Introduction
Targeting of epigenetic alterations such as DNA
methylation and histone modification is an important field for
cancer research and for anticancer drug development (1,2). One
class of promising epigenetic drugs is histone deacetylase (HDAC)
inhibitors. These inhibitors suppress the enzymatic activity of
HDAC and induce re-activation of tumor-suppressor genes or
downregulation of oncogenes to attenuate tumor growth. Among HDAC
inhibitors, suberoylanilide hydroxamic acid (SAHA) is a pioneer
drug that has been developed and is now approved for the treatment
of cutaneous T cell lymphoma (3,4). This
drug is also undergoing clinical trials for the treatment of solid
tumors. Several mechanisms are involved in the inhibition of tumor
progression by SAHA. First, SAHA induces apoptosis in cancer cells
by regulating pro-apoptotic or anti-apoptotic genes (5,6).
Second, SAHA inhibits cell cycle progression by increasing the
expression of anti-proliferative genes such as cyclin-dependent
kinase inhibitors (CDKIs) p21 and p27 to reduce cell growth
(7,8). Third, SAHA modulates the immune
response to improve anticancer immune surveillance (9,10).
Fourth, SAHA suppresses proliferation of endothelial cells or
induces their apoptosis to decrease tumor angiogenesis in
vivo(11).
Recent studies demonstrate that induction of
lymphangiogenesis is strongly associated with tumor metastasis and
poor prognosis in cancer patients (12–14).
Proliferation, maturation and remodeling of lymphatic vessels are
controlled by different signaling pathways. Accumulating evidence
suggests that angiopoietin (Ang)/Tie signaling is an important
regulator in angiogenesis and lymphangiogenesis. The Ang family
includes four ligands (Ang1, Ang2 and Ang3/4) and two cognate
receptors (Tie1 and Tie2). Results of previous studies suggest that
Ang1 mainly acts as a Tie2 receptor agonist and Ang2 normally
functions as an antagonist (15,16).
However, the role of Ang1 and Ang2 is cell context-dependent and
Ang2 may function as a partial agonist under some physiological
conditions (17,18). Upon ligand stimulation, Tie2 forms a
dimer or multimer and is autophosphorylated at tyrosine residues
near its carboxyl terminus (19).
Activated Tie2 then transmits the signaling via several downstream
molecules including downstream of tyrosine kinase-related protein
(DOKR), endothelial nitric oxide synthase (eNOS), SH2
domain-containing phosphatase (SHP2), growth factor receptor-bound
protein 2 (GRB2) and the p85 subunit of PI3K to elicit different
biological processes (20,21). Mice lacking Ang2 exhibit major
lymphatic vessel defects suggesting the importance of Ang2 in
lymphatic function (22).
Interestingly, Ang1 has also been demonstrated to promote lymphatic
vessel formation and can rescue the lymphatic defect of Ang2-mutant
mice (23). In addition, Ang1/Tie2
signaling enhances lymphatic integrity by modulating tight junction
molecule expression during inflammation (24). Although SAHA has been shown to
inhibit tumor angiogenesis, its effect on tumor lymphangiogenesis
and the Ang/Tie system is still unclear. In this study, we
addressed this issue and tried to elucidate the underlying
mechanism.
Materials and methods
Generation of the Prox1-expressing
lymphatic-like endothelial cell line and cell culture
Human endothelial EA.hy926 cells were a gift from Dr
Ming-Hong Tai (National Sun Yat-Sen University, Taiwan). These
cells were grown in DMEM/high glucose (Invitrogen Life
Technologies, Carlsbad, CA, USA) medium containing 10% FBS,
antibiotics and 25 mM HEPES. EA.hy926 cells were transfected with
the pcDNA-Prox1 expression vector (provided by Dr You-Hua Xie,
Shanghai Institute of Biological Sciences, Shanghai, China). After
48 h, cells were selected with G418 for 3 weeks, and a stable cell
line FP01 was generated as previously described (25). Human primary lymphatic endothelial
cells (LECs) were purchased from PromoCell (Heidelberg, Germany).
LECs were grown in endothelial cell growth medium (MVII) with
supplement mixture (PromoCell). All cell lines were cultured at
37°C in a CO2 incubator.
Reagents and antibodies
SAHA was purchased from LC Laboratories (Woburn, MA,
USA). MG-132, chloroquine, mithramycin A, leupeptin and the
anti-actin antibody were purchased from Sigma-Aldrich (St. Louis,
MO, USA). Antibodies against Tie2 and Sp1 were obtained from
Millipore (Billerica, MA, USA). Antibody against c-Cbl was
purchased from Gene Tex, Inc. (San Antonio, TX, USA). Antibodies
against cyclin E, cdc2, cdk2, p21 and p27 were purchased from Santa
Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Antibodies against
cyclin D1, cyclin A and cyclin B were obtained from Cell Signaling
Technology (Beverly, MA, USA). Vascular endothelial growth factor
(VEGF)-C (Cys156Ser) was purchased from R&D Systems
(Minneapolis, MN, USA).
RNA interference
c-Cbl and control luciferase shRNA interference
vectors were obtained from National RNAi Core Facility (Institute
of Molecular Biology, Academia Sinica, Taipei, Taiwan) and the
targeting sequences were: pLKO.1-shCbl (target sequence
5′-CCAGTGAGTTGGGAG TTATTA-3′) and pLKO.1-shLuc (target sequence
5′-CTTCGAAATGTCCGT TCGGTT-3′).
Transient cell transfection
Cells were seeded in 6-well plates. After overnight
incubation, the GFP-Sp1 expression vector (provided by Dr Jan-Jong
Hung, National Cheng Kung University, Taiwan) and the control
vector were transfected into cells using Lipofectamine®
2000 (Invitrogen Life Technologies). After 48 h, cells were treated
with SAHA for an additional 24 h and then harvested for different
analyses.
Construction of Tie-2 promoter and
luciferase assay
The genomic DNA was isolated from EA.hy926 cells by
using the genomic DNA extraction kit (Qiagen, Hilden, Germany), and
the −703/+232 region from transcription start site of Tie2 gene
(NM_000459) was amplified by PCR using two specific primers:
Tie2–703 forward, 5′-ATACTCGAGCTTGGGGCTA CATTGAGCAT-3′ and reverse,
5′-ATTAAGCTTCACAGA GCCTTTGCATTTCA-3′. The amplified DNA fragment
was subcloned into the luciferase reporter gene vector pGL3
(Promega, Madison, WI, USA) by XhoI and HindIII to
yield the luciferase reporter construct pGL3-Tie2-(−703/+232).
Using this construct as a template, two 5′-deletion constructs were
generated by the following primers: Tie2–357 forward, 5′-AAA
CTCGAGTACAGCAGCAGCAAAAGCAG-3′ and Tie2–138 forward,
5′-ATACTCGAGGTTCCTTCTTGCCTCTAACTT GT-3′. FP01 cells were seeded
into 6-well plates and transfected with 1 μg of serial Tie-2
promoter-luciferase plasmids. After 24 h, cells were incubated with
various concentrations of SAHA for an additional 24 h. The
luciferase activity was detected by a reporter assay system
according to the manufacturer's instructions (Promega), and the
results were normalized to the protein concentration in cell
lysates.
Reverse transcription-polymerase chain
reaction (RT-PCR)
Cells were treated with different concentrations of
SAHA for 24 h. Total RNA was isolated by using the RNeasy Mini kit
(Qiagen), and mRNAs were reverse-transcribed to cDNA by M-MLV
reverse transcriptase (Promega), using oligo-dT primers according
to the manufacturer's instructions. The conditions of the PCR
reaction included an initialization step for 5 min at 95°C, 30
cycles of amplification (1 min at 95°C for denaturation, 1 min at
60°C for annealing and 1 min at 72°C for elongation) and 7 min at
72°C for final extension. The PCR primers were: Tie2 forward,
5′-AGTTCGAGGAGAGGC AATCA-3′ and reverse,
5′-CCGAGGTGAAGAGGTTTCCT-3′; Tie1 forward,
5′-TTTAACCCTGGTGTGCATCC-3′ and reverse, 5′-CCGCAGAAAATCTAGCAGGT-3′;
Ang1 forward, 5′-TATGCCAGAACCCAAAAAGG-3′ and reverse,
5′-GGGCACATTTGCACATACAG-3′; Ang2 forward, 5′-TGG
GATTTGGTAACCCTTCA-3′ and reverse, 5′-CCTTGAGCG AATAGCCTGAG-3′;
c-Cbl forward, 5′-CCATGGCTCTGA AATCCACT-3′ and reverse
5′-GGAGAATGTTCCCATCAG CA-3′; GAPDH forward,
5′-TGGGGAAGGTGAAGGTCGGAGTC-3′ and reverse
5′-TCCCGTTCTCAGCCTTGACGG-3′.
Protein stability assay
Cells were cultured in the media containing various
concentrations of SAHA for 24 h and then treated with 10 μg/ml of
cycloheximide to block protein synthesis. Cellular proteins were
harvested at different times after cycloheximide addition, and the
Tie2 protein level was detected by western blotting. Tie2 protein
level in the cells collected at time zero was defined as 100%.
Western blotting
Cells were treated with different concentrations of
SAHA for 24 h. After treatment, cells were washed with cold PBS and
lysed by RIPA buffer (50 mM Tris-HCl, pH 7.4, 50 mM NaCl, 1 mM
EDTA, 0.5 M sucrose, 0.25% sodium deoxycholate, 10% glycerol, 1%
NP-40 and protease inhibitors) on ice for 10 min. Cell debris was
removed by centrifugation at 13,000 rpm at 4°C for 10 min. Cell
lysates were subjected to SDS-PAGE separation and subsequently
transferred onto a polyvinylidene difluoride membrane (Millipore).
The membranes were incubated with 5% non-fat milk in Tris-buffered
saline and were probed with various primary antibodies. After
extensive washing, the membranes were incubated with horseradish
peroxidase-conjugated secondary antibodies and developed by
enhanced chemiluminescence (ECL; Millipore) reagent according to
the manufacturer's instructions.
Cell proliferation and flow
cytometry
For cell proliferation assay, cells were seeded at a
density of 7,500 cells/well in 96-well plates. After overnight
incubation, cells were treated with different concentrations of
SAHA for 48 h. MTT assay was carried out according to the
manufacturer's instructions to investigate cellular growth. For
flow cytometric analysis, cells were cultured in the medium
containing various concentrations of SAHA for 48 h, washed with
cold PBS and fixed in 70% ethanol overnight. Subsequently, cells
were washed with cold PBS and stained with PI solution containing
20 μg/ml propidium iodide, 200 μg/ml RNase A and 0.1% Triton X-100
for 30 min. Cell cycle analysis was performed by using Epics XL-MCL
flow cytometry (Beckman Coulter, Miami, FL, USA).
Tube formation and sprouting assay
Tube formation and sprouting assays were performed
as previously described (25). For
the sprouting assay, 10,000 cells suspended in medium containing
VEGF-C (Cys156Ser) and different concentrations of SAHA were mixed
with Matrigel and added into 96-well plates. After 24 h, cells
sprouting in the three dimensional culture of Matrigel were
observed by a microscope. One hundred cells were counted, and the
percentage of cells with sprouting was expressed as the means ± SE.
For the tube formation assay, 20,000 cells suspended in medium
containing VEGF-C (Cys156Ser) and different concentrations of SAHA
were added into 96-well plates precoated with Matrigel. The
formation of the tube-like structures was detected at 8 and 24 h
after cell seeding. The number of vessel joints in 5 light
microscopic fields was counted by using the Angiogenesis Image
Analyzer V.2.0.0 software (Kurabo Industries, Osaka, Japan).
Results from three independent experiments are expressed as the
mean ± SE.
Statistical analysis
All data are expressed as the means ± SE. The
Student's t-test was used to evaluate the differences between
various experimental groups. P-value <0.05 was considered to
indicate a statistically significant result.
Results
SAHA inhibits the proliferation of
LEC-like FP01 cells
Culture of primary LECs is difficult as only a
limited cell number can be isolated by using specific markers such
as LYVE-1. To study LEC function, we recently established an
LEC-like cell line (FP01) by overexpressing the master LEC
transcription factor PROX1 in EA.hy926 endothelial cells (25). FP01 cells exhibited a gene
expression pattern similar to primary LECs. In addition, these
cells expressed vascular endothelial growth factor receptor 3
(VEGFR3) and their proliferation, sprouting and tube formation were
strongly stimulated by VEGF-C (25). Our results suggest that the FP01
cell line is a useful model for functional investigation of LECs.
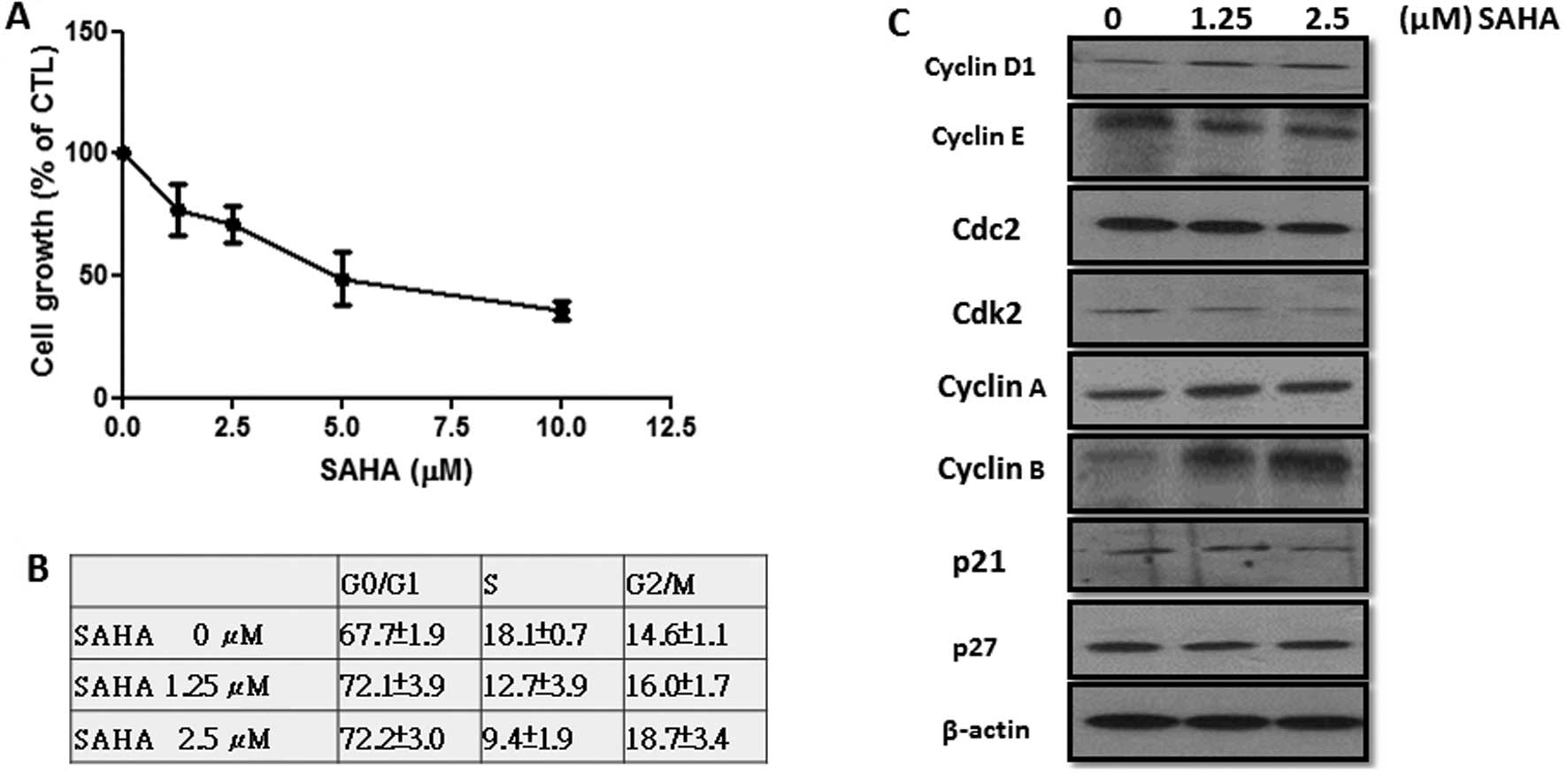
As shown in Fig. 1A, SAHA
dose-dependently inhibited the proliferation of FP01 cells and this
drug at 10 μM suppressed cell growth by 60–70%. Flow cytometric
analysis demonstrated that SAHA increased the percentage of cells
at the G0/G1 and G2/M phases (Fig.
1B). Conversely, the percentage of cells at S phase was
significantly reduced. Expression of CDKIs (p21 and p27) was not
changed by SAHA (Fig. 1C).
Interestingly, cyclin D1 and B1 were increased while cyclin E was
reduced. Since constitutive expression of cyclin D1 and B1 will
prevent G1/S progression and mitotic exit, our data suggest that
SAHA-induced G0/G1 and G2/M cell accumulation may be associated
with upregulation of cyclin D1 and B1. In addition, reduction in
cyclin E reflected the decrease of cells at S phase.
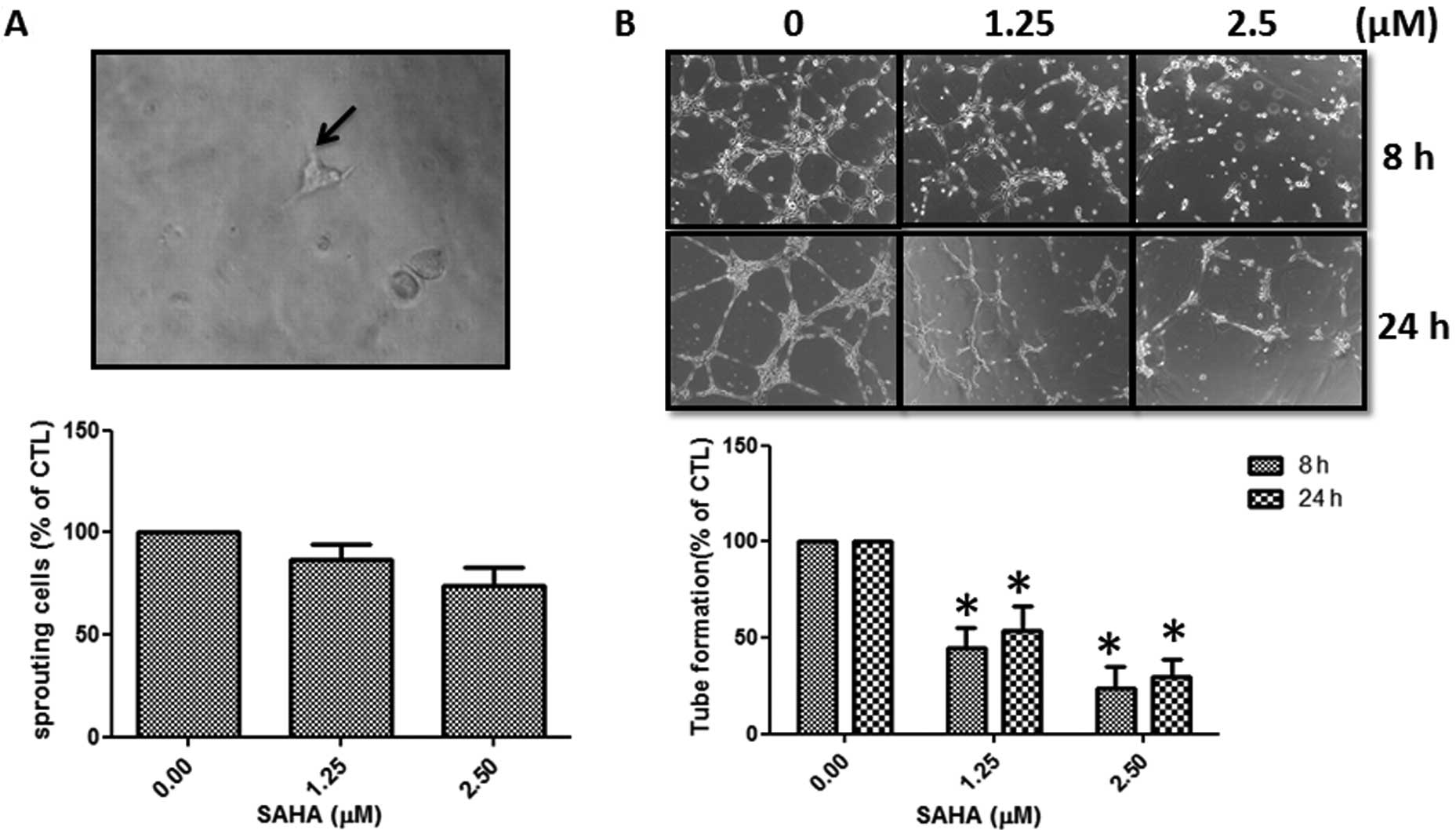
SAHA inhibits sprouting and tube
formation of FP01 cells
For induction of lymphangiogenesis, LECs need to
spout from existing lymphatic vessels and then organize into a new
tube network structure. Treatment of SAHA repressed the sprouting
of FP01 cells in the 3D culture (Fig.
2A). In addition, organization of the tube network structure of
FP01 cells on matrix-coated plates was also significantly inhibited
(Fig. 2B). Our data demonstrated
that SAHA at the concentration of 2.5 μM inhibited the number of
vessel joints by 70–80% (Fig.
2B).
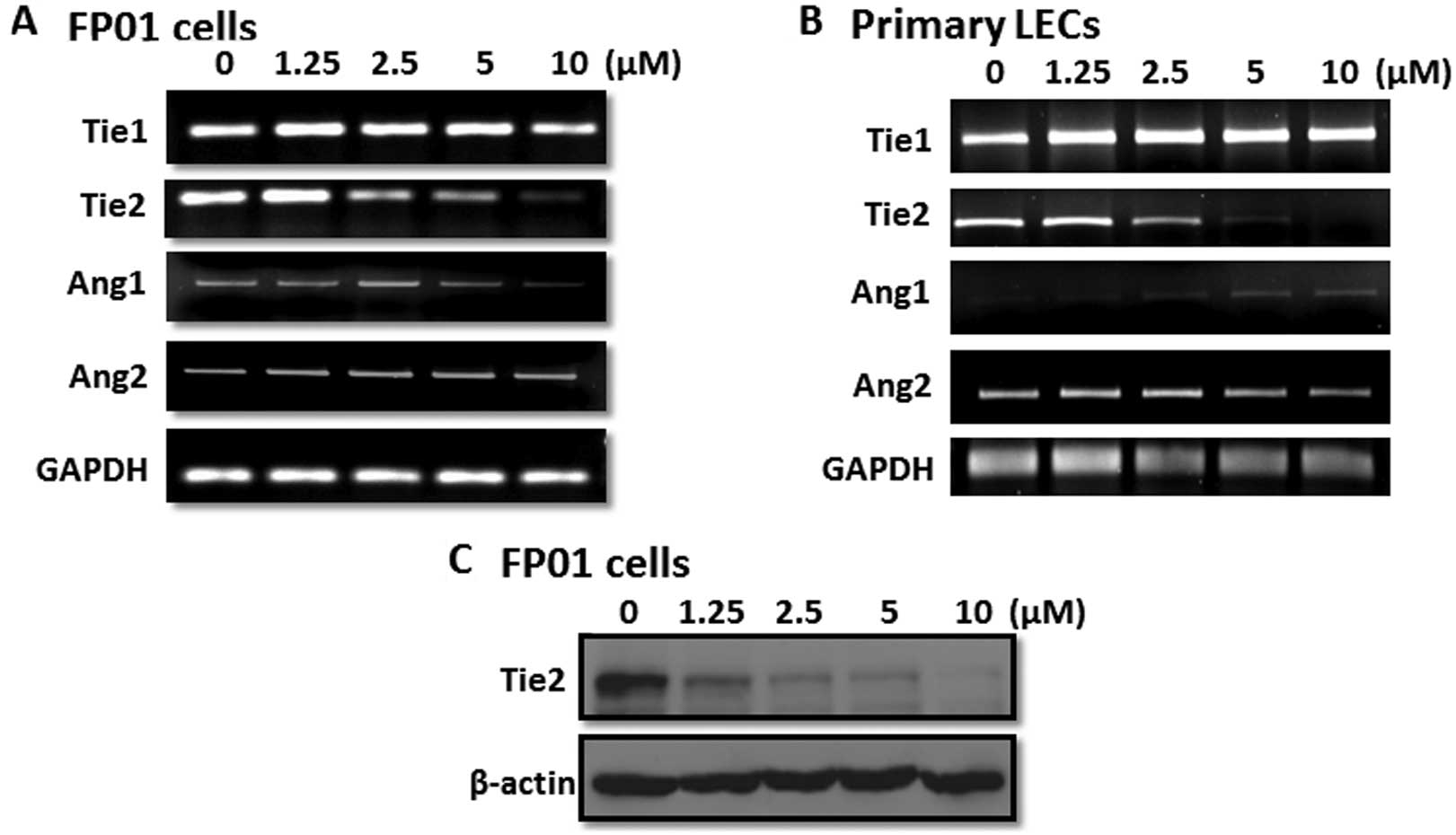
SAHA inhibits Tie2 transcription
The Ang/Tie signaling pathway is important for LEC
maturation and biological function. As shown in Fig. 3A, FP01 cells expressed Ang1, Ang2,
Tie1 and Tie2. Interestingly, we found that SAHA repressed Tie2
expression in a dose-dependent manner while Tie1 and Ang2 were not
affected and Ang1 was only marginally reduced. We also used primary
cultured LECs to confirm our results. Consistent with the data of
FP01 cells, only Tie2 expression was significantly inhibited by
SAHA (Fig. 3B). Western blot
analysis demonstrated that the Tie2 protein level was dramatically
reduced by SAHA in FP01 cells (Fig.
3C). Since Tie2 mRNA was attenuated, a direct inhibition of
Tie2 transcription by SAHA was investigated. We cloned the proximal
promoter region of the human Tie2 gene and generated a series of
deletion mutants of the promoter-luciferase reporter (Fig. 4A). The reporter containing the
−703/+232 region of the human Tie2 gene exhibited high luciferase
activity in FP01 cells indicating that this region is the major
regulatory region of Tie2 expression in endothelial cells (Fig. 4B). These data were consistent with
our RT-PCR data showing a high level of Tie2 mRNA in FP01 cells.
Deletion mutant containing the −357/+232 region was also repressed
by SAHA. Conversely, luciferase activity of the deletion mutant
containing the −138/+232 region was very low and was not affected
by SAHA suggesting that the −357/−138 promoter region is important
for Tie2 transcription in FP01 cells and is responsible for the
inhibition by SAHA (Fig. 4D).
Bioinformatics prediction revealed several transcription factor
binding sites including Sp1, Sp3, NF-κB and Ets-1 in this region.
However, ectopic expression of Sp1 or Ets-1 could not rescue
downregulation of Tie2 by SAHA (Fig.
4C). These data indicate that SAHA inhibits Tie2 transcription
via the −357/−138 promoter region and this effect is independent of
Sp1 and Ets-1.
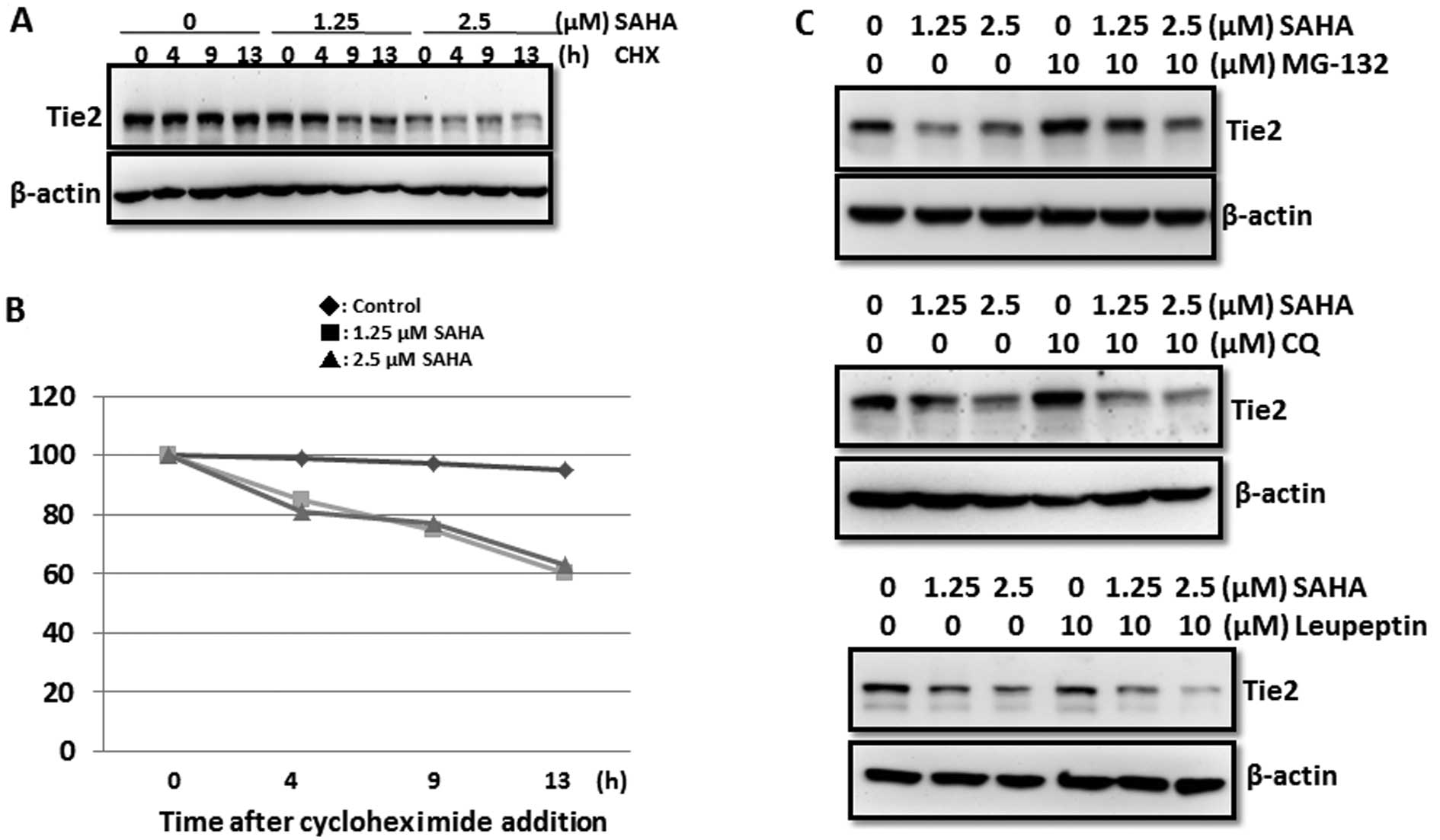
Induction of Tie2 protein degradation via
the ubiquitin/proteasome pathway by SAHA
Although SAHA directly inhibits Tie2 gene
transcription, our data demonstrated that the reduction in Tie2
protein was more significant than that of Tie2 mRNA. In addition,
SAHA at the concentration of 1.25 μM did not affect the Tie2 mRNA
level while it dramatically reduced Tie2 protein level (Fig. 3A and C). These results suggest a
transcription-independent inhibitory effect of SAHA on Tie2
expression. We blocked protein synthesis by cycloheximide and
examined Tie2 protein stability in control and SAHA-treated cells.
Our results showed that SAHA reduced Tie2 protein by 40% at 13 h
after exposure to cycloheximide (Fig.
5A and B). Conversely, no significant reduction in Tie2 protein
was found in the control group suggesting that SAHA increased Tie2
degradation. Treatment of MG132 (proteasome inhibitor) but not
chloroquine (autophagy inhibitor) or leupeptin (protease inhibitor)
rescued SAHA-induced downregulation of Tie2 protein. These data
suggest the involvement of the ubiquitin/proteasome pathway in Tie2
degradation (Fig. 5C). A previous
study demonstrated that the E3 ligase responsible for the
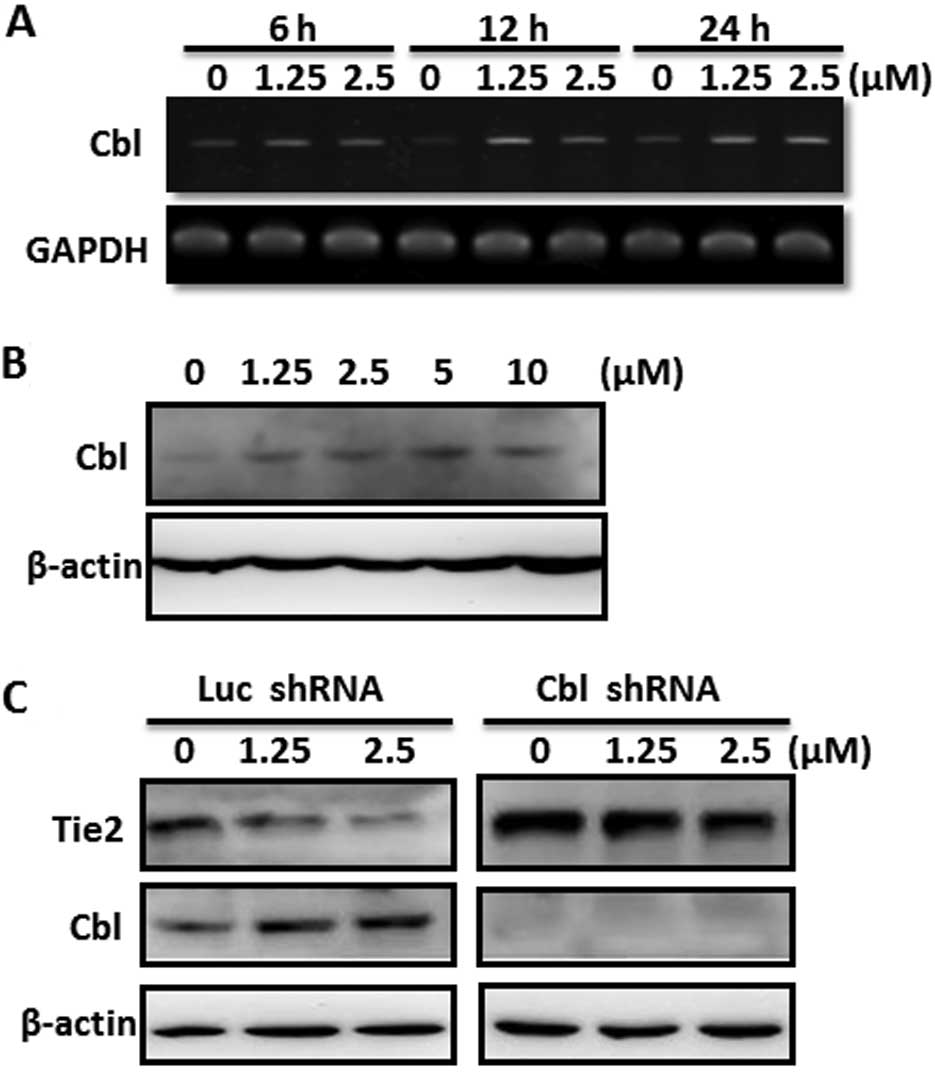
ubiquitination of Tie2 protein is c-Cbl (26). We found that SAHA increased the mRNA
level of c-Cbl in a dose-dependent manner (Fig. 6A). The induction was rapidly
detected at 6 h after SAHA treatment and high expression of c-Cbl
was persistently found at 24 h (Fig.
6A). c-Cbl protein was also upregulated dose-dependently in
FP01 cells (Fig. 6B). More
importantly, knockdown of c-Cbl increased the basal Tie2 protein
level and effectively reversed SAHA-induced downregulation of Tie2
(Fig. 6C). Collectively, we
demonstrated that SAHA upregulated c-Cbl expression and induced Tie
protein degradation via the ubiquitin/proteasome pathway.
Discussion
Solid tumors including breast, lung, gastric, head
and neck and prostate carcinomas are dependent on angiogenesis and
lymphangiogenesis to promote tumor growth and to increase cancer
metastasis. In the present study, we provide evidence that the
clinically used HDAC inhibitor SAHA suppresses proliferation,
sprouting and tube formation of LECs. The anti-angiogenic activity
of SAHA was firstly demonstrated by Deroanne et al(27). By using human umbilical cord
endothelial cells (HUVEC) as a model, they showed that
Trichostatin-A (TSA) and SAHA prevented VEGF-stimulated HUVECs from
invading a type I collagen gel and forming capillary-like
structures. In addition, they also found that TSA inhibited the
expression of VEGFR1, VEGFR2 and neuropilin-1 induced by VEGF. The
in vivo effect of SAHA on angiogenesis was shown by Ugur
et al(28). The authors
demonstrated that SAHA repressed tumor growth of glioma in an
orthotopic animal model. They found that a 30% reduction in the
angiogenesis rate was detected in SAHA-treated animals.
Subsequently, Mühlethaler-Mottet et al(29) also showed that SAHA and TSA strongly
impaired hypoxia-induced VEGF expression and secretion in
neuroblastoma cells. In addition to the inhibition of
pro-angiogenic factors, HDAC inhibitors also stimulated the
expression of anti-angiogenic factors to attenuate tumor
angiogenesis. For example, a disintegrin and metalloproteinase with
thrombospondin motifs 1 (ADAMTS1), an anti-angiogenic proteinase,
was upregulated by HDAC inhibitors in lung cancer cells (30). These results support the
anti-angiogenic activity of HDAC inhibitors. However, whether HDAC
inhibitors can inhibit lymphangiogenesis is unclear. Induction of
tumor lymphangiogenesis consists of two main steps. First, cancer
cells secrete pro-lymphangiogenic factors including VEGF-A, VEGF-C
and fibroblast growth factor-2 to stimulate the proliferation of
quiescent LECs. Second, the stimulated LECs sprout from the
lymphatic vessels, migrate toward tumors and form a new tube
structure. Thus, both cancer cells and LECs are important for
lymphangiogenesis. We recently demonstrated that SAHA attenuated
the expression and production of VEGF-C in breast cancer cells
(31). We now demonstrate that SAHA
also suppresses the proliferation, sprouting and tube formation of
LECs. Our results suggest that SAHA is a potent
anti-lymphangiogenic drug by inhibiting both cancer cells and
LECs.
Another important finding of this study is the
identification of Tie2 as a molecular target of SAHA. The Ang/Tie
signaling pathway is known to play a crucial role in angiogenesis
and lymphangiogenesis. Therefore, several strategies have been
developed to inhibit this signaling pathway. The first strategy is
targeting the Tie2 receptor. To date, a number of Tie2 kinase
inhibitors have been tested in cell-based assays or in animal
studies (32). However, only a few
compounds have advanced to clinical trials. For example, CEP-11981
is now undergoing Phase I study in patients with advanced solid
tumors. However, this drug inhibits Tie2, VEGFRs and FGFRs. Thus,
it is not truly Tie2 selective. The second strategy is Ang1 or Ang2
traps. For example, AMG-386 is a peptibody developed to target both
Ang1 and Ang2 (33). Peptibodies,
an alternative therapeutic format to monoclonal antibodies, consist
of biologically active peptides grafted onto an Fc domain. This
design has benefits to increase selectivity by the active peptide
domain and to retain the desirable features of antibodies to
prolong plasma residency time (34,35).
AMG-386 is now undergoing Phase I and II trials, either alone or in
combination with chemotherapeutic drugs, for the treatment of
various cancers (36). In the
present study, we explored SAHA as a new Tie2 inhibitor by
downregulating its expression in LECs. In addition, we elucidated
the underlying mechanisms and found that SAHA suppressed Tie2 via
repression of gene transcription and promotion of protein
degradation via the ubiquitination/proteasome pathway.
Acknowledgements
This study was supported by the grants: DOH
101-TD-C- 111-002 and DOH 101-TD-C-111-004 from the Department of
Health, Taiwan, to W.-C. H.
References
|
1
|
Minucci S and Pelicci PG: Histone
deacetylase inhibitors and the promise of epigenetic (and more)
treatments for cancer. Nat Rev Cancer. 6:38–51. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Inche AG and La Thangue NB: Chromatin
control and cancer-drug discovery: realizing the promise. Drug
Discov Today. 11:97–109. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Olsen EA, Kim YH, Kuzel TM, et al: Phase
IIb multicenter trial of vorinostat in patients with persistent,
progressive, or treatment refractory cutaneous T-cell lymphoma. J
Clin Oncol. 25:3109–3115. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Duvic M and Vu J: Vorinostat: a new oral
histone deacetylase inhibitor approved for cutaneous T-cell
lymphoma. Expert Opin Investig Drugs. 16:1111–1120. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Emanuele S, Lauricella M and Tesoriere G:
Histone deacetylase inhibitors: apoptotic effects and clinical
implications (Review). Int J Oncol. 33:637–646. 2008.PubMed/NCBI
|
|
6
|
Zhao Y, Tan J, Zhuang L, et al: Inhibitors
of histone deacetylases target the Rb-E2F1 pathway for apoptosis
induction through activation of proapoptotic protein Bim. Proc Natl
Acad Sci USA. 102:16090–16095. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gui CY, Ngo L, Xu WS, et al: Histone
deacetylase (HDAC) inhibitor activation of p21WAF1
involves changes in promoter-associated proteins, including HDAC1.
Proc Natl Acad Sci USA. 101:1241–1246. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Richon VM, Sandhoff TW, Rifkind RA and
Marks PA: Histone deacetylase inhibitor selectively induces
p21WAF1 expression and gene-associated histone
acetylation. Proc Natl Acad Sci USA. 97:10014–10019. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Woan KV, Sahakian E, Sotomayor EM, Seto E
and Villagra A: Modulation of antigen-presenting cells by HDAC
inhibitors: implications in autoimmunity and cancer. Immunol Cell
Biol. 90:55–65. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Blanchard F and Chipoy C: Histone
deacetylase inhibitors: new drugs for the treatment of inflammatory
diseases? Drug Discov Today. 10:197–204. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Deroanne CF, Bonjean K, Servotte S, et al:
Histone deacetylases inhibitors as anti-angiogenic agents altering
vascular endothelial growth factor signaling. Oncogene. 21:427–436.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Alitalo K: The lymphatic vasculature in
disease. Nat Med. 17:1371–1380. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Achen MG, McColl BK and Stacker SA: Focus
on lymphangiogenesis in tumor metastasis. Cancer Cell. 7:121–127.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cao R, Björndahl MA, Religa P, et al:
PDGF-BB induces intratumoral lymphangiogenesis and promotes
lymphatic metastasis. Cancer Cell. 6:333–345. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Augustin HG, Koh GY, Thurston G and
Alitalo K: Control of vascular morphogenesis and homeostasis
through the angiopoietin-Tie system. Nat Rev Mol Cell Biol.
10:165–177. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Maisonpierre PC, Suri C, Jones PF, et al:
Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo
angiogenesis. Science. 277:55–60. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yuan HT, Khankin EV, Karumanchi SA and
Parikh SM: Angiopoietin 2 is a partial agonist/antagonist of Tie2
signaling in the endothelium. Mol Cell Biol. 29:2011–2022. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim I, Kim JH, Moon SO, Kwak HJ, Kim NG
and Koh GY: Angiopoietin-2 at high concentration can enhance
endothelial cell survival through the phosphatidylinositol
3′-kinase/Akt signal transduction pathway. Oncogene. 19:4549–4552.
2000.PubMed/NCBI
|
|
19
|
Murray BW, Padrique ES, Pinko C and
McTigue MA: Mechanistic effects of autophosphorylation on receptor
tyrosine kinase catalysis: enzymatic characterization of Tie2 and
phospho-Tie2. Biochemistry. 40:10243–10253. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jones N, Chen SH, Sturk C, Master Z, Tran
J, Kerbel RS and Dumont DJ: A unique autophosphorylation site on
Tie2/Tek mediates Dok-R phosphotyrosine binding domain binding and
function. Mol Cell Biol. 23:2658–2668. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jones N, Master Z, Jones J, Bouchard D,
Gunji Y, Sasaki H, Daly R, Alitalo K and Dumont DJ: Identification
of Tek/Tie2 binding partners. Binding to a multifunctional docking
site mediates cell survival and migration. J Biol Chem.
274:30896–30905. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dellinger M, Hunter R, Bernas M, Gale N,
Yancopoulos G, Erickson R and Witte M: Defective remodeling and
maturation of the lymphatic vasculature in Angiopoietin-2 deficient
mice. Dev Biol. 319:309–320. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shimoda H, Bernas MJ, Witte MH, Gale NW,
Yancopoulos GD and Kato S: Abnormal recruitment of periendothelial
cells to lymphatic capillaries in digestive organs of
angiopoietin-2-deficient mice. Cell Tissue Res. 328:329–337. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kajiya K, Kidoya H, Sawane M,
Matsumoto-Okazaki Y, Yamanishi H, Furuse M and Takakura N:
Promotion of lymphatic integrity by angiopoietin-1/Tie2 signaling
during inflammation. Am J Pathol. 180:1273–1282. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pan MR, Chang TM, Chang HC, Su JL, Wang HW
and Hung WC: Sumoylation of Prox1 controls its ability to induce
VEGFR3 expression and lymphatic phenotypes in endothelial cells. J
Cell Sci. 122:3358–3364. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wehrle C, Van Slyke P and Dumont DJ:
Angiopoietin-1-induced ubiquitylation of Tie2 by c-Cbl is required
for internalization and degradation. Biochem J. 423:375–380. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Deroanne CF, Bonjean K, Servotte S, et al:
Histone deacetylases inhibitors as anti-angiogenic agents altering
vascular endothelial growth factor signaling. Oncogene. 21:427–436.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ugur HC, Ramakrishna N, Bello L, et al:
Continuous intracranial administration of suberoylanilide
hydroxamic acid (SAHA) inhibits tumor growth in an orthotopic
glioma model. J Neurooncol. 83:267–275. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mühlethaler-Mottet A, Meier R, Flahaut M,
et al: Complex molecular mechanisms cooperate to mediate histone
deacetylase inhibitors anti-tumour activity in neuroblastoma cells.
Mol Cancer. 7:552008.PubMed/NCBI
|
|
30
|
Chou CW and Chen CC: HDAC inhibition
upregulates the expression of angiostatic ADAMTS1. FEBS Lett.
582:4059–4065. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cheng HT and Hung WC: Inhibition of
lymphangiogenic factor VEGF-C expression and production by the
histone deacetylase inhibitor suberoylanilide hydroxamic acid in
breast cancer cells. Oncol Rep. 29:1238–1244. 2013.
|
|
32
|
Huang H, Bhat A, Woodnutt G and Lappe R:
Targeting the ANGPT-TIE2 pathway in malignancy. Nat Rev Cancer.
10:575–585. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Oliner J, Min H, Leal J, et al:
Suppression of angiogenesis and tumor growth by selective
inhibition of angiopoietin-2. Cancer Cell. 6:507–516. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shimamoto G, Gegg C, Boone T and Quéva C:
Peptibodies: A flexible alternative format to antibodies. MAbs.
4:586–591. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Beck A and Reichert JM: Therapeutic
Fc-fusion proteins and peptides as successful alternatives to
antibodies. MAbs. 3:415–416. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Herbst RS, Hong D, Chap L, et al: Safety,
pharmacokinetics and antitumor activity of AMG 386, a selective
angiopoietin inhibitor, in adult patients with advanced solid
tumors. J Clin Oncol. 27:3557–3565. 2009. View Article : Google Scholar : PubMed/NCBI
|