Introduction
Hepatocyte growth factor (HGF) and its receptor,
c-Met, regulate a wide variety of cellular functions. HGF was
originally identified and cloned as a mitogenic polypeptide for
hepatocytes (1). Subsequent studies
revealed that HGF-induced c-Met tyrosin kinase activation is
involved in multiple biological effects, including mitogenic,
motogenic, morphogenic and anti-apoptotic activities (2–5). In
tumor tissues, however, various malignant cells express c-Met
receptor and utilize the biological actions of HGF/c-Met pathway
for their dissociative, invasive and metastatic behaviors (6–9). HGF
also binds to the receptor expressed on endothelial cells that
stimulates angiogenesis, a process critical to continued growth of
solid tumors (5,10).
In vivo tumor growth is enhanced by the
coexistence of stromal fibroblasts. Host stromal-derived factor is
a key molecule that enhances progressive potential of tumor cells.
In several types of cancer, the HGF/c-Met pathway is regulated
through the paracrine loop (11).
Tumor cells secrete a variety of HGF-inducers, such as
interleukin-1, basic fibroblast growth factor (bFGF),
platelet-derived growth factor, transforming growth factor-α and
prostaglandin E2, through which HGF production from stromal
fibroblasts is upregulated (12).
These observations suggest a mutual interaction between tumor cells
and stromal fibroblasts; tumor cells secrete HGF-inducers for
stromal cells, while stromal cells secrete HGF that may stimulate
tumor growth and angiogenesis.
NK4, a product of proteolytic digestion of HGF, has
been identified as a competitive antagonist for HGF and c-Met
receptor and inhibits HGF-induced c-Met tyrosine phosphorylation
(13). Furthermore, subsequent
studies showed that NK4 has bifunctional actions to suppress not
only HGF/c-Met-dependent tumor progression and angiogenesis but
also HGF/c Met-independent angiogenesis induced by bFGF and
vascular endothelial growth factor (VEGF) (14,15).
In previous experiments using animal models, administration of NK4
protein or NK4 gene therapy was demonstrated to suppress tumor
invasion, metastasis, and angiogenesis, effectively transforming
malignant tumors into benign-like dormant tumors. In this effect,
anti-angiogenesis is considered to be predominant, especially in
vivo(16,17).
We previously demonstrated that HGF/c-Met pathway
activation in CT26 tumor cells promoted HGF-inducers and subsequent
stroma (fibroblast)-derived HGF secretion, resulting in amplifying
HGF paracrine loop (18).
Furthermore, some authors have shown that HGF stimulates
endothelial cells directly and indirectly by facilitating
expression of other angiogenic factors, represented by VEGF
(19–21). Therefore, considering the angiogenic
activity of HGF and tumor-stromal interaction, the mechanism of
anti-angiogenesis by NK4 seems to be more complicated.
In the present study, we investigated the
cooperation of HGF and VEGF in tumor-stromal interaction consisting
of HGF paracrine loop using a co-culture system with CT26 tumor
cells and fibroblasts and we described in vivo tumor
anti-angiogenic activities of NK4 in detail.
Materials and methods
Cell lines and culture
CT26 is an undifferentiated colon adenocarcinoma
cell line originally derived by intrarectal injections of
N-nitroso-N-methylurethamine in a female BALB/c mouse. The genetic
modification of CT26 to produce NK4 was previously described
(18). The transfectant expressing
the highest amount of NK4 was designated as CT26-NK4. Cells
transfected with the neomycin-resistance gene (pSVneo) alone were
used as a control (CT26-NEO). Primary mouse fibroblasts were
obtained from dermal tissues in culture where fibroblasts initially
proliferated outward from the tissues.
The CT26 transfectants and the fibroblasts were
maintained in RPMI-1640 and MEM (both from Nacalai Tesque, Kyoto,
Japan), respectively, supplemented with 100 IU/ml penicillin, 100
mg/ml streptomycin (Sigma, Welwyn Garden, UK) and 10%
heat-inactivated fetal bovine serum (FBS; JRH Biosciences, Lenexa,
KS, USA) at 37ºC in a humidified atmosphere containing 5%
CO2.
Determination of murine VEGF and HGF in a
separate co-culture system
To evaluate regulation through soluble factors
released by cancer cells and fibroblasts, each well of a 24-well
plate was divided into two compartments using inner wells with 8 μm
pores (Cell Culture Insert; BD Falcon, Franklin Lakes, NJ, USA).
Mouse fibroblasts were plated on 24-well culture plates at a
density of 1×105 cells/well and were cultured for 24 h.
After replacing fresh media, tumor cells were seeded at
5×104 cells on each inner well and co-cultured for 24 h.
Cells were subsequently given fresh media with 0.2% FBS and were
co-cultured for a further 32 h. The cultured supernatants were
collected every 8 h and the concentration of murine VEGF and HGF
was determined by ELISA (VEGF; R&D Systems, Minneapolis, MN,
USA) (HGF; Institute of Immunology Co., Tokyo, Japan) according to
the manufacturer’s instructions. For time course analysis of VEGF
or HGF secretion in the separate co-culture system, the cultured
supernatants were collected 8, 16, 24 and 32 h after replacing
fresh media with 0.2% FBS.
Real-time polymerase chain reaction
analyses
Total RNA was isolated from tumor cells (the inner
chamber) in the separate co-culture system using RNeasy Mini kit
(Qiagen, Valencia, CA, USA) according to the manufacturer’s
instructions. The PCR primers used were: VEGF-A,
5′-CTGGATATGTTTGACTGCTGTGGA-3′ (sense) and
5′-GTTTCTGGAAGTGAGCCAATGTG-3′ (anti-sense). For normalization, the
18S ribosomal protein served as the housekeeping gene. Rps18,
5′-TTCTGGCCAACGGTCTAGACAAC-3′ (sense) and
5′-CCAGTGGTCTTGGTGTGCTGA-3′ (anti-sense). Real-time RT-PCR was
performed with a LightCycler1.5 (Roche, Basel, Switzerland) using
SYBR-Green I (Qiagen). The RT-PCR protocol consisted of a 50ºC
reverse transcription step for 20 min and 95ºC PCR initial
activation step for 15 min followed by 40 cycles with a 94ºC
denaturation for 15 sec, 60ºC annealing for 30 sec, 72ºC extension
for 30 sec. To confirm specific amplification, the PCR products
were subjected to a melting curve analysis. The results of
real-time RT-PCR were normalized with Rps18 and expressed as the
relative value of control (CT26-NEO alone). Relative expression
levels were calculated with the ΔΔCt method. Repeated experiments
were performed for two or three times and similar results were
obtained.
Animal experiments
Eight-week-old BALB/c female mice were purchased
from Shimizu Laboratory Animal Center (Kyoto, Japan). All mice were
maintained under specific-pathogen-free conditions. To generate
in vivo tumors, 5×105 cells of CT26-NEO or
CT26-NK4 with/without 1×106 cells of primary mouse
fibroblasts co-cultured for 24 h were inoculated subcutaneously
(s.c.) into syngeneic BALB/c mice in the right lower flank (n=10
for each group). Two perpendicular diameters of resulting tumors
were measured every 2–3 days using a caliper. Tumor volumes were
calculated using the formula, tumor volume (mm3) = 0.52
× (width)2 × (length). The investigation protocols were
approved by the Ethics Committee of Kyoto Prefectural University of
Medicine. The mice were sacrificed by excess administration of
general anesthesia before the tumor weight exceeded 1/3 of body
weight.
Immunohistochemistry
BALB/c female mice were euthanized 14 days after the
s.c. co-inoculation mentioned above. Tissues were fixed in 10%
neutral-buffered formalin, embedded in paraffin. For staining of
microvessels, tissue sections fixed in 10% formalin were subjected
to immunostaining with a rat antibody against mouse CD31 (platelet
endothelial cell adhesion molecule, or PECAM-1; Pharmingen, San
Diego, CA, USA). The microvessel density was evaluated by counting
CD31-positive vessels using a light microscope at 200-fold
magnification in ≥10 randomly selected fields at the periphery of
each section.
Statistical analysis
Statistical evaluation was performed with the
two-tailed Student’s t-test unless mentioned otherwise in the text.
Differences with a P-value of <0.05 were considered
statistically significant.
Results
VEGF and HGF secretion induced by a
separate co-culture system with fibroblasts
To investigate whether mutual tumor-stromal
interaction induces VEGF expression, we determined VEGF
concentration of separately co-cultured supernatants. As shown in
Fig. 1A, fibroblasts alone secreted
little VEGF, indicating that VEGF in co-cultured supernatants was
derived from CT26 cells. VEGF secretion of CT26 transfectants
increased time-dependently and was enhanced by separate co-culture
with fibroblasts. The concentration of VEGF in CT26-NK4 was clearly
lower than that in CT26-NEO. NK4 gene transfer strongly inhibited
VEGF secretion of CT26 cells even in the co-culture system with
fibroblasts. This suggested that soluble mediators, which were
enhanced in the presence of fibroblasts, promoted VEGF secretion of
CT26 cells through tumor-stromal interaction, and one of the
soluble mediators seemed to be HGF, as VEGF secretion was
suppressed by NK4, an HGF antagonist.
We then examined HGF concentration in the separate
co-culture system. As shown in Fig.
1B, CT26 transfectants scarcely produced HGF, indicating that
HGF in the co-culture system was derived from fibroblasts. The HGF
production from fibroblasts increased time-dependently and was
enhanced by co-culture with CT26-NEO. However, NK4 gene transfer
suppressed HGF production down to the same level as that in
fibroblasts alone. This result was similar to our previous study
that demonstrated an obviously inductive effect when the
conditioned media from CT26 transfectants were added to
primary-cultured fibroblasts (18).
VEGF mRNA expression induced by a
separate co-culture system with fibroblasts
We evaluated VEGF mRNA expression of CT26
transfectants in the co-culture system by real-time RT-PCR. VEGF
mRNA of CT26-NEO in a co-culture system with fibroblasts was
evidently higher than that of CT26-NEO alone. On the other hand,
VEGF mRNA expression of CT26-NK4 was inhibited as compared to that
of control. Although fibroblasts promoted VEGF mRNA expression of
CT26-NK4, the change was smaller than that of CT26-NEO (Fig. 2). The changes of VEGF secretion in
cultured supernatants were supported at the mRNA level of the tumor
cells.
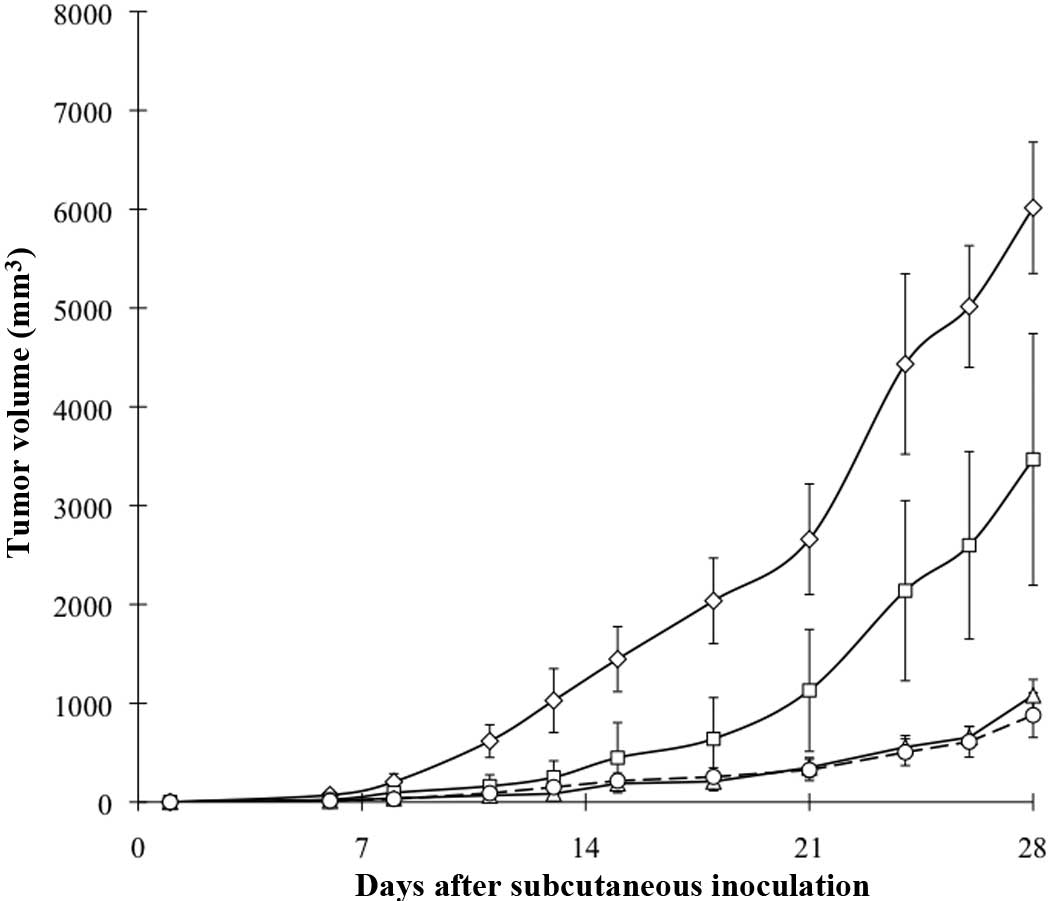
Tumor growth kinetics induced by
co-inoculation with fibroblasts
Fig. 3 shows the
growth curves of CT26-NEO and CT26-NK4 with/without fibroblast
co-inoculation. CT26-NEO tumor growth was markedly enhanced by
fibroblasts, while the effect of fibroblast co-inoculation was not
observed in CT26-NK4. This result revealed that NK4 gene transfer
blocked the enhancing effect of fibroblasts on in vivo CT26
s.c. tumor growth.
Implanted tumor vascularity induced by co
-inoculation with fibroblasts
We next examined whether co-inoculation with
fibroblasts would promote angiogenesis of homografts. Similar to
our previous study, the microvessel density of CT26-NK4 homografts
was evidently lower than that of CT26-NEO. Co-inoculation of
CT26-NEO with fibroblasts showed a significant increase in
microvessel density as compared with CT26-NEO cells alone, however,
this effect was not observed in CT26-NK4 (Fig. 4). Thus, NK4 gene transfer completely
canceled the enhancing effect of co-inoculation with fibroblasts on
CT26 tumor vascularity.
Discussion
In the present study, we found that tumor
angiogenesis involved co-operation between HGF and VEGF in CT26
tumor cells and tumor-stromal interaction consisting of the HGF
paracrine loop intricately. Furthermore, we gained understanding of
the importance of fibroblasts in tumor angiogenesis. Recent studies
have shown that interactions between tumor and stromal cells create
a unique microenvironment that is essential for tumor growth
(22,23). Tumor stroma contains several types,
such as activated fibroblasts, endothelial cells, and inflammatory
cells including macrophages. It has become clear that activated
fibroblasts in cancer stroma are prominent modifiers of tumor
progression.
It has been reported that normal fibroblasts inhibit
cancer progression (24). The
transdifferentiation of fibroblasts into cancer-associated
fibroblasts is modulated by cancer cell-derived cytokines, such as
transforming growth factor-β (TGF-β), platelet-derived growth
factor (PDGF), and bFGF (25–27),
which have been identified as HGF inducers (12,28).
Therefore, we used the fibroblasts activated by pre co-culture with
CT26 cells in both our in vitro and in vivo studies.
In the in vitro separate co-culture system,
fibroblast-derived HGF secretion was verified to increase
time-dependently as compared with fibroblast alone. Our previous
study revealed that the HGF/c-Met pathway activation promoted
secretion of HGF inducers and subsequent fibroblast-derived HGF.
Collectively, the results of our in vitro studies may
demonstrate the process of CT26 tumor progression mediated by HGF
paracrine loop that is amplified like a malignant cycle. On the
other hand, HGF promoted by paracrine loop amplification naturally
acts not only on tumor cells but also on endothelial cells and
induces angiogenesis. In fact, serum HGF level and/or c-Met
expression on tumor cells are reported to correlate tumor
vascularity with clinical prognosis (29,30).
Similar to fibroblast-derived HGF, tumor-derived
VEGF was also promoted in the separate co-culture system with
fibroblasts. HGF/c-Met pathway activation is reported to induce the
VEGF expression in cancer cells in addition to stimulating
endothelial cells to proliferate and migrate, inducing blood
formation (21,31). We also previously showed in an in
vitro study using the CT26 transfectants that VEGF expression
was induced by HGF and this action was effectively inhibited by
anti-HGF neutralizing antibody and HGF antagonist NK4. Furthermore,
we have revealed that tumor-derived VEGF expression also decreased
in in vivo CT26 tumor and this action involved HIF-1α
synthesis through HGF/c-Met pathway downstream; PI3K, MAPK, STAT3
signaling (32). From these
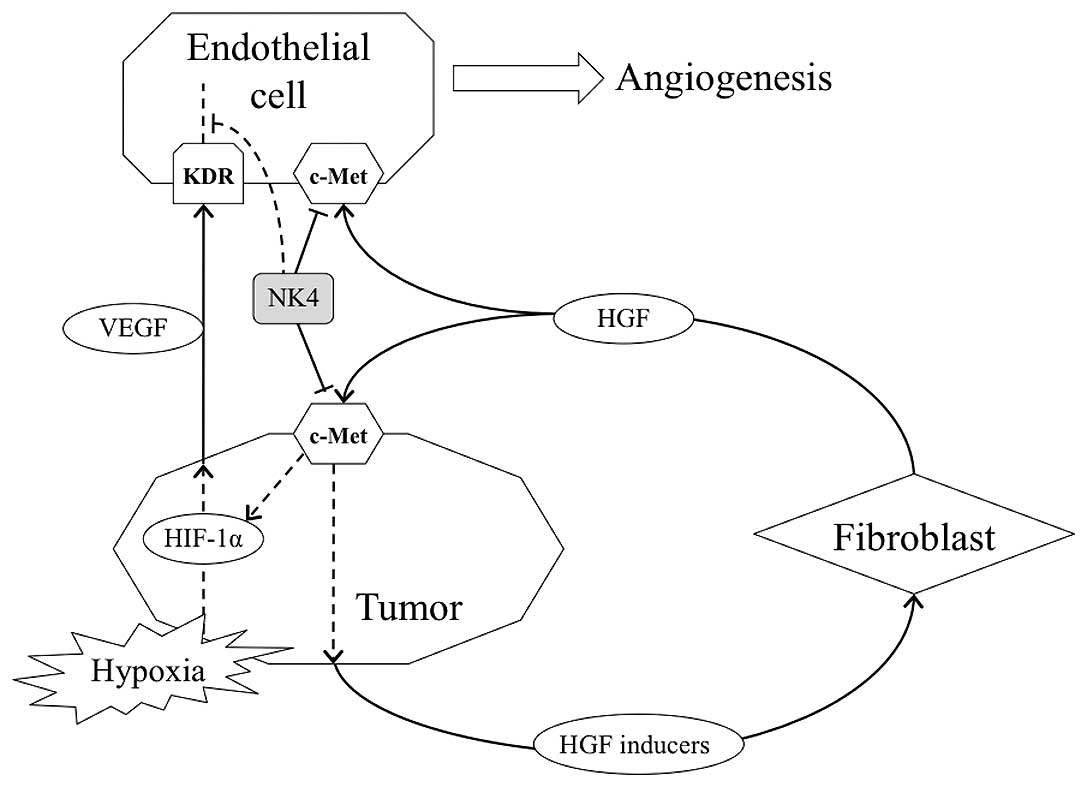
findings, HGF amplified by malignant cycle of the paracrine loop
possibly stimulates endothelial cells directly and indirectly via
increased secretion of tumor-derived VEGF, resulting in
accelerating tumor angiogenesis. Therefore, interruption of the
malignant cycle may hold the key to exert anti-angiogenesis for
VEGF-expressing and c-Met positive cancer. In in vivo
experiments of the present study, NK4 gene transfer completely
blocked angiogenesis and tumor growth which were enhanced by
co-inoculation with fibroblasts.
Since angiogenesis is critical for tumor growth,
increased angiogenesis coincides with increased tumor cell entry
into blood circulation and thus facilitates metastasis (33). Therapeutic approaches using
angiogenesis inhibitors have received much attention. NK4, an HGF
antagonist, is also known to have anti-angiogenic activity, which
is independent of its activity as an HGF antagonist (15). Accumulating evidence has shown that
NK4 suppresses primary tumor growth predominantly through
inhibition of tumor angiogenesis. Our results showed that
fibroblast-derived HGF and tumor-derived VEGF, induced by
co-culture of CT26 cells with fibroblasts, were inhibited by NK4
gene transfer. This was due to interruption of the malignant cycle
of the HGF paracrine loop by blockade of the HGF/c-Met pathway;
that is, the activity of NK4 as an HGF antagonist. Furthermore, the
findings in our in vivo study are consistent with previous
studies. Thus, anti-angiogenesis strategy by NK4 is expected to
exert additional effects on c-Met positive tumor grown up depending
on HGF paracrine loop through intricate tumor-stromal interaction
(Fig. 5).
In conclusion, we have demonstrated that the
HGF/c-Met pathway regulated VEGF expression of CT26 cells through
the HGF paracrine loop. For anti-tumor strategy, the interruption
of HGF paracrine loop by NK4 possibly exerts potent anti-angiogenic
activity via inhibition of tumor-derived VEGF expression in
addition to the previously reported mechanisms, such as suppression
of endothelial cell proliferation by antagonizing HGF/c-Met pathway
or blockade of intracellular signaling downstream of VEGF and bFGF.
Therefore, the HGF/c-Met pathway may be a significant candidate for
molecular targeting strategy against tumor angiogenesis.
References
|
1
|
Nakamura T, Nishizawa T, Hagiya M, Seki T,
Shimonishi M, Sugimura A, Tashiro K and Shimizu S: Molecular
cloning and expression of human hepatocyte growth factor. Nature.
342:440–443. 1989. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Matsumoto K, Okazaki H and Nakamura T:
Up-regulation of hepatocyte growth factor gene expression by
interleukin-1 in human skin fibroblasts. Biochem Biophys Res
Commun. 188:235–243. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brinkmann V, Foroutan H, Sachs M, Weidner
KM and Birchmeier W: Hepatocyte growth factor/scatter factor
induces a variety of tissue-specific morphogenic programs in
epithelial cells. J Cell Biol. 131:1573–1586. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schmidt C, Bladt F, Goedecke S, Brinkmann
V, Zschiesche W, Sharpe M, Gherardi E and Birchmeier C: Scatter
factor/hepatocyte growth factor is essential for liver development.
Nature. 373:699–702. 1995. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bussolino F, Di Renzo MF, Ziche M,
Bocchietto E, Olivero M, Naldini L, Gaudino G, Tamagnone L, Coffer
A and Comoglio PM: Hepatocyte growth factor is a potent angiogenic
factor which stimulates endothelial cell motility and growth. J
Cell Biol. 119:629–641. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Di Renzo MF, Olivero M, Giacomini A, Porte
H, Chastre E, Mirossay L, Nordlinger B, Bretti S, Bottardi S,
Giordano S, Plebani M, Gespach C and Comoglio PM: Overexpression
and amplification of the met/HGF receptor gene during the
progression of colorectal cancer. Clin Cancer Res. 1:147–154.
1995.PubMed/NCBI
|
|
7
|
Matsumoto K and Nakamura T: Emerging
multipotent aspects of hepatocyte growth factor. J Biochem.
119:591–600. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Birchmeier C, Birchmeier W, Gherardi E and
Vande Woude GF: Met, metastasis, motility and more. Nat Rev Mol
Cell Biol. 4:915–925. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Miller CT, Lin L, Casper AM, Lim J, Thomas
DG, Orringer MB, Chang AC, Chambers AF, Giordano TJ, Glover TW and
Beer DG: Genomic amplification of MET with boundaries within
fragile site FRA7G and upregulation of MET pathways in esophageal
adenocarcinoma. Oncogene. 25:409–418. 2006.PubMed/NCBI
|
|
10
|
Grant DS, Kleinman HK, Goldberg ID,
Bhargava MM, Nickoloff BJ, Kinsella JL, Polverini P and Rosen EM:
Scatter factor induces blood vessel formation in vivo. Proc Natl
Acad Sci USA. 90:1937–1941. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Di Renzo MF, Poulsom R, Olivero M,
Comoglio PM and Lemoine NR: Expression of the Met/hepatocyte growth
factor receptor in human pancreatic cancer. Cancer Res.
55:1129–1138. 1995.PubMed/NCBI
|
|
12
|
Nakamura T, Matsumoto K, Kiritoshi A, Tano
Y and Nakamura T: Induction of hepatocyte growth factor in
fibroblasts by tumor-derived factors affects invasive growth of
tumor cells: in vitro analysis of tumor-stromal interactions.
Cancer Res. 57:3305–3313. 1997.
|
|
13
|
Date K, Matsumoto K, Shimura H, Tanaka M
and Nakamura T: HGF/NK4 is a specific antagonist for pleiotrophic
actions of hepatocyte growth factor. FEBS Lett. 420:1–6. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kuba K, Matsumoto K, Date K, Shimura H,
Tanaka M and Nakamura T: HGF/NK4, a four-kringle antagonist of
hepatocyte growth factor, is an angiogenesis inhibitor that
suppresses tumor growth and metastasis in mice. Cancer Res.
60:6737–6743. 2000.PubMed/NCBI
|
|
15
|
Matsumoto K and Nakamura T: Mechanisms and
significance of bifunctional NK4 in cancer treatment. Biochem
Biophys Res Commun. 333:316–327. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Saimura M, Nagai E, Mizumoto K, Maehara N,
Minamishima YA, Katano M, Matsumoto K, Nakamura T and Tanaka M:
Tumor suppression through angiogenesis inhibition by SUIT-2
pancreatic cancer cells genetically engineered to secrete NK4. Clin
Cancer Res. 8:3243–3249. 2002.
|
|
17
|
Tomioka D, Maehara N, Kuba K, Mizumoto K,
Tanaka M, Matsumoto K and Nakamura T: Inhibition of growth,
invasion, and metastasis of human pancreatic carcinoma cells by NK4
in an orthotopic mouse model. Cancer Res. 61:7518–7524.
2001.PubMed/NCBI
|
|
18
|
Kubota T, Fujiwara H, Amaike H, Takashima
K, Inada S, Atsuji K, Yoshimura M, Matsumoto K, Nakamura T and
Yamagishi H: Reduced HGF expression in subcutaneous CT26 tumor
genetically modified to secrete NK4 and its possible relation with
antitumor effects. Cancer Sci. 95:321–327. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gerritsen ME, Tomlinson JE, Zlot C, Ziman
M and Hwang S: Using gene expression profiling to identify the
molecular basis of the synergistic actions of hepatocyte growth
factor and vascular endothelial growth factor in human endothelial
cells. Br J Pharmacol. 140:595–610. 2003. View Article : Google Scholar
|
|
20
|
Wojta J, Kaun C, Breuss JM, Koshelnick Y,
Beckmann R, Hattey E, Mildner M, Weninger W, Nakamura T, Tschachler
E and Binder BR: Hepatocyte growth factor increases expression of
vascular endothelial growth factor and plasminogen activator
inhibitor-1 in human keratinocytes and the vascular endothelial
growth factor receptor flk-1 in human endothelial cells. Lab
Invest. 79:427–438. 1999.
|
|
21
|
Dong G, Chen Z, Li ZY, Yeh NT, Bancroft CC
and Van Waes C: Hepatocyte growth factor/scatter factor-induced
activation of MEK and PI3K signal pathways contributes to
expression of proangiogenic cytokines interleukin-8 and vascular
endothelial growth factor in head and neck squamous cell carcinoma.
Cancer Res. 61:5911–5918. 2001.
|
|
22
|
Mantovani A, Allavena P, Sica A and
Balkwill F: Cancer-related inflammation. Nature. 454:436–444. 2008.
View Article : Google Scholar
|
|
23
|
Whiteside TL: The tumor microenvironment
and its role in promoting tumor growth. Oncogene. 27:5904–5912.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kuperwasser C, Chavarria T, Wu M, Magrane
G, Gray JW, Carey L, Richardson A and Weinberg RA: Reconstruction
of functionally normal and malignant human breast tissues in mice.
Proc Natl Acad Sci USA. 101:4966–4971. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
De Wever O and Mareel M: Role of tissue
stroma in cancer cell invasion. J Pathol. 200:429–447.
2003.PubMed/NCBI
|
|
26
|
Pietras K, Sjöblom T, Rubin K, Heldin CH
and Ostman A: PDGF receptors as cancer drug targets. Cancer Cell.
3:439–443. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kitadai Y: Cancer-stromal cell interaction
and tumor angiogenesis in gastric cancer. Cancer Microenviron.
3:109–116. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Matsumoto K, Date K, Shimura H and
Nakamura T: Acquisition of invasive phenotype in gallbladder cancer
cells via mutual interaction of stromal fibroblasts and cancer
cells as mediated by hepatocyte growth factor. Jpn J Cancer Res.
87:702–710. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ren Y, Cao B, Law S, Xie Y, Lee PY, Cheung
L, Chen Y, Huang X, Chan HM, Zhao P, Luk J, Vande Woude G and Wong
J: Hepatocyte growth factor promotes cancer cell migration and
angiogenic factors expression: a prognostic marker of human
esophageal squamous cell carcinomas. Clin Cancer Res. 11:6190–6197.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kaposi-Novak P, Lee JS, Gòmez-Quiroz L,
Coulouarn C, Factor VM and Thorgeirsson SS: Met-regulated
expression signature defines a subset of human hepatocellular
carcinomas with poor prognosis and aggressive phenotype. J Clin
Invest. 116:1582–1595. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Moriyama T, Kataoka H, Hamasuna R,
Yokogami K, Uehara H, Kawano H, Goya T, Tsubouchi H, Koono M and
Wakisaka S: Up-regulation of vascular endothelial growth factor
induced by hepatocyte growth factor/scatter factor stimulation in
human glioma cells. Biochem Biophys Res Commun. 249:73–77. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Matsumura A, Kubota T, Taiyoh H, Fujiwara
H, Okamoto K, Ichikawa D, Shiozaki A, Komatsu S, Nakanishi M, Kuriu
Y, Murayama Y, Ikoma H, Ochiai T, Kokuba Y, Nakamura T, Matsumoto K
and Otsuji E: HGF regulates VEGF expression via the c-Met receptor
downstream pathways, PI3K/Akt, MAPK and STAT3, in CT26 murine
cells. Int J Oncol. 42:535–542. 2013.PubMed/NCBI
|
|
33
|
Hanahan D and Folkman J: Patterns and
emerging mechanisms of the angiogenic switch during tumorigenesis.
Cell. 86:353–364. 1996. View Article : Google Scholar : PubMed/NCBI
|