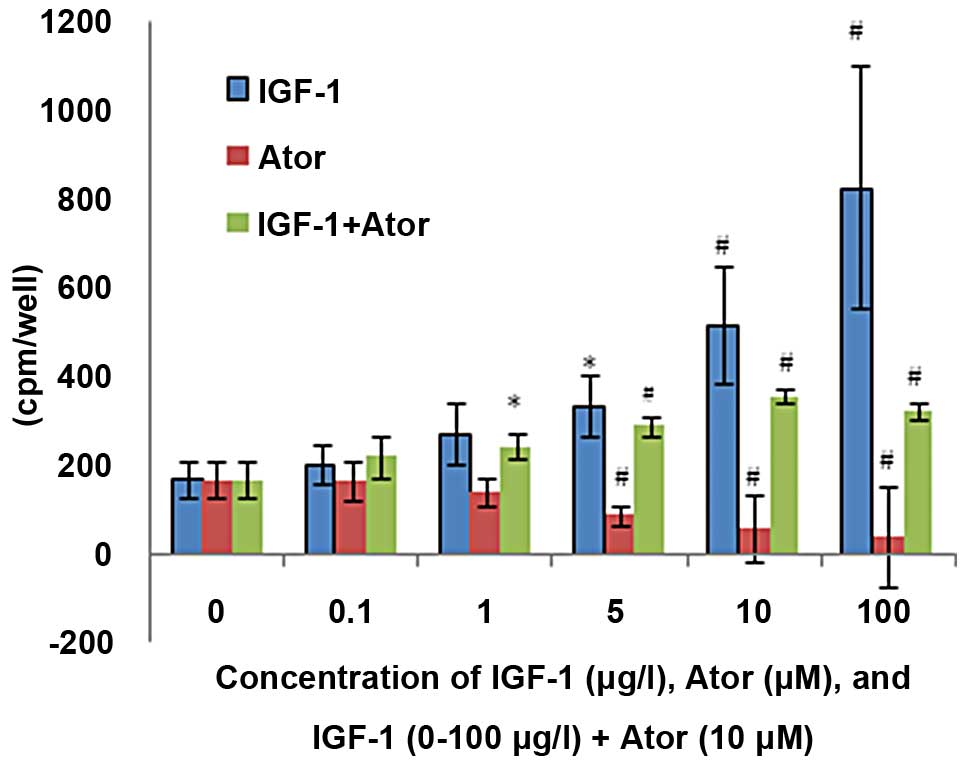
IGF-1 caused a 2.0- to 4.9-fold increase in
[3H]thymidin incorporation in a concentration-dependent
mode (5–100 μg/l, p<0.05), which was abolished by atorvastatin.
Atorvastatin decreased the constitutive proliferation of CM cells
dose-dependently (5–100 μM, p<0.05) by 49–77% (Fig. 1).
Effect of IGF-1 and atorvastatin on the
expression of PTEN and PHLPPs in CM cells
IGF-1, atorvastatin, and IGF-1 plus atorvastatin did
not affect the protein expression of PTEN, PHLPP1 and PHLPP2 after
stimulated for 24 h (Fig. 2).
Effect of IGF-1 on the activity of PTEN
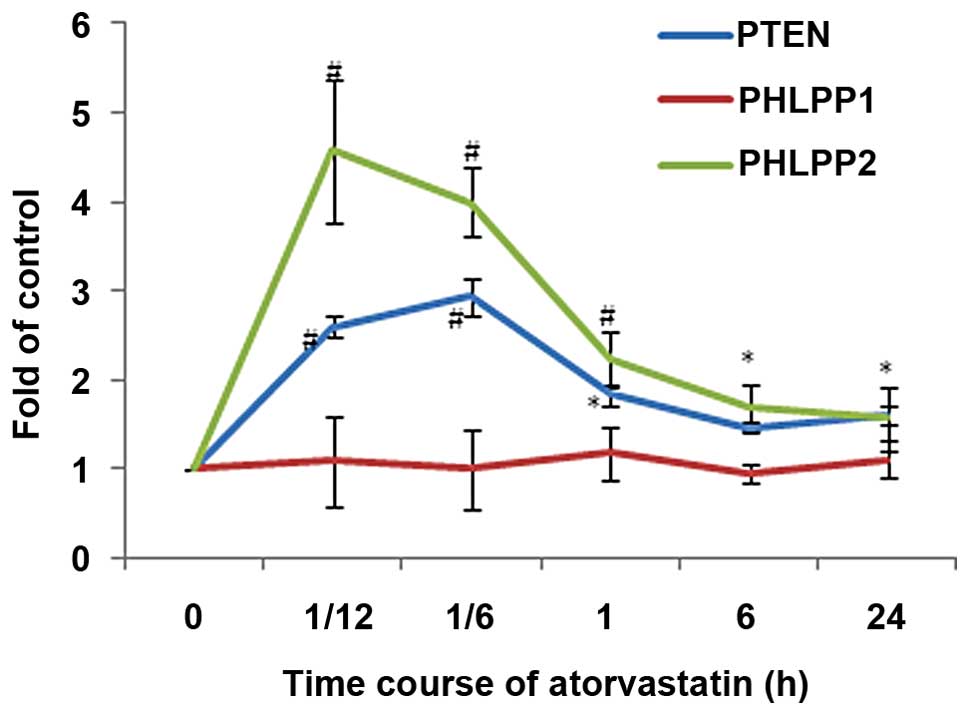
and PHLPPs in CM cells
IGF-1 deceased both PTEN and PHLPP2 but not PHLPP1
activity by 22–44% and 18–69% separately (p<0.01) in a
time-dependent mode, with a maximum depressive effect noted at 5
min and lasting at least for 24 h (Fig.
3). IGF-1 inhibited both PTEN and PHLPP2 but not PHLPP1
activity by 14–54% and 14–70% separately in a
concentration-dependent mode (5–100 μg/l, p<0.01) (Fig. 4). These findings suggest that IGF-1
signaling was via the PTEN and PHLPP2 pathway in CM cells.
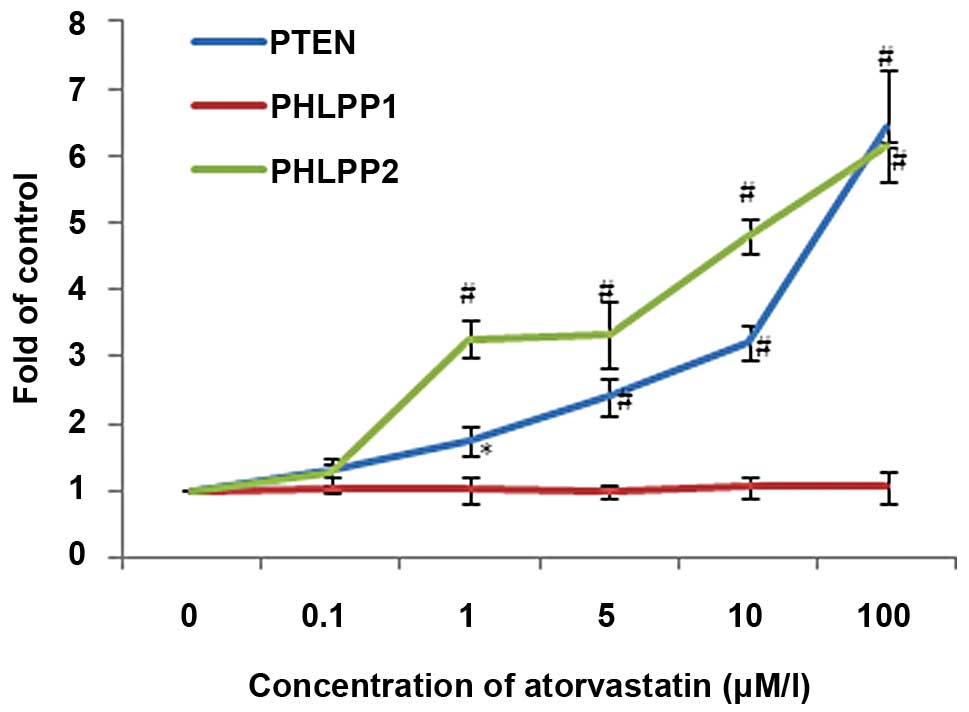
Effect of atorvastatin on the activity of
PTEN and PHLPPs in CM cells
Atorvastatin increased the constitutive phosphatase
activity of PTEN and PHLPP2 but not PHLPP1 to 1.83- to 2.9-fold and
1.7- to 4.57-fold separately (p<0.01) in a time-dependent mode
which lasted for 1 and 6 h separately (Fig. 5). Atorvastatin increased
constitutive phosphatase activity of PTEN and PHLPP2 but not PHLPP1
to 1.73- to 6.43-fold and 3.27- to 6.17-fold separately in a
concentration-dependent mode (5–100 μM, p<0.05) (Fig. 6).
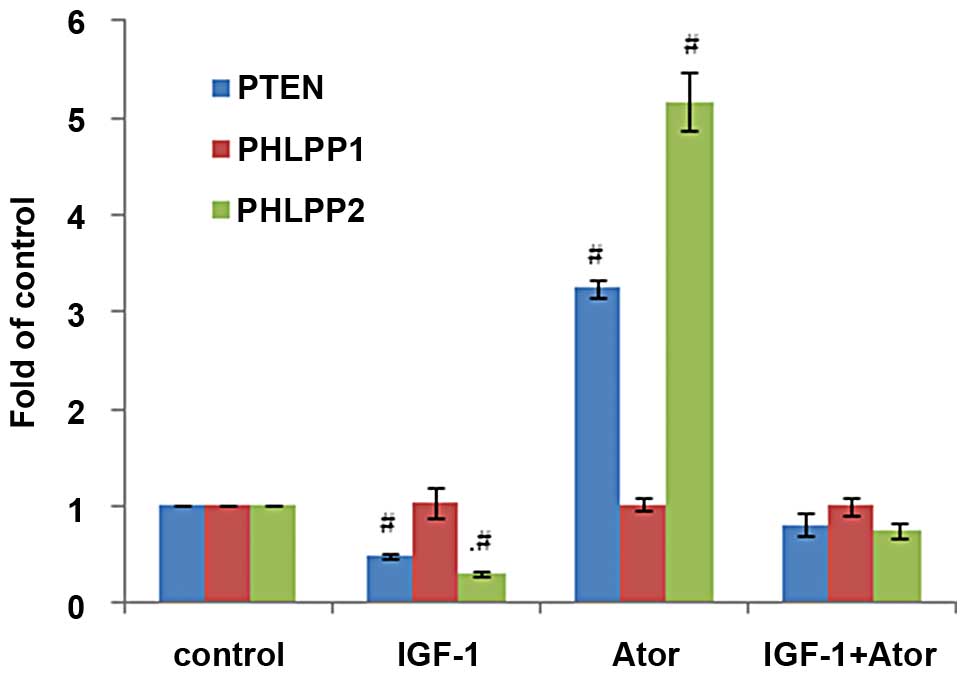
Effect of atorvastatin on the activity of
PTEN and PHLPPs in CM cells induced by IGF-1
Atorvastatin (10 μM) increased PTEN and PHLPP2
phosphatase activity inhibited by IGF-1 (100 μg/l) by 32 and 45%
separately and reversed them nearly to the level of the control
(both p<0.01) (Fig. 7).
Discussion
To elucidate the molecular mechanism of CM, a wide
spectrum of molecules and molecular markers has been studied, which
include higher expression of membrane-associated MUC1 gene, matrix
metalloproteinases, vascular endothelial growth factor and its
receptors, basic fibroblast growth factor and its receptor,
endothelin and its precursor, monocyte chemotactic protein-1,
thymidine phosphorylase, transforming growth factor β, epidermal
growth factor, interleukin-6, interleukin-8 and growth-related
oncogenes (5–10). It was reasoned that abnormalities in
the expression levels of any of these molecules can potentially
contribute to cancer pathogenesis, but the signaling pathways are
poorly understood and targeting drugs for sporadic cardiac myxoma
are unknown.
Recent experiments revealed that PKB/Akt and protein
and lipid dual phosphatase PTEN were involved in the survival and
proliferation of various cancer cell lines. It was also believed
that IGF-1 may play an important role in the carcinogenesis of
certain malignancies (18,26), but IGF-1 has rarely been
investigated in CM cells.
Previous data from our laboratory and other centers
have shown that IGF-1 activated PI3K/Akt. In turn it phosphorylates
an array of substrates to regulate cell survival, proliferation,
growth, metabolism and motility (11,27).
The Akts are indirectly downregulated from upstream by the lipid
phosphatase PTEN (21–25), and inactivated by PHLPP directly
from downstream. PHLPPs selectively regulate Akt isoforms. PHLPP1
dephosphorylates Akt-2, whereas PHLPP2 dephosphorylates Akt-1. By
specifically dephosphorylating the hydrophobic motif, PHLPP1
controls the degree of agonist-evoked signaling by Akt and the
cellular levels of PKC. The aberrant regulation of either kinase
and phosphatase was noted in many diseases, notably diabetes and
cancer (28,40–43).
Recently, Wahdan-Alaswad et al reported that
IGF-1 is a critical regulator of prostate tumor cell growth, which
is mediated by its ability to suppress bone morphogenetic
protein-induced apoptosis and Smad-mediated gene expression through
a mechanism dependent on the PI3K, Akt, Raptor and Rictor signaling
pathway (17). It has not yet been
elucidated whether PTEN and PHLPPs are expressed in CM cells
constitutively or following stimulation by IGF-1.
In the present study, we found that IGF-1 increased
the proliferation of CM cells to 2.0 to -4.9 times that of the
control in a concentration- and time-dependent manner. This was
similar to our previous study on VSMCs in which the proliferation
was increased significantly by IGF-1 (27). The signal mechanistic study revealed
that PTEN, PHLPP1 and PHLPP2 were constitutively expressed in CM
cells. Furthermore, IGF-1 stimulation resulted in a significant
decrease in PTEN and PHLPP2 activity but not PHLPP1 activity,
suggesting that IGF-1 primarily utilizes Akt-1 to transmit its
signal in CMs.
For the drug interference study, we selected
atorvastatin, since statins have been used for dozens of years for
the treatment of cardiovascular diseases and have been reported to
be associated with a significant reduction in the risk of cancer
and lymphoma (32–39). Research of relative molecular
mechanisms have demonstrated that statins inhibited the synthesis
of mevalonate or of downstream isoprenoids, decreased the
availability of dolichol, impeded the glycosylation of nascent
IGF-1 receptors, prevented their transfer to the cell surface, and
eventually exerted cancer-retardant efficacy (30–32).
Miraglia et al recently reported that long-term treatment of
non-small cell lung cancer A549 cells with high concentrations of
statins increased PTEN expression, enhanced PHLPP2 expression,
decreased PHLPP1 expression and inhibited downstream pAkt signaling
(32).
In the present study, atorvastatin decreased the DNA
synthesis of CMs by 49–77% concentration-dependently and reversed
the proliferative effect of IGF-1. This result was similar to that
of Brown et al who reported that lipophilic statins
including atorvastatin reduced the migration and colony formation
of PC-3 cells in human bone marrow stroma by inhibiting
geranylgeranyl pyrophosphate production, reducing the formation and
the spread of metastatic prostate colonies (37). An in vivo study using Wistar
rats by Parada et al also found that atorvastatin had a
clear inhibitory effect on bladder cancer development, probably due
to its antioxidant, antiproliferative and anti-inflammatory
properties. Regarding protein expression, our findings were not
consistent with those of Miraglia et al, who found
significant changes in the three proteins following treatment with
atorvastatin (32). These
discrepancies perhaps due to the different dose and incubation
period used.
It is meaningful to point out that in the present
study atorvastatin possessed a strong and lasting inhibitory effect
on the proliferation of CM cells even at a concentration (1 μM)
relevant for lowering cholesterol levels and for preventing
cardiovascular disease. In addition, this study demonstrated that
atorvastatin not only increased the phosphatase activity of PTEN
and PHLPP2 constitutively but also significantly restored their
phosphatase activity inhibited by IGF-1, which suggests that
atorvastatin may exert its anticancer effect by positive regulation
of phosphatase activity as previously reported in pancreatic cancer
and prostate cancer cells (41–44).
In conclusion, our study supports the hypothesis
that altered phosphatase signaling plays a role in the
tumorigenesis of CM, and statins may have chemopreventive effects
on CM by regulating the regulator of the Akt pathway. This finding
is important for the management of CM, since patients with this
disorder often first present at the cardiology department, and
statins are widely used in cardiovascular disease prevention by
physicians who are familiar with its usage and side effects. This
may lead to an easy and affordable strategy for the post-operative
treatment of CM, however further evidence from large clinical
trials is warranted.
Acknowledgements
We thank Tianran Wu from Melbourne University in
Australia for editing the manuscript.
Abbreviations:
|
Ator
|
atorvastatin
|
|
CMs
|
cardiac myxomas
|
|
IGF-1
|
insulin-like growth factor-1
|
|
PTEN
|
phosphatases and tensin homolog
deleted on chromosome ten
|
|
PHLPP1 and 2
|
pleckstrin homology domain
leucine-rich repeat phosphatase 1 and 2
|
References
|
1
|
Roschkov S, Rebeyka D, Mah J and Urquhart
G: The dangers of cardiac myxomas. Prog Cardiovasc Nurs. 22:27–30.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wilkes D, McDermott DA and Basson CT:
Clinical phenotypes and molecular genetic mechanisms of Carney
complex. Lancet Oncol. 6:501–508. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Figueroa-Torres Y, Martínez-Ojeda JA,
Franqui-Rivera H, et al: Benign cardiac neoplasms: the experience
at the Cardiovascular Center of Puerto Rico and the Caribbean. PR
Health Sci J. 27:373–376. 2008.PubMed/NCBI
|
|
4
|
Wu X, Yang D, Yang Z, et al: Clinical
characteristics and long term post-operative outcome of cardiac
myxoma. EXCLI J. 11:240–249. 2012.
|
|
5
|
Barh D, Kumar A, Chatterjee S and Liloglou
T: Molecular features, markers, drug targets, and prospective
targeted therapeutics in cardiac myxoma. Curr Cancer Drug Targets.
9:705–716. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chu PH, Jung SM, Yeh TS, et al: MUC1, MUC2
and MUC5AC expressions in cardiac myxoma. Virchows Arch. 446:52–55.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Orlandi A, Ciucci A, Ferlosio A, et al:
Increased expression and activity of matrix metalloproteinases
characterize embolic cardiac myxomas. Am J Pathol. 166:1619–1628.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sakamoto H, Sakamaki T, Kanda T, et al:
Vascular endothelial growth factor is an autocrine growth factor
for cardiac myxoma cells. Circ J. 68:488–493. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fujisawa H, Koide N, Kono T, et al:
Expression of basic fibroblast growth factor and its receptor-1 in
cardiac myxoma. J Cardiovasc Surg. 43:589–594. 2002.PubMed/NCBI
|
|
10
|
Amano J, Kono T, Wada Y, et al: Cardiac
myxoma: its origin and tumor characteristics. Ann Thorac Cardiovasc
Surg. 9:215–221. 2003.PubMed/NCBI
|
|
11
|
Uzoh CC, Holly JMP, Biernacka KM, et al:
Insulin-like growth factor-binding protein-2 promotes prostate
cancer cell growth via IGF-dependent or -independent mechanisms and
reduces the efficacy of docetaxel. Br J Cancer. 104:1587–1593.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rzucidlo EM: Signaling pathways regulating
vascular smooth muscle cell differentiation. Vascular. 17(Suppl 1):
S15–S20. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Perks C: The role of insulin-like growth
factor binding proteins. Neuroendocrinology. 83:154–160. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mateus C, Palangié A, Franck N, et al:
Heterogeneity of skin manifestations in patients with Carney
complex. J Am Acad Dermatol. 59:801–810. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Raff SB, Carney JA, Krugman D, et al:
Prolactin secretion abnormalities in patients with the ‘syndrome of
spotty skin pigmentation, myxomas, endocrine overactivity and
schwannomas’ (Carney complex). J Pediatr Endocrinol Metab.
13:373–379. 2000.
|
|
16
|
Kurtkaya-Yapicier O, Scheithauer BW,
Carney JA, et al: Pituitary adenoma in Carney complex: an
immunohistochemical, ultrastructural, and immunoelectron
microscopic study. Ultrastruct Pathol. 26:345–353. 2002. View Article : Google Scholar
|
|
17
|
Wahdan-Alaswad RS, Song K, Krebs T, et al:
Insulin-like growth factor I suppresses bone morphogenetic protein
signaling in prostate cancer cells by activating mTOR signaling.
Cancer Res. 70:9106–9117. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Grimberg A: Mechanisms by which IGF-I may
promote cancer. Cancer Biol Ther. 2:630–635. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu B, Lee KW, Anzo M, et al: Insulin-like
growth factor-binding protein-3 inhibition of prostate cancer
growth involves suppression of angiogenesis. Oncogene.
26:1811–1819. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Waalkes S, Simon P, Hennenlotter J, et al:
Altered expression of Akt signaling pathway parameters in prostate
needle biopsies derived from benign, adjacent and cancerous tissue.
Oncol Rep. 23:1257–1260. 2010.PubMed/NCBI
|
|
22
|
Falbo V, Floridia G, Censi F, et al: Three
cases of rare salivary gland tumours: a molecular study of TP53,
CDKN2A/ARF, RAS, BRAF, PTEN, MAPK2 and EGFR genes. Oncol
Rep. 26:3–11. 2011.PubMed/NCBI
|
|
23
|
Tserga A, Michalopoulos NV, Levidou G, et
al: Association of aberrant DNA methylation with
clinicopathological features in breast cancer. Oncol Rep.
27:1630–1638. 2012.PubMed/NCBI
|
|
24
|
Bouali S, Chrétien AS, Ramacci C, et al:
PTEN expression controls cellular response to cetuximab by
mediating PI3K/AKT and RAS/RAF/MAPK downstream signaling in
KRAS wild-type, hormone refractory prostate cancer cells.
Oncol Rep. 21:731–735. 2009.PubMed/NCBI
|
|
25
|
Jang K, Kim M, Seo HS, et al: PTEN
sensitizes MDA-MB-468 cells to inhibition of MEK/Erk signaling for
the blockade of cell proliferation. Oncol Rep. 24:787–793.
2010.PubMed/NCBI
|
|
26
|
Garcia JA and Danielpour D: Mammalian
target of rapamycin inhibition as a therapeutic strategy in the
management of urologic malignancies. Mol Cancer Ther. 7:1347–1354.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu XL, Yang DY, Yang ZS, et al: Effect of
IGF-1 on PI3K/PTEN signal pathway in vascular smooth muscle cell.
Zhongguo Ying Yong Sheng Li Xue Za Zhi. 20:259–262. 2004.(In
Chinese).
|
|
28
|
Brognard J, Sierecki E, Gao T, et al:
PHLPP and a second isoform, PHLPP2, differentially attenuate the
amplitude of Akt signaling by regulating distinct Akt isoforms. Mol
Cell. 25:917–931. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gao T, Brognard J and Newton AC: The
phosphatase PHLPP controls the cellular levels of protein kinase C.
J Biol Chem. 283:6300–6311. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
McCarty MF: Suppression of dolichol
synthesis with isoprenoids and statins may potentiate the
cancer-retardant efficacy of IGF-I down-regulation. Med Hypotheses.
56:12–16. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Martínez-González J, Viñals M, Vidal F, et
al: Mevalonate deprivation impairs IGF-I/insulin signaling in human
vascular smooth muscle cells. Atherosclerosis. 135:213–223.
1997.PubMed/NCBI
|
|
32
|
Miraglia E, Högberg J and Stenius U:
Statins exhibit anticancer effects through modifications of the
pAkt signaling pathway. Int J Oncol. 40:867–875. 2012.PubMed/NCBI
|
|
33
|
Duncan RE, El-Sohemy A and Archer MC:
Statins and cancer development. Cancer Epidemiol Biomarkers Prev.
14:1897–1898. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bonovas S, Filioussi K, Tsavaris N, et al:
Statins and cancer risk: a literature-based meta-analysis and
meta-regression analysis of 35 randomized controlled trials. J Clin
Oncol. 24:4808–4817. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chan KK, Oza AM and Siu LL: The statins as
anticancer agents. Clin Cancer Res. 9:10–19. 2003.
|
|
36
|
Lutski M, Shalev V, Porath A and Chodick
G: Continuation with statin therapy and the risk of primary cancer:
a population-based study. Prev Chronic Dis. 9:E1372012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Brown M, Hart C, Tawadros T, et al: The
differential effects of statins on the metastatic behaviour of
prostate cancer. Br J Cancer. 106:1689–1696. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Parada B, Reis F, Pinto A, et al:
Chemopreventive efficacy of atorvastatin against
nitrosamine-induced rat bladder cancer: antioxidant,
anti-proliferative and anti-inflammatory properties. Int J Mol Sci.
13:8482–8499. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lai SW, Liao KF, Lai HC, et al:
Atorvastatin correlates with decreased risk of esophageal cancer: a
population-based case-control study from Taiwan. Libyan J Med.
7:188302012.PubMed/NCBI
|
|
40
|
Miyamoto S, Purcell NH, Smith JM, et al:
PHLPP-1 negatively regulates Akt activity and survival in the
heart. Circ Res. 107:476–484. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nitsche C, Edderkaoui M, Moore RM, et al:
The phosphatase PHLPP1 regulates Akt2, promotes pancreatic cancer
cell death, and inhibits tumor formation. Gastroenterology.
142:377–387. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mulholland DJ, Tran LM, Li Y, et al: Cell
autonomous role of PTEN in regulating castration-resistant prostate
cancer growth. Cancer Cell. 19:792–804. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lee JK, Edderkaoui M, Truong P, et al:
NADPH oxidase promotes pancreatic cancer cell survival via
inhibiting JAK2 dephosphorylation by tyrosine phosphatases.
Gastroenterology. 133:1637–1648. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mistafa O, Ghalali A, Kadekar S, et al:
Purinergic receptor-mediated rapid depletion of nuclear
phosphorylated Akt depends on pleckstrin homology domain
leucine-rich repeat phosphatase, calcineurin, protein phosphatase
2A, and PTEN phosphatases. J Biol Chem. 285:27900–27910. 2010.
View Article : Google Scholar
|