Introduction
Chronic myelogenous leukemia (CML) is a malignancy
arising from hematopoietic stem cells. CML has a typical
progressive course with transition from a chronic phase to a
terminal blast crisis phase (1,2). The
therapeutic effects following treatment of blastic phase CML
(CML-Bp) are currently disappointing. Prolonging the chronic phase
and preventing the blastic phase constitute the main goals of CML
treatment. However, the mechanisms that lead to disease progression
remain unclear.
Bone marrow mesenchymal stem cells (BMMSCs)
constitute a subset of non-hematopoietic adult stem cells
originating from the mesoderm (3,4).
BMMSCs possess a self-renewal ability and are characterized by
multilineage differentiation into not only mesoderm-lineage, such
as chondrocytes, osteocytes, adipocytes, myocytes and astrocytes,
but also ectodermic and endodermic cells (5–10). The
bone marrow microenvironment supports and regulates the
proliferation and differentiation of hematopoietic cells. BMMSCs
are rare residents of the bone marrow microenvironment, but play
important roles in maintaining the bone marrow microenvironment.
Abnormal hematopoiesis in CML is related, at least in part, to
abnormalities in the hematopoietic cells themselves leading to
abnormal interactions between CML progenitors and the marrow
microenvironment. The role of the microenvironment itself in CML
has not been well characterized. It was found that BMMSCs regulate
the proliferation of leukemia cells through the secretion of
cytokines (11). In addition, the
interaction between leukemia cells and BMMSCs affects the
hematopoietic microenvironment (12).
In the present study, the biological characteristics
of BMMSCs were determined including proliferation, apoptosis and
the secretion of cytokines during CML-Bp. The effects of BMMSCs in
CML-Bp on human CML K562 cells and the CML-Bp original generation
leukemia cells were also investigated.
Materials and methods
Patients and healthy donors
Thirty CML patients who were treated at the
Department of Hematology, The Second Hospital, Hebei Medical
University (Shijiazhuang, China) were included in the present
study. These patients included 20 (12 males, 9 females) chronic
phase CML (CML-Cp) patients and 10 (6 males, 4 females) CML-Bp
patients. Ten healthy donors were included in the study as the
control group. All the individuals provided written informed
consent prior to enrollment. This study was approved by the Medical
Ethics Committee of the Second Hospital of Hebei Medical
University.
Cell culture
For primary CML-Bp leukemia cells, after informed
consent was obtained from all the subjects, blood mononuclear cells
were collected by bone marrow aspiration (heparinized bone marrow).
The heparinized bone marrow was diluted twice with
phosphate-buffered saline (PBS). Nucleated cells were then isolated
by density-gradient centrifugation and cultured in RPMI-1640
supplemented with 100 U/ml penicillin, 100 U/ml streptomycin and
10% fetal bovine serum (FBS) at 37°C with 5% CO2.
K562 cells, obtained from a patient with chronic
myeloid leukemia in blast crisis, were constantly preserved in our
laboratory. The culture conditions were identical to primary CML-Bp
leukemia cells.
The blood mononuclear cells were isolated by
density-gradient centrifugation, incubated and cultured in BMMSC
culture medium supplemented with 100 U/ml penicillin, 100 U/ml
streptomycin and 10% FBS at 37°C with 5% CO2. The medium
was replaced twice per week and non-adherent cells were discarded.
When BMMSCs were 80–90% confluent, the cells were digested with
trypsin and harvested in the medium. BMMSCs of the 4th or 5th
passage were used in this study.
MTT assay
The cells were seeded at a density of
5×103/well in 96-well plates and cultured in a 5%
CO2 incubator. The cells were treated uner the indicated
conditions, such as with adriamycin (ADM; Zhejiang Pharmaceutical
Co., Ltd., China). For the indicated time periods, 20 μl of MTT
solution (Amresco, Solon, OH, USA) was added to each well. After 4
h of incubation, the medium was discarded and 150 μl of dimethyl
sulfoxide (DMSO) was added into each well. The cells were shaken in
the dark for 10 min. The absorbance reading for each well was
performed at 490 nm using a microplate reader. Each assay was
repeated at least 3 times.
Apoptosis assay by flow cytometry
The cells were treated with ADM or Dickkopf-1 (DKK1,
recombinant human DKK1 protein; R&D Systems, Minneapolis, MN,
USA). Both suspension and adherent cells were collected, and washed
twice with ice-cold PBS. The cells were then suspended in 200 μl of
binding buffer and 10 μl of Annexin V-FITC for 15 min in the dark.
Subsequently, 300 μl of binding buffer and 5 μl of propidium iodide
(PI) were added to each sample. Finally, the cells were analyzed
using BD FACSDiva flow cytometry (BD FACS Canto™ II) with CellQuest
software.
RNA extraction and reverse
transcription-polymerase chain reaction (RT-PCR) analysis
RT-PCR was used to determine the expression of
BCR/ABL, thombopoietin (TPO), interleukin (IL)-6, IL-12, stem cell
factor (SCF) and granulocyte-colony stimulating factor (G-CSF).
Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad,
CA, USA) according to the manufacturer’s instructions. The primers
used are listed in Table I. The
conditions for PCR were as follows: denaturation at 94°C for 4 min,
denaturation at 94°C for 50 sec, annealing at 52°C for 45 sec, and
extension at 72°C for 45 sec, 35 cycles, and a final 10 min at
72°C. PCR products were visualized by gel electrophoresis on 1.5%
agarose (w/v) gel, and then viewed using an ultraviolet photometry
(UVP) bioimaging system.
 | Table IList of primers used in RT-PCR. |
Table I
List of primers used in RT-PCR.
| Gene | Primer | Size (bp) |
|---|
| IL-6 | F:
5′-CACACAGACAGCCACTCACC-3′ | 330 |
| R:
5′-TCTTTGGAAGGTTCAGGTTGT-3′ | |
| IL-12 | F:
5′-TTCTCCCTGACATTCTGCG-3′ | 356 |
| R:
5′-CCATTCGCTCCAAGATGAG-3′ | |
| SCF | F:
5′-GGAAAGAAGACAACGACACG-3′ | 143 |
| R:
5′-GGGTCAGGAATAAACCTCAAGT-3′ | |
| TPO | F:
5′-GACCTCCGAGTCCTCAGTAAAC-3′ | 125 |
| R:
5′-GAATGTCCTGTGCCTTGGT-3′ | |
| G-CSF | F:
5′-GACCCAAGAGCAGTTTCC-3′ | 138 |
| R:
5′-AGTCACAGCGGAGATAGTGC-3′ | |
| BCL/ABL | F:
5′-GCTTCTCCCTGACATCCGTG-3′ | 232 |
| R:
5′-CGAGCGGCTTCACTCAGACC-3′ | |
| F:
5′-CTCCAGACTGTCCACAGCATTCCG-3′ | 165 |
| R:
5′-CAGACCCTGAGGCTCAAAGTCAGA-3′ | |
| GAPDH | F:
5′-TGAACGGGAAGCTCACTGG-3′ | 120 |
| R:
5′-GCTTCACCACCTTCTTGATGTC-3′ | |
Western blot analysis
Total protein was extracted and quantified according
to the manufacturer’s protocol. Each equal amount of protein was
loaded on sodium dodecyl sulfate-polyacrylamide gel at 100 V for 2
h, and then the protein was transferred to polyvinylidene fluoride
(PVDF)-membranes. The membranes were blocked in 5% fat-free milk at
room temperature for 2 h, and the blots were stained with specific
primary antibodies, including anti-bcl-2 (BioWorld, Atlanta, GA,
USA), anti-bax (BioWorld), anti-active caspase-3 (Cell Signaling
Technology, Inc., Danvers, MA, USA), anti-β-catenin (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) and anti-GAPDH antibodies
(Santa Cruz Biotechnology, Inc.) overnight at 4°C. The membranes
were then washed and incubated with goat anti-rabbit HRP-IgG (Santa
Cruz Biotechnology, Inc.) for 1 h at room temperature, and observed
with a chemiluminescent substrate. Bound immunoglobulins were
removed from the membranes by washing twice with Restore™ Western
Blot Stripping Buffer, and the signal was visualized by enhanced
chemiluminescence and detected using the ChemiDoc XRS+ system
(Bio-Rad, Hercules, CA, USA); the signal was analyzed by the Image
Lab (ECL). GAPDH was used as an internal control.
Statistical analysis
Data are presented as means ± standard deviation
(SD). All the statistical analyses were performed using SPSS 13.0
software. The statistical analysis of results was carried out using
Student’s t-test and one-way ANOVA analysis. P<0.05 was
considered to indicate a statistically significant difference.
Results
Growth, proliferation and passage of
CML-Bp BMMSCs
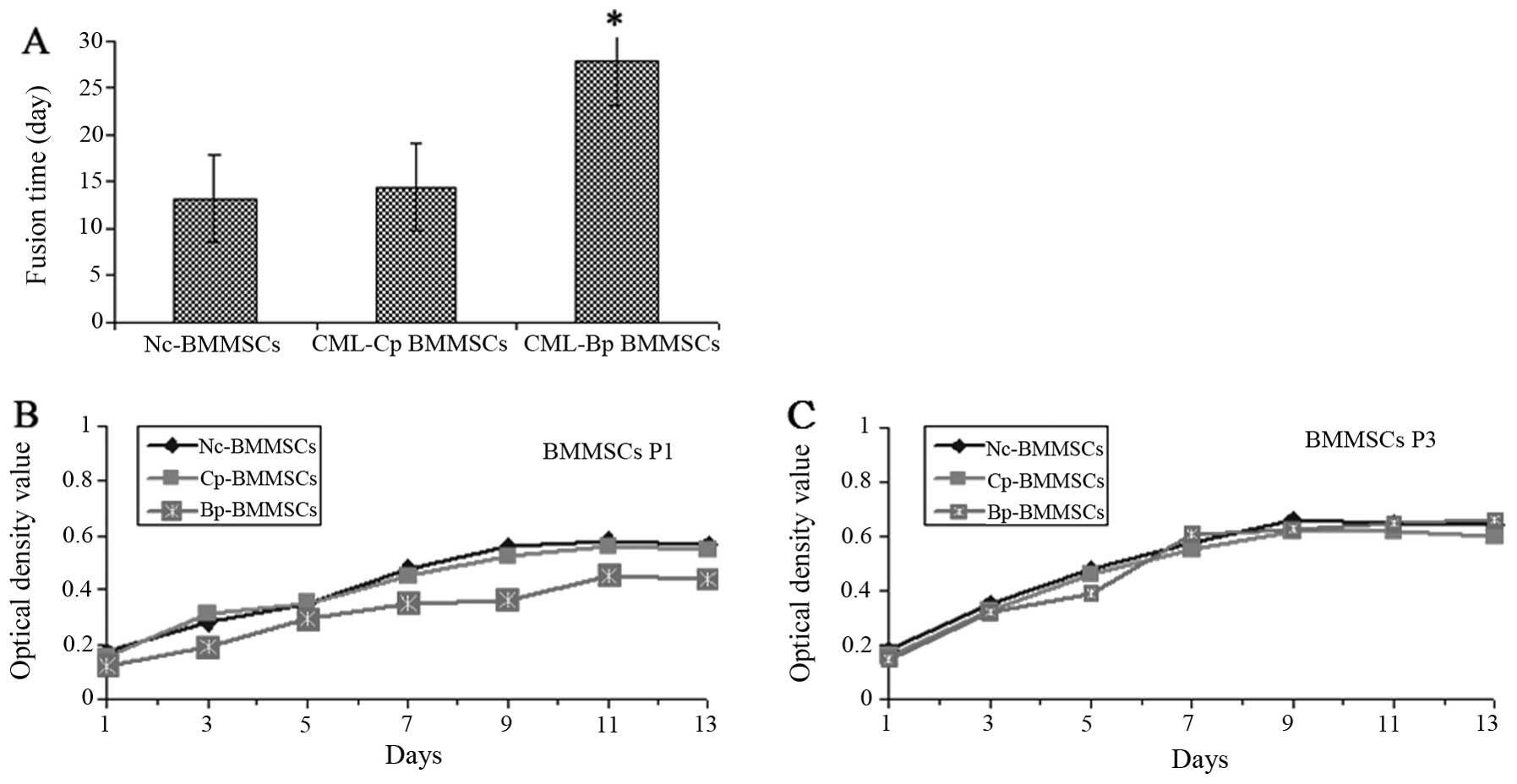
As shown in Fig. 1A,
the 90% fusion time of primary CML-Bp BMMSCs was significantly
longer compared with Nc-BMMSCs and CML-Cp BMMSCs (27.75±2.29 vs.
13.18±1.31 and 14.46±1.56, P<0.05). CML-Bp BMMSCs at first
passage grew slowly with a few colonies and adherent cells
(Fig. 1B). At the third
generations, the cell proliferative capacity of CML-Bp BMMSCs was
enhanced, similarly to CML-Cp BMMSCs and Nc-BMMSCs (Fig. 1C).
Apoptotic rate of CML-Bp BMMSCs
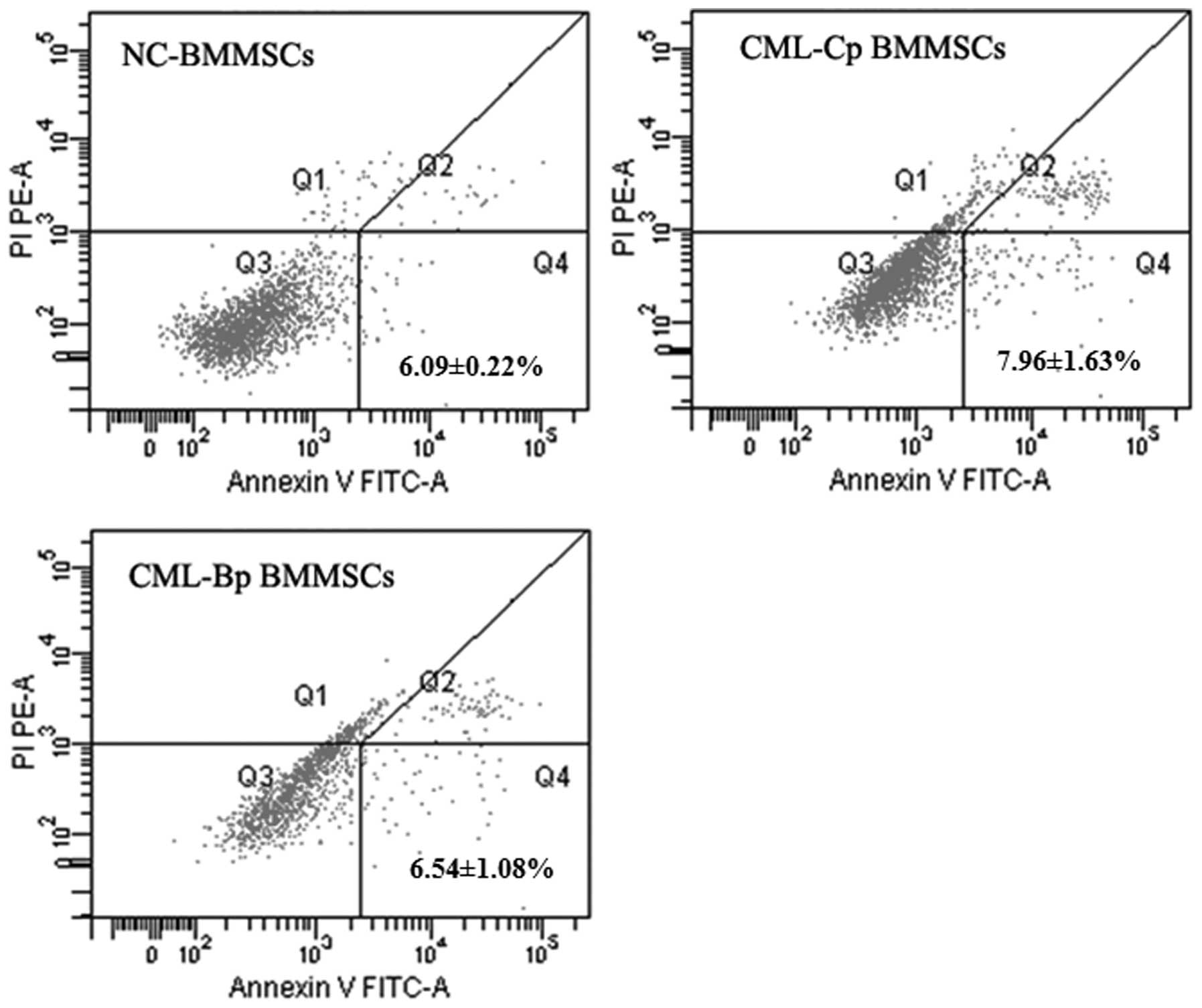
As shown in Fig. 2,
the apoptotic rate of Nc-BMMSCs, CML-Cp BMMSCs and CML-Bp BMMSCs
was 6.09±0.22, 7.96±1.63 and 6.54±1.08%, respectively. No
significant difference was observed among the three groups
(P>0.05).
Expression of Bcr/abl fusion gene and
cytokine secretion
As shown in Fig. 3A,
the Bcr/abl fusion gene was expressed in human CML K562 cells and
CML-Bp primary leukemia cells, while it was not expressed in CML-Bp
BMMSCs, Nc-BMMSCs and CML-Cp BMMSCs. The expression of TPO, IL-6,
IL-12 and SCF in the CML-Bp BMMSC group was significantly lower
compared with the CML-Cp BMMSC group (P<0.05). However, G-CSF
secretion was not significantly different between these two groups
(P>0.05) (Fig. 3B and C).
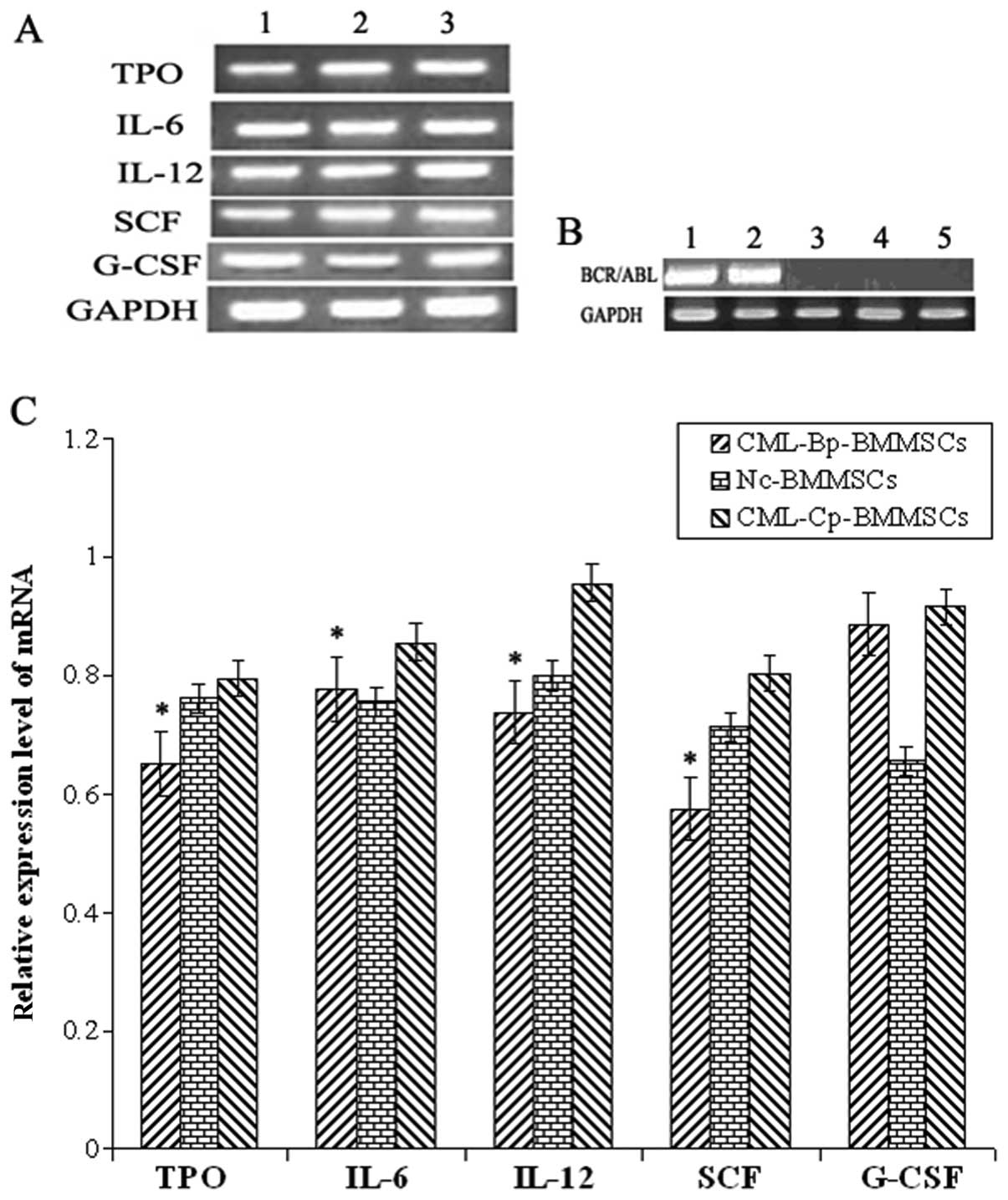 | Figure 3Expression of BCR/ABL and cytokines
detected using RT-PCR. (A and C) Expression of TPO, IL-6, IL-12,
SCF and G-CSF mRNA detected by RT-PCR. Lane 1, CML-Bp-BMMSCs; lane
2, Nc-BMMSCs; lane 3, CML-Cp-BMMSCs. *P<0.05 compared
with CML-Cp BMMSCs. (B) Expression of BCR/ABL mRNA detected by
RT-PCR. Lane 1, K562 cells; lane 2, CML-Bp primary leukemia cells;
lane 3, Nc-BMMSCs; lane 4, CML-Cp BMMSCs; lane 5, CML-Bp BMMSCs.
CML, chronic myelogenous leukemia; Cp, chronic phase; Bp, blastic
phase; BMMSCs, bone marrow mesenchymal stem cells. |
Effects of BMMSCs on the proliferation of
K562 and primary CML-Bp cells
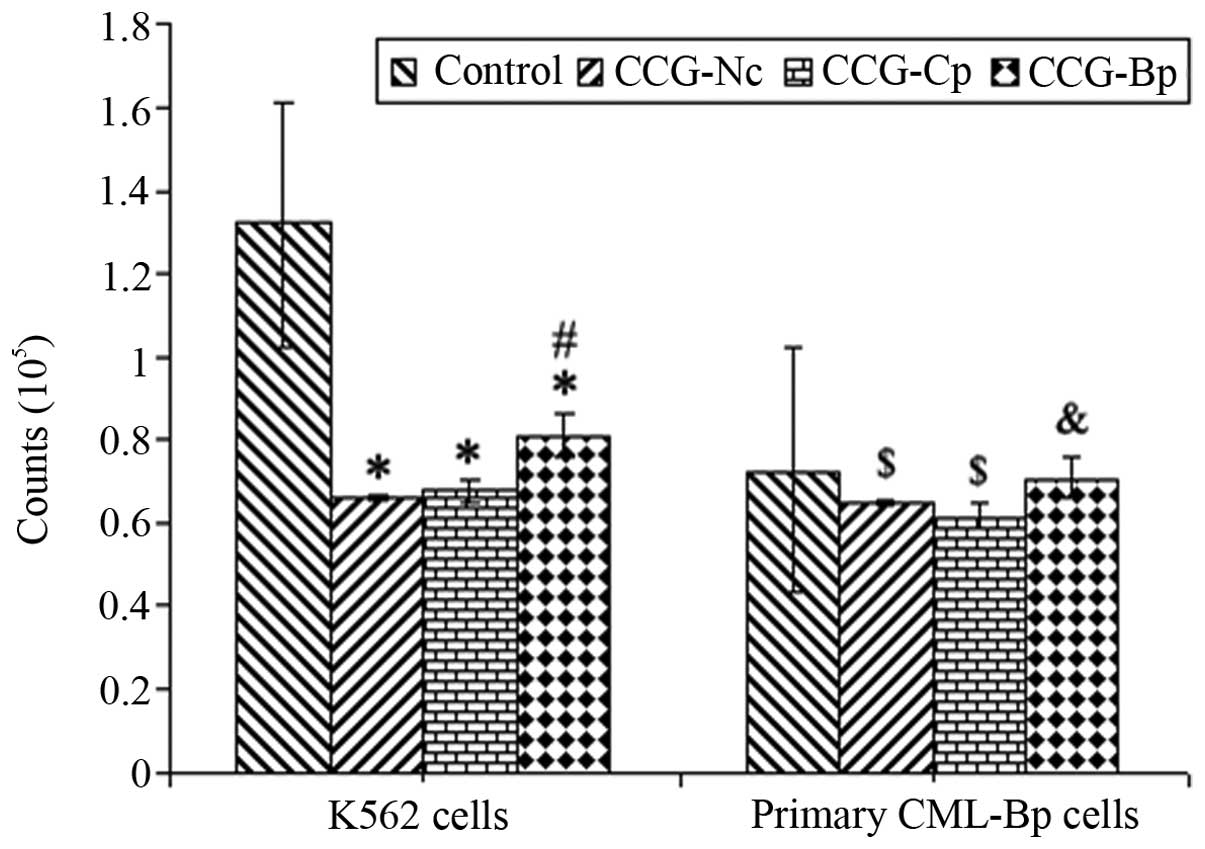
As shown in Fig. 4,
K562 cells and primary CML-Bp cells were co-cultured with Nc-BMMSCs
(CCG-Nc group), CML-Bp BMMSCs (CCG-Bp group) and CML-Cp BMMSCs
(CCG-Cp group) for 48 h, respectively. The ratio of BMMSCs to K562
or primary CML-Bp cells was 1:10. The growth of K562 cells was
significantly decreased following co-culture with BMMSCs, with a
slight decrease in the CML-Bp BMMSC group (P<0.05). However, the
growth of primary CML-Bp cells was not significantly altered
following co-culture with BMMSCs, with a slight decrease in the
CML-Bp BMMSC group (P>0.05). These results to some extent
indicate that CML-Bp BMMSCs only slightly inhibited K562 cell
proliferation, while they did not inhibit the growth of primary
CML-Bp leukemia cells.
Effects of BMMSCs on the proliferation of
K562 and primary CML-Bp cells following ADM treatment
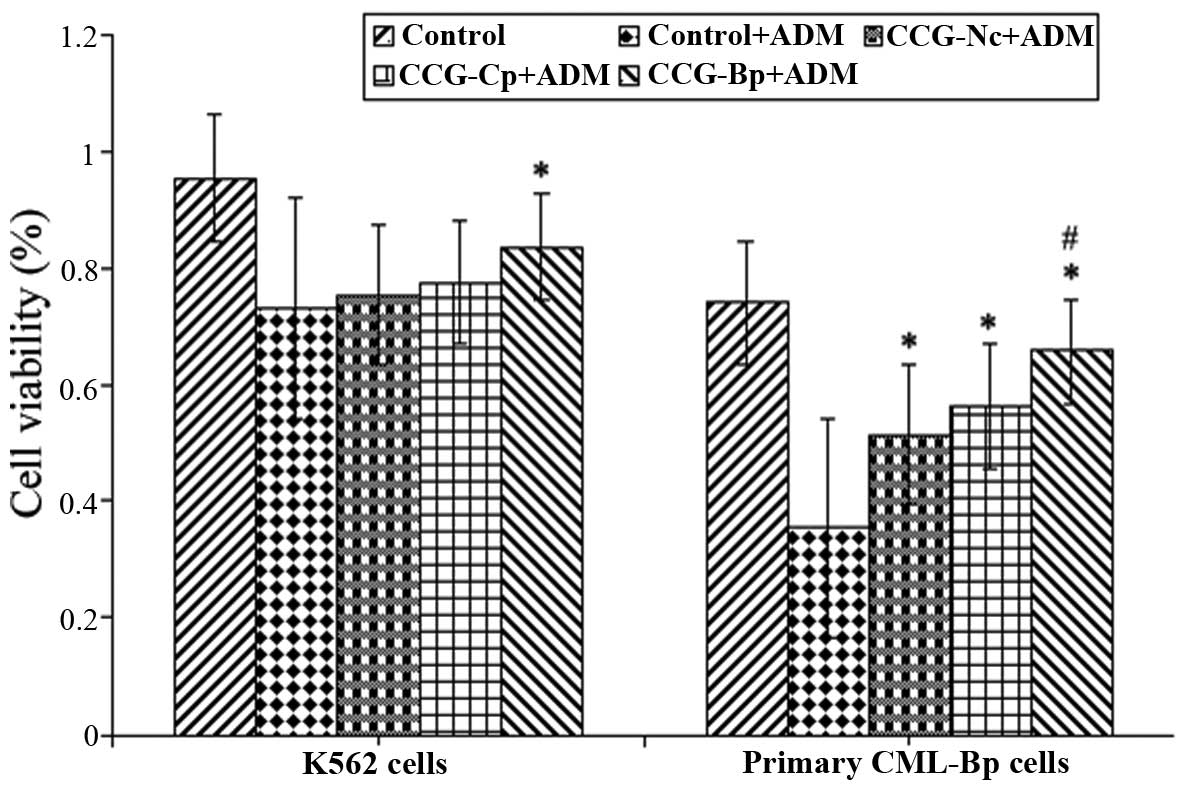
As shown in Fig. 5,
K562 and primary CML-Bp cells were co-cultured with Nc-BMMSCs,
CML-Bp BMMSCs and CML-Cp BMMSCs for 48 h, respectively. The cells
were then treated with ADM for 48 h. The cell viability of the
CML-Bp BMMSC group was significantly increased when compared with
the ADM control group (83.78±5.17 vs. 73.13±2.42%, P<0.05). The
cell viability of the Nc-BMMSC and CML-Cp BMMSC groups was
75.45±3.27 and 77.56±3.11%, respectively, which was not
significantly different compared with the ADM control group
(P>0.05). Thus, these data indicate that CML-Bp BMMSCs, but not
CML-Cp BMMSCs or Nc BMMSC, protected K562 cells against apoptosis.
In addition, CML-Bp BMMSCs were shown to protect the primary CML-Bp
leukemia cells.
Effects of BMMSCs on ADM-induced
apoptosis of K562 and primary CML-Bp cells
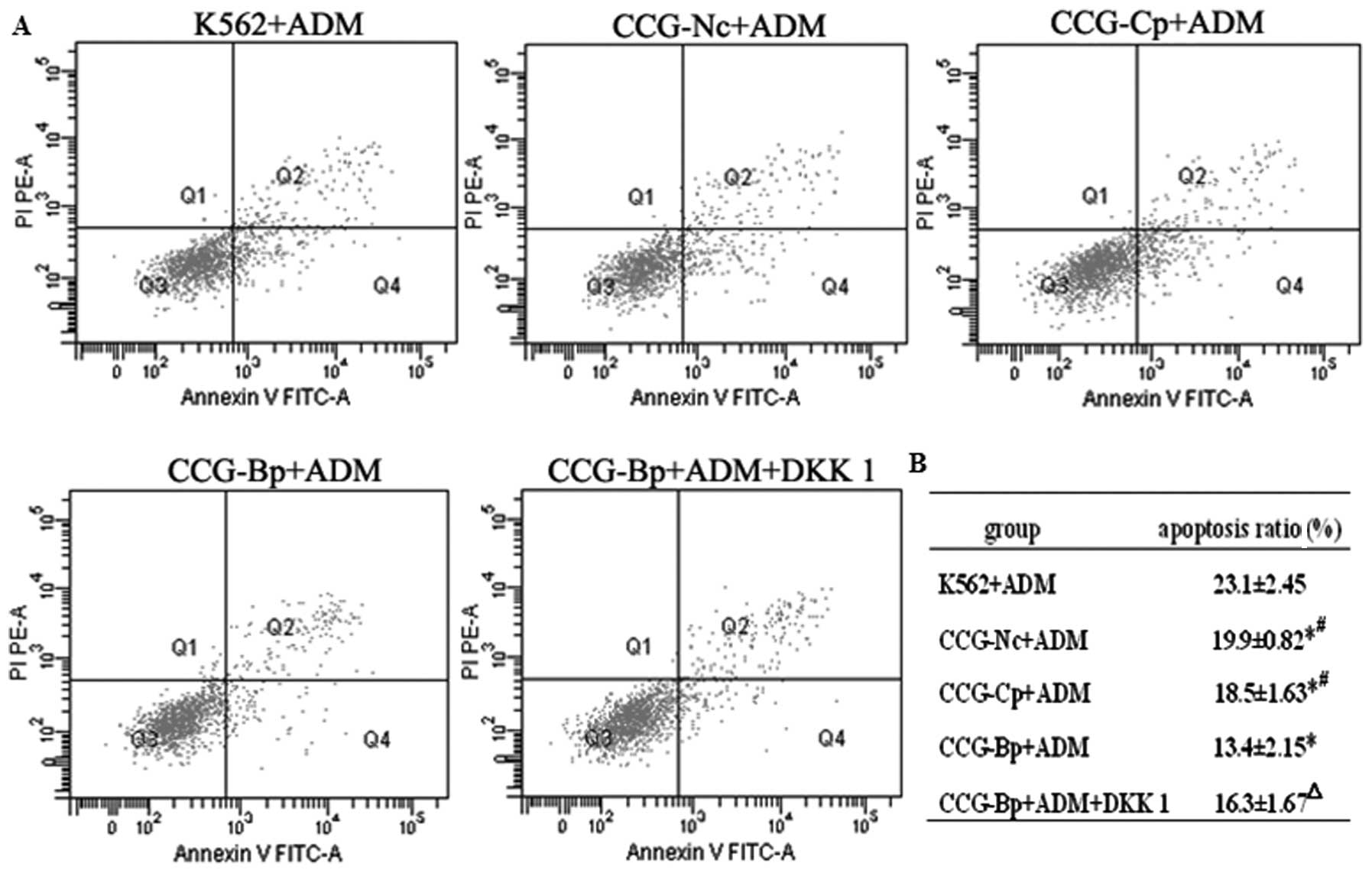
As shown in Fig. 6,
K562 cells were co-cultured with Nc-BMMSCs, CML-Bp BMMSCs and
CML-Cp BMMSCs for 48 h, respectively. The cells were then treated
with ADM for 48 h. The apoptotic rate of the Nc-BMMSC, CML-Cp BMMSC
and CML-Bp BMMSC groups was 19.9±0.82, 18.5±1.63 and 13.4±2.15%,
respectively, which was significantly decreased compared with the
apoptotic rate of the control group (23.1±2.45%, P<0.05). The
most significantly decreased apoptotic rate was observed in the
CML-Bp BMMSC group.
As shown in Fig. 7,
primary CML-Bp leukemia cells were co-cultured with Nc-BMMSCs,
CML-Bp BMMSCs and CML-Cp BMMSCs for 48 h, respectively. The cells
were then treated with ADM for 48 h. The apoptotic rate of the
Nc-BMMSC, CML-Cp BMMSC and CML-Bp BMMSC groups was 45.9±2.82,
40.1±5.63 and 30.5±2.33%, respectively, which was significantly
decreased compared with the control group (60.9±2.52%, P<0.05).
The most significantly decreased apoptotic rate was observed in the
CML-Bp BMMSC group. These results indicate that CML-Bp BMMSCs
reduced ADM-induced leukemia cell apoptosis.
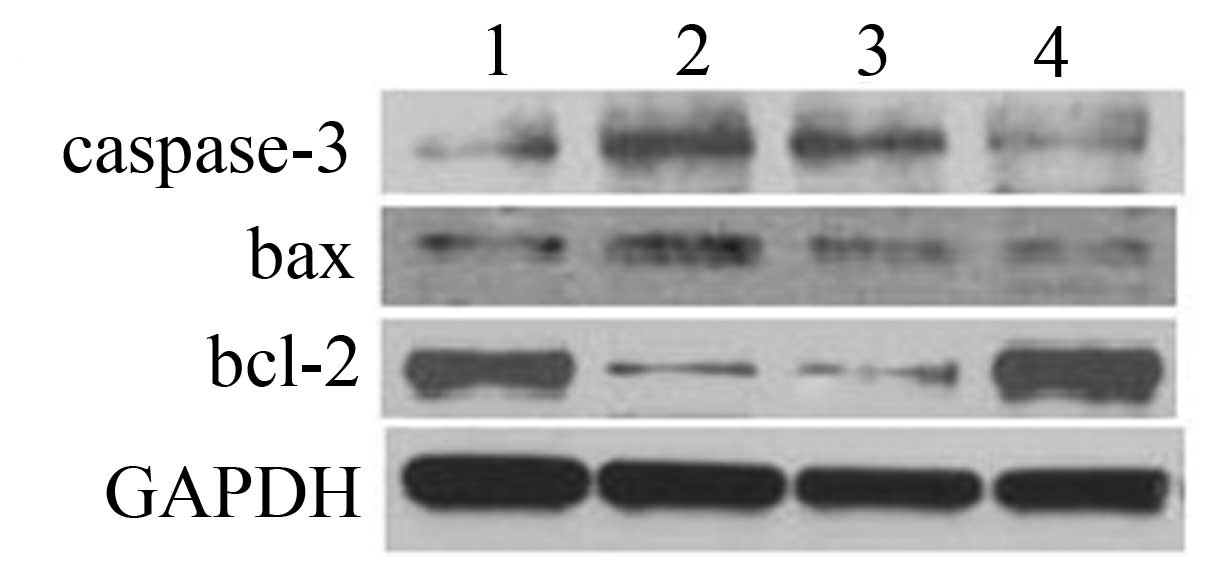
Expression of bcl-2, bax and
caspase-3
The expression of caspase-3 and bax of K562 cells at
the protein level was significantly increased following ADM
treatment, while the expression of bcl-2 was significantly
decreased (Fig. 8), suggesting that
K562 cells underwent apoptosis in response to ADM treatment. In the
CML-Bp BMMSC group, the expression of caspase-3 and bax were
significantly decreased, while bcl-2 expression was significantly
increased compared with the ADM control group. There was no
significant change in the CML-Cp BMMSC group compared with the ADM
control group. These results suggest that CML-Bp BMMSCs inhibited
ADM-induced K562 cell apoptosis.
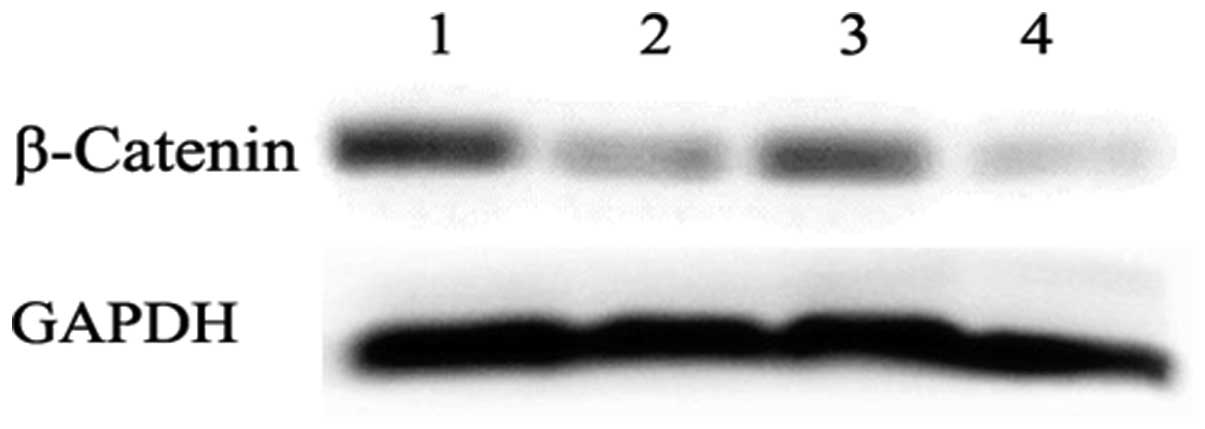
DKK1 protein reverses the protective
effect of CML-Bp BMMSCs on K562 cells
DKK1 is a secretory antagonist of the Wnt/β-catenin
signaling pathway and has been shown to play a crucial role in
carcinogenesis. Our results showed that the ADM-induced cell
apoptosis of K562 cells in the CML-Bp BMMSC group recovered after
DKK1 treatment (13.4±2.15 vs. 16.3±1.67%, P<0.05; Fig. 6). The results from western blot
analysis showed that β-catenin expression of K562 cells was
significantly decreased following ADM treatment. In the ADM-treated
CML-Bp BMMSC group, the expression of β-catenin was downregulated
following DKK1 treatment (Fig. 9).
These results to some extent suggest that CML-Bp BMMSCs antagonize
ADM-induced K562 cell apoptosis, potentially through the activation
of the Wnt pathway.
Discussion
To elucidate the correlation between BMMSCs and
CML-Bp, the determination of the biological characteristics of
BMMSCs from CML-Bp and their effect on leukemia cells is required.
In the present study, we found that the growth of primary CML-Bp
BMMSCs was significantly slower when compared with the growth of
CML-Cp BMMSCs. After three passages, the proliferation of CML-Bp
BMMSCs was increased. The spontaneous apoptosis of CML-Bp BMMSCs
was similar to that of the Nc-BMMSCs and CML-Cp BMMSCs. Our results
showed that the expression levels of IL-6, IL-12, SCF and TPO in
CML-Bp BMMSCs were significantly lower when compared with CML-Cp
BMMSCs (P<0.05). SCF has a positive regulatory effect on
hematopoiesis (13), and IL-6
affects the early proliferation and differentiation of bone marrow
hematopoietic stem cells (14). The
downregulation of IL-12 has been suggested to result in defects of
the immune surveillance system of tumor cells in leukemia patients,
and the IL-12 expression levels to increase after treatment
(15). These results suggested that
the expression levels of certain cytokines were altered during the
proliferation of CML-Bp BMMSCs. In addition, BCR/ABL expression was
not observed in CML-Bp BMMSCs and CML-Cp BMMSCs. BCR/ABL expression
is a marker of CML malignant clones, suggesting that CML malignant
clones might not accumulate during CML-Bp.
It has been shown that BMMSCs regulate the
proliferation of leukemia cells, displaying an inhibitory or
promotive effect on the growth of tumor cells (16–18).
Contradictory results might have been obtained due to the
heterogeneity of MSCs and the different response to various tumor
cells under different experimental conditions. The present study
demonstrated that CML-Cp BMMSCs significantly inhibited the growth
of leukemia K562 cells in the absence of chemotherapeutic agents,
while CML-Bp BMMSCs had a weak inhibitory effect on K562 cells and
no inhibitory effect on primary CML-Bp leukemia cells. However, no
promotive effect of CML-Bp BMMSCs on the proliferation of tumor
cells was observed.
BMMSCs constitute an important part of the bone
marrow microenvironment and play an important role in the
development and drug-resistance of hematopoietic malignancies. It
has been reported that imatinib resistance is associated with the
protective effect of the bone marrow microenvironment on leukemia
stem cells (19). Nefedova et
al(20) and Konopleva et
al(21) found that BMMSCs are
involved in the chemotherapy resistence of leukemia cells, a fact
that led to the investigation of the protective effect of BMMSCs on
tumor cells. The results of the present study showed that BMMSCs
decreased the ADM-induced K562 apoptosis, suggesting that BMMSCs
protect K562 cells and antagonize ADM-induced apoptosis. CML-Bp
BMMSCs were found to have the strongest protective effect on
leukemia cells when compared with CML-Cp BMMSCs and Nc-BMMSCs.
Blockade of tumor cell apoptosis is an important
process underlying chemotherapy resistance. Bcl-2, Bax, survivin,
p53 and C-myc are involved in the regulation of the anti-apoptotic
ability of leukemia cells, in addition to the P38MAPK, Wnt,
PI3K/Akt and NF-κB pathways (22–24).
Our results showed that CML-Bp BMMSCs upregulated the expression of
bcl-2, downregulated the expression of bax and active caspase-3
protein, suggesting that CML-Bp BMMSCs protected K562 cells against
apoptosis by inhibiting the apoptotic pathway. The Bcl-2 family was
found to be involved in this process.
However, in the present study, DKK1 increased the
apoptosis of K562 cells, and downregulated the expression of
β-catenin, suggesting that CML-Bp BMMSCs activate the Wnt pathway
and reduce the apoptosis of K562 cells. As a negative regulator of
the Wnt pathway, DKK1 inhibited the activation of the Wnt pathway
in K562 cells, leading to increased apoptosis, further suggesting
that the Wnt pathway is involved in this process. The essence of
CML blastic change is the proliferation of original naive cells.
CML blastic change has been suggested to be related to the
activation of the Wnt pathway (25,26).
Wnt/β-catenin, as an important self-renewal pathway (27), regulated the transcription of
survivin, p53 and c-myc. Subsequent experiments showed that
blocking the protective effect of BMMSCs, thereby restoring the
sensitivity to chemotherapy drugs, could be used as a novel
potential treatment strategy. However, DKK1 did not completely
reverse the protective effect of BMMSCs on K562 cells, suggesting
the involvement of additional protective mechanisms.
In conclusion, the levels of the cytokines secreted
by CML-Bp BMMSCs were altered when compared with CML-Cp BMMSCs.
CML-Bp BMMSCs protect tumor cells and increase the anti-apoptotic
ability through regulating the expression of apoptosis-related
proteins and activating the Wnt pathway. Further studies are needed
in order to elucidate whether there is a correlation between CML-Bp
BMMSCs and CML.
References
|
1
|
Uehara E, Takeuchi S, Yang Y, et al:
Aberrant methylation in promoter-associated CpG islands of multiple
genes in chronic myelogenous leukemia blast crisis. Oncol Lett.
3:190–192. 2012.PubMed/NCBI
|
|
2
|
Cortes J and Kantarjian H: How I treat
newly diagnosed chronic phase CML. Blood. 120:1390–1397. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Deans RJ and Moseley AB: Mesenchymal stem
cells: biology and potential clinical uses. Exp Hematol.
28:875–884. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jiang Y, Jahagirdar BN, Reinhardt RL, et
al: Pluripotency of mesenchymal stem cells derived from adult
marrow. Nature. 418:41–49. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Barry FP and Murphy JM: Mesenchymal stem
cells: clinical applications and biological characterization. Int J
Biochem Cell Biol. 36:568–584. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bianchi G, Borgonovo G, Pistoia V and
Raffaghello L: Immunosuppressive cells and tumour microenvironment:
focus on mesenchymal stem cells and myeloid derived suppressor
cells. Histol Histopathol. 26:941–951. 2011.PubMed/NCBI
|
|
7
|
Prockop DJ: Marrow stromal cells as stem
cells for nonhematopoietic tissues. Science. 276:71–74. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Granero-Molto F, Weis JA, Longobardi L and
Spagnoli A: Role of mesenchymal stem cells in regenerative
medicine: application to bone and cartilage repair. Expert Opin
Biol Ther. 8:255–268. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Salem HK and Thiemermann C: Mesenchymal
stromal cells: current understanding and clinical status. Stem
Cells. 28:585–596. 2010.PubMed/NCBI
|
|
10
|
Dezawa M, Ishikawa H, Itokazu Y, et al:
Bone marrow stromal cells generate muscle cells and repair muscle
degeneration. Science. 309:314–317. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Oostendorp RA and Dörmer P: VLA-4-mediated
interactions between normal human hematopoietic progenitors and
stromal cells. Leuk Lymphoma. 24:423–435. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Colmone A, Amorim M and Pontier AL:
Leukemic cells create bone marrow niches that disrupt the behavior
of normal hematopoietic progenitor cells. Science. 322:1861–1865.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Duarte RF and Frank DA: SCF and G-CSF lead
to the synergistic induction of proliferation and gene expression
through complementary signaling pathways. Blood. 96:3422–3430.
2000.PubMed/NCBI
|
|
14
|
Frick JS, Zahir N, Mulier M, et al:
Colitogenic and non-colitogenic commensal bacteria differentially
trigger DC maturation and Th cell polarization: an important role
for IL-6. Eur J Immunol. 36:1537–1547. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bandini G, Zuffa E, Rosti G, et al:
Long-term outcome of adults with acute myelogenous leukaemia:
results of a prospective, randomized study of chemotherapy with a
minimal follow-up of 7 years. Br J Haematol. 77:486–490.
1991.PubMed/NCBI
|
|
16
|
Zhu Y, Sun Z, Han Q, et al: Human
mesenchymal stem cells inhibit cancer cell proliferation by
secreting DKK-1. Leukemia. 23:925–933. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Moshaver B, van der Pol MA, Westra AH, et
al: Chemotherapeutic treatment of bone marrow stromal cells
strongly affects their protective effect on acute myeloid leukemia
cell survival. Leuk Lymphoma. 49:134–148. 2008. View Article : Google Scholar
|
|
18
|
Gaundar SS, Bradstock KF and Bendall LJ:
p38MAPK inhibitors attenuate cytokine production by bone marrow
stromal cells and reduce stroma-mediated proliferation of acute
lymphoblastic leukemia cells. Cell Cycle. 8:2975–2983. 2009.
View Article : Google Scholar
|
|
19
|
Konopleva M and Andreeff M: Targeting the
leukemia microenvironment. Curr Drug Targets. 8:685–701. 2007.
View Article : Google Scholar
|
|
20
|
Nefedova Y, Landowski TH and Dalton WS:
Bone marrow stromal-derived soluble factors and direct cell contact
contribute to de novo drug resistance of myeloma cells by distinct
mechanisms. Leukemia. 17:1175–1182. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Konopleva M, Konoplev S, Hu W, et al:
Stromal cells prevent apoptosis of AML cells by up-regulation of
anti-apoptotic proteins. Leukemia. 16:1713–1724. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yeung J, Esposito MT, Gandillet A, et al:
β-Catenin mediates the establishment and drug resistance of MLL
leukemic stem cells. Cancer Cell. 18:606–618. 2010.
|
|
23
|
Sheth K, Friel J, Nolan B and Bankey P:
Inhibition of p38 mitogen activated protein kinase increases
lipopolysaccharide induced inhibition of apoptosis in neutrophils
by activating extracellular signal-regulated kinase. Surgery.
130:242–248. 2001. View Article : Google Scholar
|
|
24
|
Grandage VL, Gale RE, Linch DC and Khwaja
A: PI3-kinase/Akt is constitutively active in primary acute myeloid
leukaemia cells and regulates survival and chemoresistance via
NF-kappaB, Mapkinase and p53 pathways. Leukemia. 19:586–594.
2005.PubMed/NCBI
|
|
25
|
Valencia A, Román-Gómez J, Cervera J, et
al: Wnt signaling pathway is epigenetically regulated by
methylation of Wnt antagonists in acute myeloid leukemia. Leukemia.
23:1658–1666. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Radich JP, Dai H, Mao M, et al: Gene
expression changes associated with progression and response in
chronic myeloid leukemia. Proc Natl Acad Sci USA. 103:2794–2799.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hu Y, Chen Y, Douglas L and Li S:
Beta-Catenin is essential for survival of leukemic stem cells
insensitive to kinase inhibition in mice with BCR-ABL-induced
chronic myeloid leukemia. Leukemia. 23:109–116. 2009. View Article : Google Scholar : PubMed/NCBI
|