Introduction
Antifolates are the first class of antimetabolites
introduced to the clinic approximately 60 years ago (1). Methotrexate (MTX), a folic acid
antagonist, competitively inhibits dihydrofolate reductase (DHFR)
to disrupt cellular folate metabolism. MTX suppresses synthesis of
purine and pyrimidine by inhibiting its target enzyme, DHFR
(2). MTX reversibly inhibits the
proliferation of cells in the late G1 phase and may cause
cytotoxicity of cells in the S phase (3). MTX also promotes adenosine release to
cause adenosine-mediated suppression of inflammation (4). The adenosine-mediated
anti-inflammatory effect of MTX is now supported by clinical data
(5). MTX has been widely used since
1985 for the treatment of rheumatoid arthritis (RA) via its
presumed anti-proliferative properties (6–8). MTX
has also been used in dermatology for more than 5 decades. MTX was
introduced to treat severe psoriasis vulgaris in 1951 (9). The anti-psoriatic effect is based on
its anti-proliferative, anti-inflammatory and possibly
immunosuppressive properties (9).
Due to its cytotoxicity, MTX was also demonstrated to be a potent
and effective therapy for cancers including leukemia (1) and head and neck cancers (10). After years of use of antifolates
against malignancies, particularly leukemia, the full understanding
of the mechanisms of action of these agents remain unclear
(1). A recent report suggests that
low-dose MTX is promising for tumor dormancy therapy in patients
with osteosarcoma and lung metastasis (11). However, combination treatment of MTX
and PUVA may induce cancer (12).
To date, much effort has been given to investigate whether MTX
combined with traditional chemotherapy drugs and/or radiotherapy
produces a synergistic effect in the treatment of various types of
cancers.
Aspirin (ASA), a cyclooxygenase (COX)-1/2 inhibitor,
has been successful during the past century for its clinical use
for anti-inflammatory conditions. Moreover, high doses of ASA
(2.5–3.9 g/day) are sometimes used to treat diseases, such as RA
(13,14). ASA also nonselectively blocks COX-1
and COX-2 via irreversible acetylation. COX-2 regulates many
physiological functions such as augmentation of apoptosis,
inhibition of angiogenesis and cell motility. Thus, high COX-2
expression in tumors has been associated with poor survival, and
intake of ASA is associated with a decreased risk of various types
of cancer including those of the colorectum, stomach, oesophagus,
breast, ovary and lung (15–17).
ASA may also possess the potential for combination use with
standard chemotherapy or radiation therapy. However, a reduction in
renal clearance of MTX was observed in patients receiving a
maintenance dose of MTX with nonsteroidal anti-inflammatory drugs
(NSAIDs) (18,19). The combination of MTX and
salicylates was found to greatly increased the frequency of
abnormal liver enzyme values (20).
Due to its spectrum of effects, the increased toxic side effect of
MTX was found to be caused by concomitant administration with ASA
in patients (9).
In the present study, we demonstrated that ASA does
not increase the anti-proliferative activity of MTX against cancer
cells in vitro, rather, ASA antagonizes the therapeutic
efficacy of MTX in human lung cancers via preserving cell
proliferation and survival. The mechanism involved in the
antagonism between MTX and ASA was also investigated.
Materials and methods
Cell culture
The human lung adenocarcinoma cell lines were
maintained in RPMI-1640 (CL1-0 cells) (21) or DMEM (A549 cells) supplemented with
10% fetal bovine serum and 2 mM L-glutamine, 100 μg/ml streptomycin
and 100 U/ml penicillin, in a humidified 5% CO2
atmosphere.
Reagents
ASA, MTX and ibuprofen (IBU) were purchased from
Sigma Chemical Co. (St. Louis, MO, USA). Celecoxib (CXB) was
purchased from Calbiochem (an affiliate of Merck, Germany). The
final concentrations of the drug vehicle (DMSO) added to the cell
cultures were all <0.1%.
Cell viability assay
Cell viability was assayed by SRB staining as
described previously (22). In
brief, cells (1.5×103/well) were seeded on 96-well
plates, followed 24 h later by treatment with drugs (or vehicle
control) for 72 h. Absorbance at 562 nm was measured with an ELISA
reader. Cell viability was expressed as the percentage of
absorbance of the drug-treated cells relative to that of the
vehicle-treated cells. The combination index (CI) was evaluated by
the method of Chou and Talalay (23,24).
Clonogenicity assay
One hundred cells were seeded in a 10-cm culture
dish, followed 24 h later by incubation with the drugs (or vehicle
control) for 2 weeks (CL1-0) or 3 weeks (A549). Colonies consisting
of >20 cells were counted. Colonies were washed with
phosphate-buffered saline (PBS), air dried and stained with 0.4%
crystal violet for 1 min, rinsed in water, air dried and
photographed.
Cell cycle analysis
Cells (2×105/dish) were plated in 10-cm
dishes, followed 24 h later by treatment with the drugs (or vehicle
control) for the intervals indicated. At harvest, cells were
trypsinized, washed in PBS and fixed in ice-cold 70% ethanol in
PBS. Cell cycle was assayed by propidium iodide staining, followed
by FACScan analysis. Cell cycle profiles were determined using
ModFit LT software (Becton-Dickinson, San Diego, CA, USA).
Western blot analysis
At harvest, total protein extracts were prepared,
and the concentration was determined using the Bradford method.
Aliquots containing 20 μg of total protein each were subjected to
western blot analysis. Antibodies against cyclin A (sc-239), FAS
(sc-8009), caspase-3 (sc-7272) and Bcl-2 (sc-509) were purchased
from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Antibody
against DHFR (15194-1-AP) was purchased from Proteintech Group,
Inc. (Chicago, IL, USA).
Reverse transcription (RT)-polymerase
chain reaction (PCR) analysis
Total RNA was extracted from cells using TRIzol
reagent (Invitrogen, Carlsbad, CA, USA). RT was performed using 1
μg of total RNA as a template and random hexamer as a primer. The
cDNA was amplified by PCR using FAS- and DHFR-specific primer pairs
(DHFR forward primer, GAATCACCCAGGCCATCTTA and reverse,
GCCTTTCTCCTCCTGGACAT; Fas forward primer, ACGGAGTTGGGGAAGCTCTT and
reverse, TGTCAGTCACTTGGGCATTAACA). Actin was used as an internal
control (actin forward primer: AGCGAGCATCCCCCAAAGTT and reverse,
GGGCACGAAGGCTCATCATT). PCR was performed by denaturing the DNA at
94°C for 5 min, followed by 30 cycles of amplification: 94°C for 30
sec, 60°C for 30 sec, 72°C for 60 sec and a final extension step at
72°C for 10 min. Amplified fragments were separated on a 1.0%
agarose gel and visualized with ethidium bromide staining.
Statistical analysis
All data are expressed as means ± SE. All
statistical analyses were performed using the paired t-test
(SigmaPlot 2001 software). P<0.05 was considered to indicate a
statistically significant result.
Results
MTX and ASA exert an antagonistic
anticancer effect
The anticancer effects of the two drugs, the DHFR
inhibitor MTX and the COX-1/2 enzyme inhibitor ASA, were tested
individually and in combination, in two human non-small cell lung
cancer cell lines, CL1-0 (Fig.
1A–C) and A549 (Fig. 1D–F). As
shown in Fig. 1A, while treatment
of CL1-0 cells with ASA 2 mM or MTX 12.5 nM alone resulted in ~40
and 75% decreases in cell numbers (i.e., 60 and 25% viability
remained), respectively, when compared to the control, treatment
with the combination of ASA + MTX only resulted in a 46% decrease
(i.e., 54% viability) instead of a predicted 85% decrease (=100% -
60% × 25%) if the combination treatment would have been additive. A
decrease in more than 85% would be expected if the combination
effect was synergistic. Of note, the antagonistic effect of the MTX
+ ASA combination was also shown by the upper-right shift in the
MTX survival curves following the addition of increasing doses of
ASA to MTX (Fig. 1B). To
quantitatively determine the magnitude of the antagonistic effect
of the MTX + ASA combination treatment, the CI method of Chou and
Talalay (23,24) was employed. Virtually all CI values
in the CI plots were significantly >1 (Fig. 1C and F), indicating a strong
antagonism. Similar results were also observed for the A549 cells
(Fig. 1D–F).
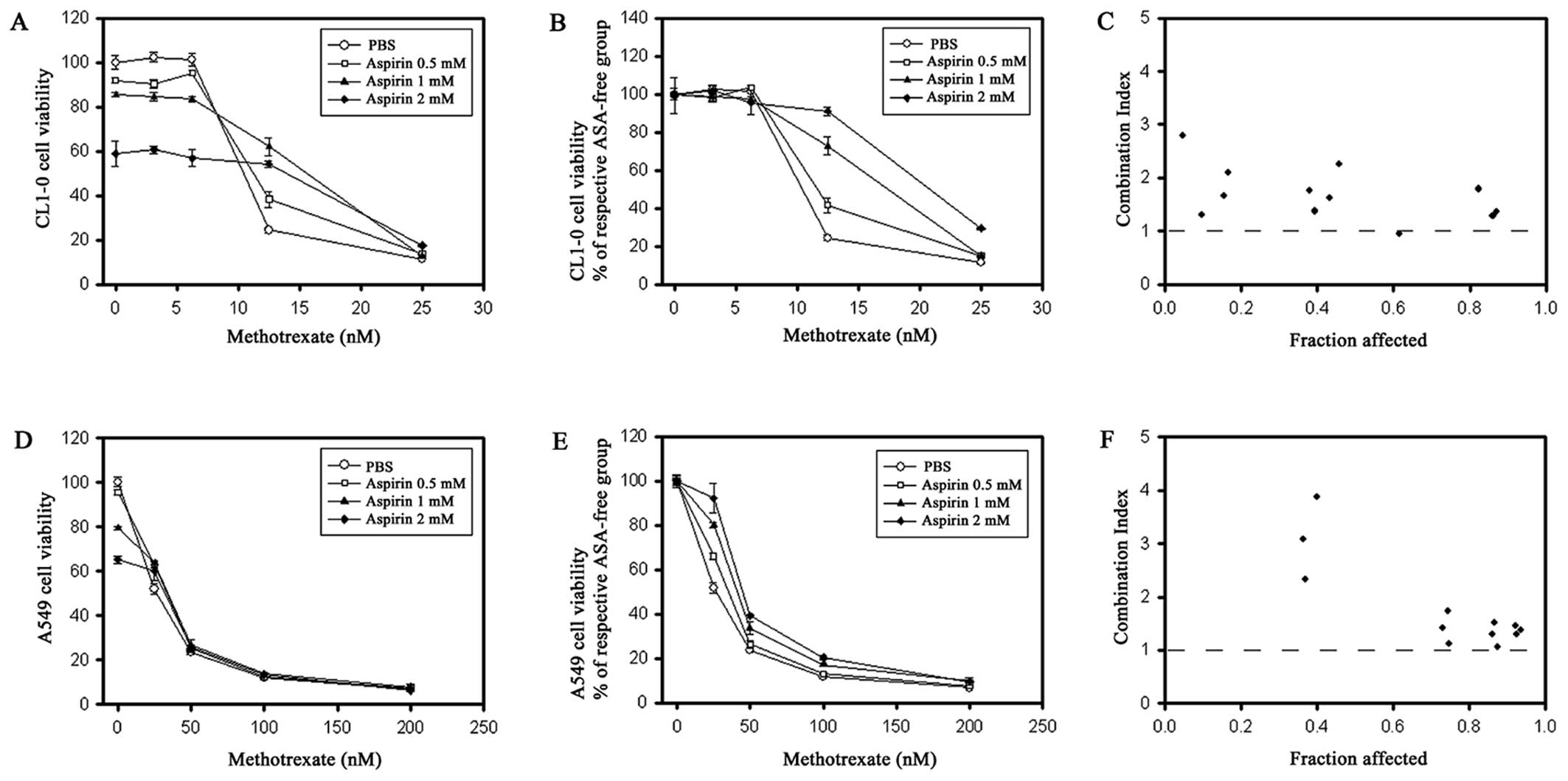 | Figure 1Antagonism of methotrexate (MTX) and
aspirin (ASA) in CL1-0 (A–C) and A549 (D–F) cells, as evaluated by
SRB staining assay. Cells were treated with various concentrations
of MTX in the presence of PBS (circle), 0.5 mM ASA (square), 1 mM
ASA (triangle), or 2 mM ASA (rhombus) for 72 h and then assayed by
SRB staining. Relative cell viability (% of PBS control) is
expressed as mean ± SE. Viability curves (A and D) were further
normalized to the viability of MTX-free cells treated with each ASA
concentration, respectively (B and E). Combination index (CI) plots
of MTX + ASA were generated in relation to cell survival inhibition
(fraction affected) (C and F), where CI >1, <1, and =1
indicate antagonism, synergism, and additive effect, respectively.
PBS, phosphate-buffered saline. |
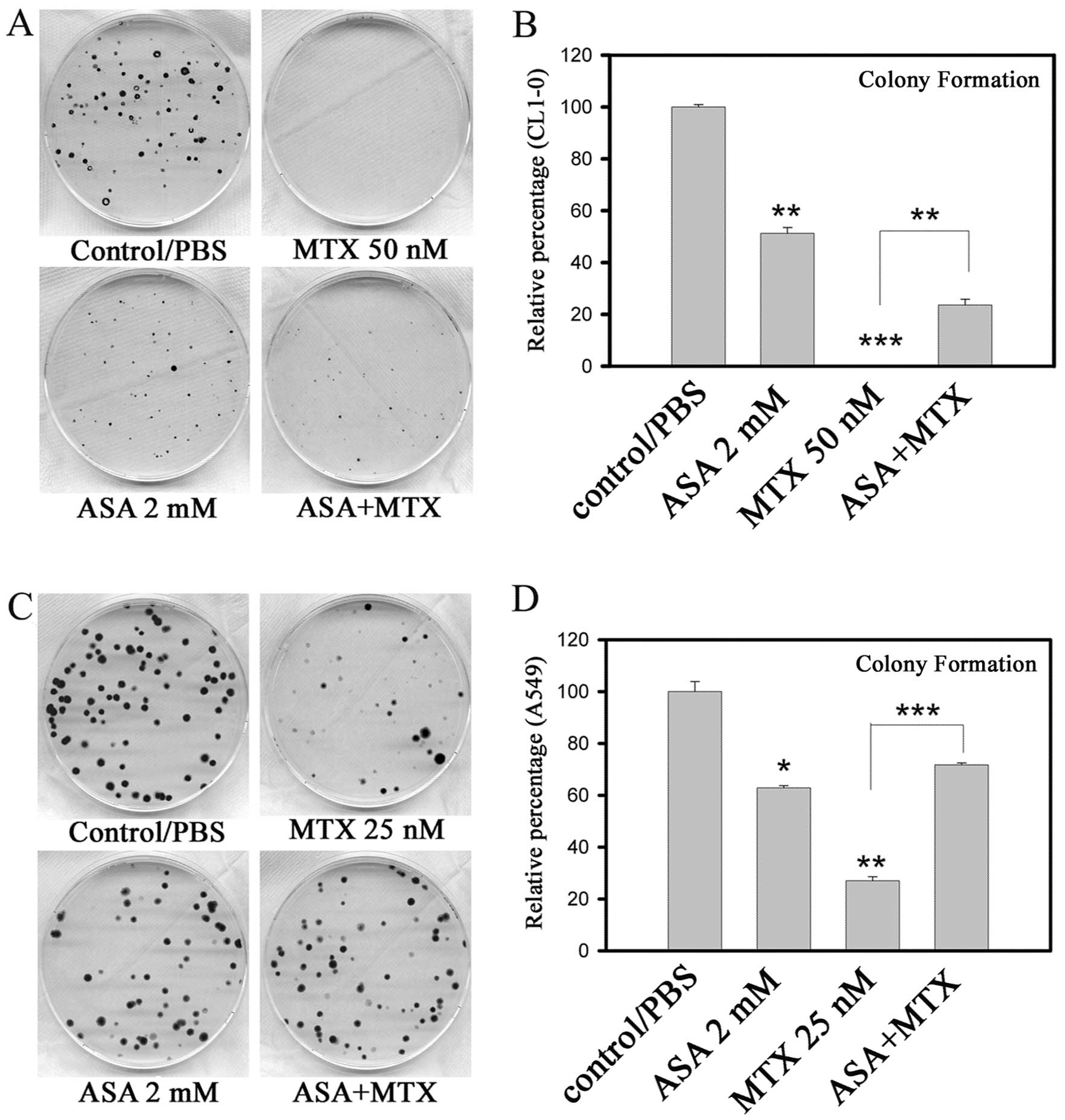
Colony formation assays were performed to further
demonstrate the effects of the combination treatment of MTX and
ASA. Images of the colonies grown in the presence or absence of
various drugs are shown in Fig. 2A
(CL1-0) and Fig. 2C (A549). While
CL1-0 and A549 cells were different in sensitivity to ASA, marked
antagonistic effects were observed in both CL1-0 and A549 cells
when the two drugs were combined (Fig.
2B and D).
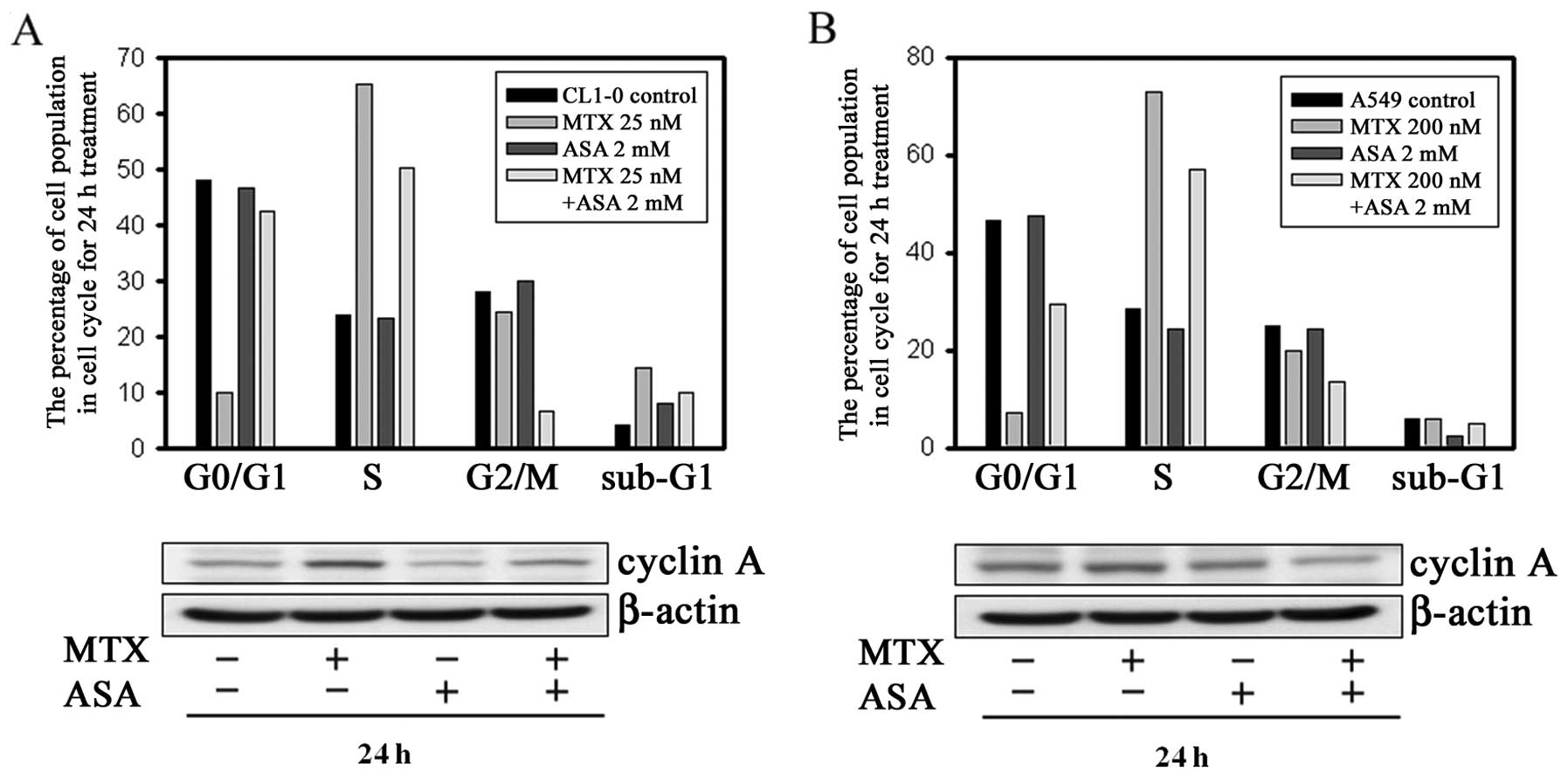
MTX-mediated S phase arrest is
antagonized by ASA
The effects of ASA on the cell cycle were examined
in our previous study (25). Here,
we assessed the effects of ASA on MTX-mediated alterations in the
cell cycle. As shown in Fig. 3A
(upper panel), while MTX alone induced a significant accumulation
of CL1-0 cells in the S phase accompanied by a marked decrease in
the number of cells in the G0/G1 phase, co-treatment with ASA
effectively reversed these changes. Cyclin A is an S phase-specific
regulatory protein that functions to induce mitosis. Expression of
cyclin A is normally low in G1 phase but increases in S phase.
Consistent with the blockade of S to G2/M phase transition, MTX
treatment resulted in a marked accumulation of cyclin A protein
(Fig. 3A, lower panel). Moreover,
the MTX-mediated cyclin A accumulation was reversed by co-treatment
with ASA. Notably, combination treatment with MTX + ASA resulted in
a marked decrease in the number of CL1-0 cells in the G2/M phase.
Similar results were obtained using A549 cells (Fig. 3B).
ASA prevents MTX-mediated apoptosis via
inhibition of caspase-3 activation and upregulation of Bcl-2
expression
We next examined the effects of MTX and ASA
treatments, individually and in combination, on cell cycle
progression and apoptosis after 72 h. Consistent with the 24-h
treatment results in the CL1-0 cells, concomitant addition of ASA
resulted in a significant reversal of MTX-mediated S phase arrest
and depletion of G0/G1 phase cells (Fig. 4A). Importantly, the number of
MTX-mediated apoptotic A549 cells was significantly reduced by ASA
(Fig. 4B, left). The antagonistic
effect of ASA against MTX was further supported by the result of
the western blot analysis (Fig. 4B,
right). While MTX treatment caused a reduction in Bcl-2 and
pro-caspase 3, ASA treatment had an opposite effect. More
importantly, following MTX + ASA co-treatment, the MTX-mediated
effects on Bcl-2 and pro-caspase 3 were also reversed.
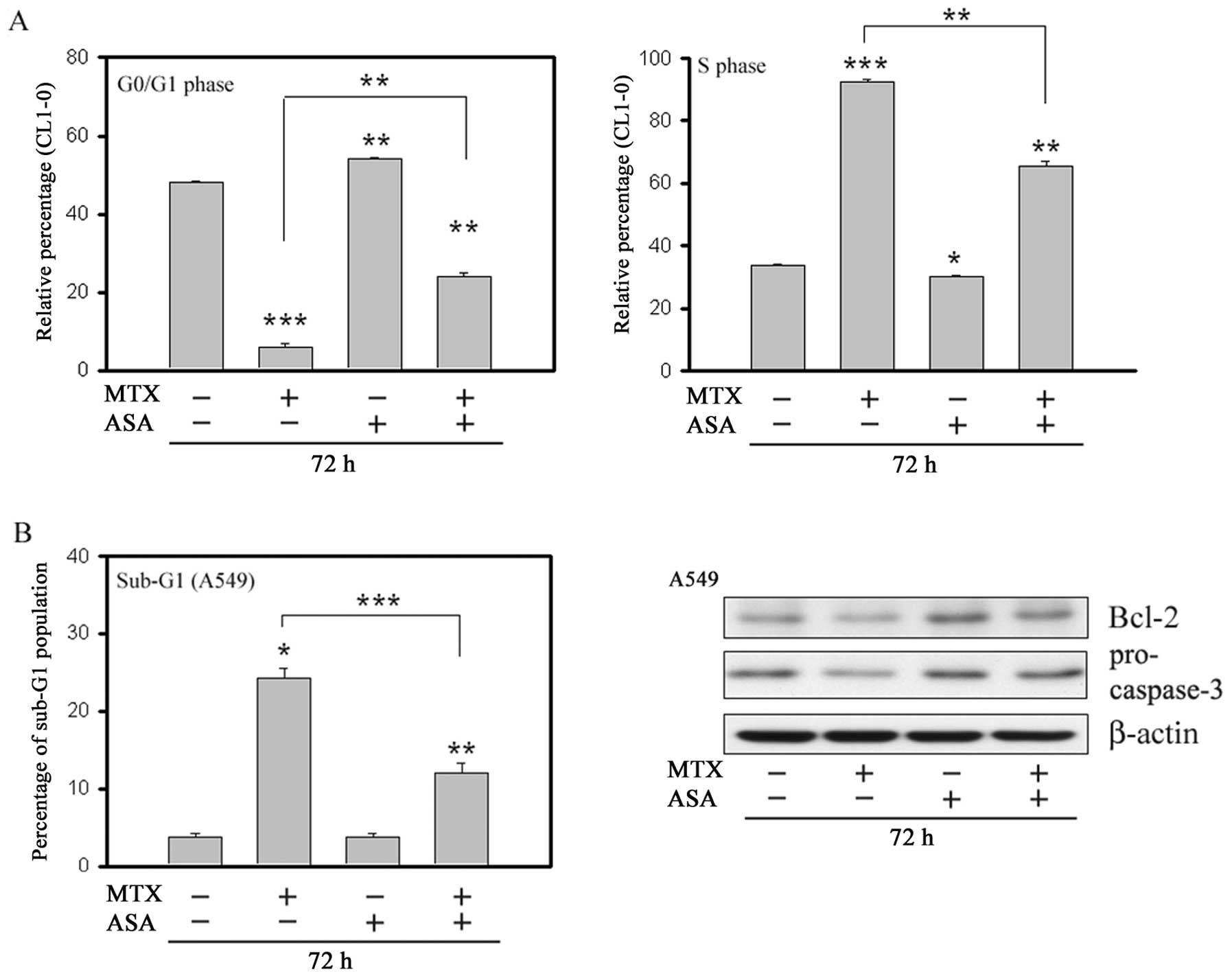 | Figure 4Effects of aspirin (ASA) (2 mM) and
methotrexate (MTX) (CL1-0, 25 nM; A549, 200 nM), alone or in
combination, on the cell cycle were determined after treatment for
72 h. The percentage of CL1-0 cells in the G0/G1 phase (A, left)
and S phase (A, right) was determined by flow cytometry after PI
staining. The percentage of A549 cells in sub-G1 phase was
determined by flow cytometry after PI staining (B, left). Cell
lysates of A549 were analyzed by western blotting for Bcl-2 and
pro-caspase-3 (B, right). β-actin served as the loading control.
Error bars, SEM (n=3). *P<0.05,
**P<0.01 and ***P<0.001 for the
comparisons indicated. |
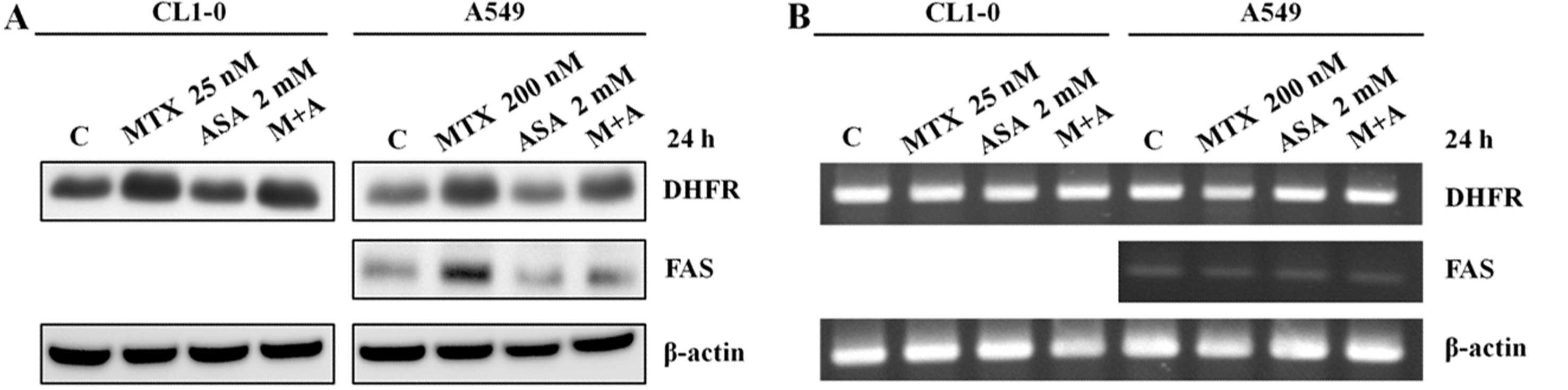
ASA antagonizes the MTX-mediated FAS
protein level, but not DHFR
To assess whether expression of DHFR and FAS is
correlated with the antagonism between ASA and MTX, the treated
cells were harvested and analyzed by western blotting and RT-PCR
(Fig. 5). DHFR protein levels were
upregulated by MTX in both CL1-0 and A549 cells. Notably, while ASA
alone had no effect on the DHFR and FAS protein levels, MTX-induced
upregulation of FAS, but not DHFR, was reversed by ASA in A549
cells (Fig. 5A, right). This
indicates that MTX-mediated apoptosis of A549 cells depends on an
increased level of FAS. DHFR and FAS were not affected at the mRNA
level (Fig. 5B).
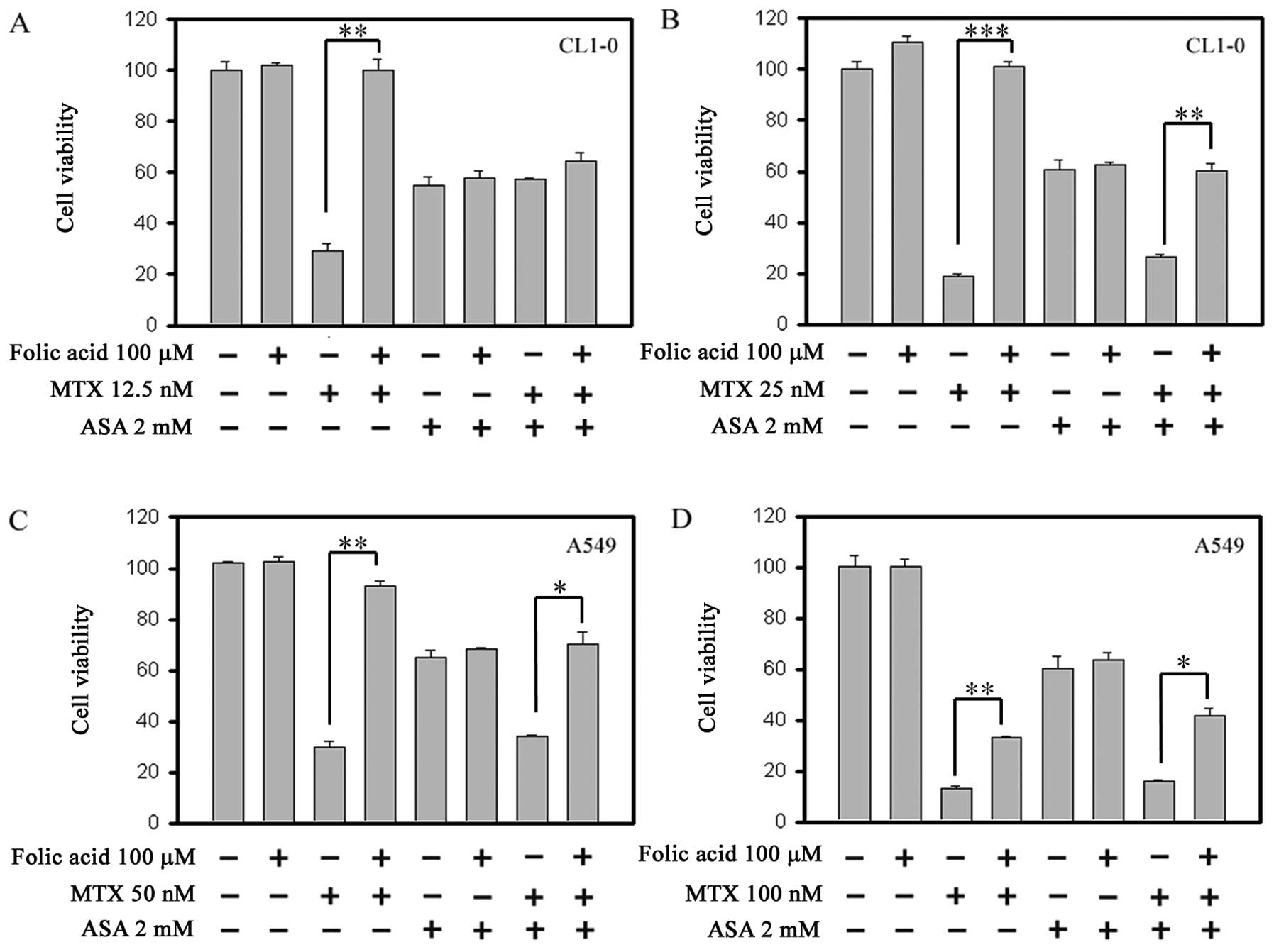
DHFR and COX-1/COX-2 are involved in
MTX-mediated cancer cell growth inhibition
We aimed to ascertain whether DHFR activity is
involved in the antagonistic effect of MTX and ASA co-treatment
with folate. As shown in Fig. 6A–D,
both the MTX-mediated growth inhibition (comparison of
3rd and 4th bar) and the MTX + ASA
co-treatment-mediated growth inhibition (comparison of the
7th and 8th bar) were reversed, completely or
partially, by folic acid in both cell lines. Folic acid did not
affect the ASA-induced growth inhibition (comparison of the
5th and 6th bar). MTX achieves its cytotoxic
effect through inhibition of the folate-dependent enzyme, DHFR, and
our results indicate that inhibition of DHFR was involved not only
in the MTX-mediated growth inhibition but also in the antagonism
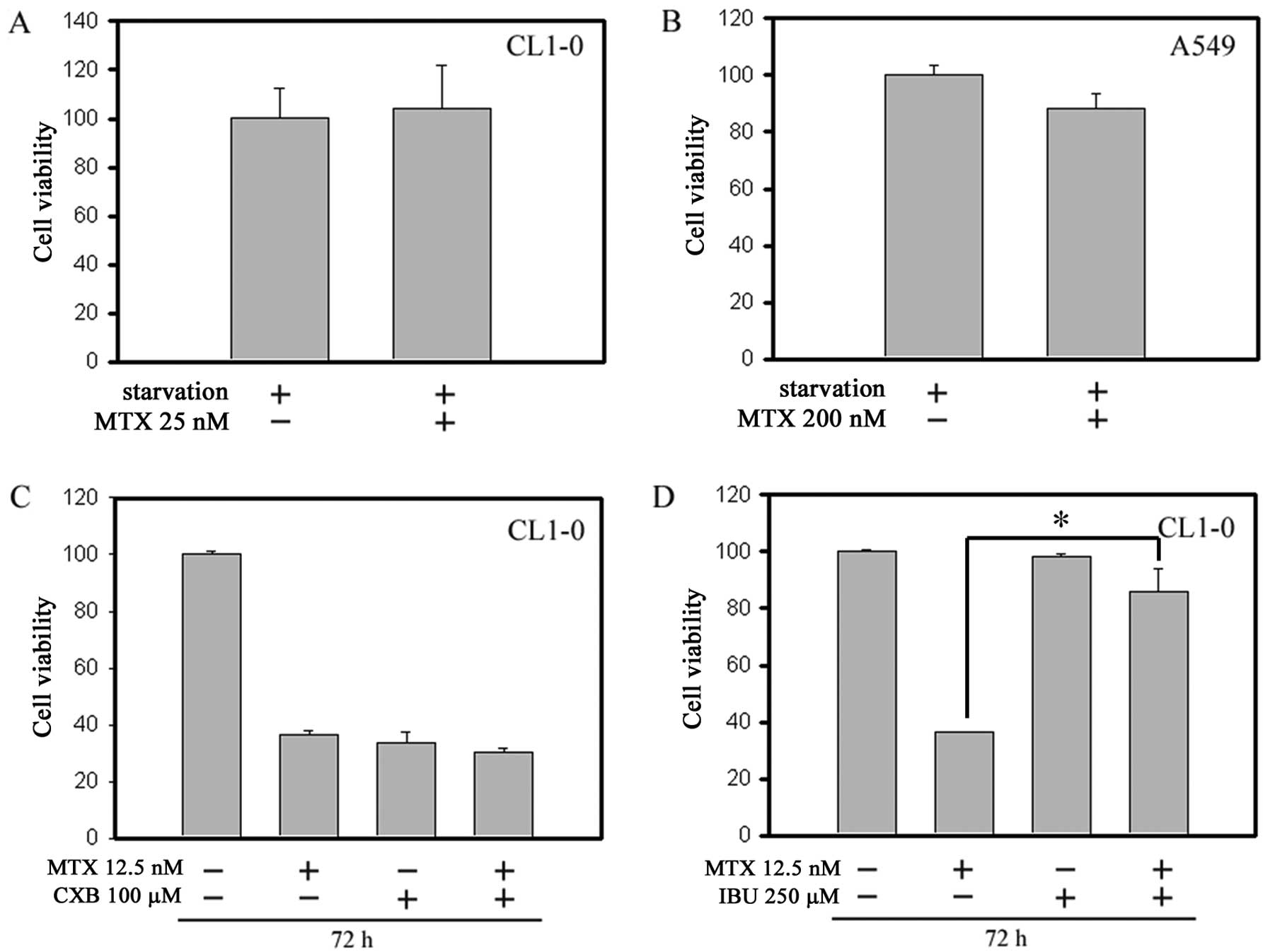
between MTX and ASA. Moreover, Fig. 7A
and B shows that starvation pretreatment protected the cells
from MTX treatment, presumably due to starvation-caused G1 phase
arrest. The G1 phase synchronization can protect cancer cells from
MTX-mediated cytotoxicity in the S phase. In order to elucidate
whether these ASA effects were mediated through COX-1 or COX-2
inhibition, we compared the COX-2-selective inhibitor CXB and the
non-selective inhibitor IBU. As shown in Fig. 7C and D, while CXB also exhibited
antagonism with MTX to some extent, IBU exerted a significantly
more potent antagonism with MTX, indicating that both COX-1 and
COX-2 are involved.
Discussion
While toxicity is common when ASA or NSAIDs are used
with MTX to treat RA (26), MTX and
ASA are prescribed as the cornerstone of therapy for RA (6,7,14,27,28).
In addition, MTX is a clinically useful blocker of DHFR (2,29), and
shows efficacy in the treatment of acute leukemias and a number of
types of solid tumors. MTX or ASA may be used in monotherapy or in
combination with other agents. In the present study, the
combination treatment with MTX + ASA was compared with treatment of
each drug alone in lung cancer cells by evaluating changes in cell
survival, cell cycle progression, cell proliferation and apoptotic
cell death. Individually ASA and MTX are effective in the treatment
of cancer; however, drug antagonism may pose a major obstacle to
their effectiveness when they are combined.
MTX acts specifically in the S phase, and therefore
exerts its activity in a cell cycle-specific manner (3). Recently, we reported that ASA induces
G1 phase accumulation in CL1-0 and A549 lung cancer cells (25), indicating that ASA controls cell
cycle progression, and this may be the underlying mechanism that
affects the therapeutic efficacy of MTX. Additionally, results of
this study found that serum-starved cells were also resistant to
MTX (Fig. 7), presumably through
synchronizing cells in the G1 phase and inducing growth arrest, an
effect similar to that induced by ASA. The observation that both
COX-1 and COX-2 inhibitors antagonized the antineoplastic effect of
MTX is important. COX-1/2 inhibitors, including IBU and CXB, have
been shown to induce a blockade of the G1 to S phase transition and
E2F inhibition (30,31), and these effects may have
contributed to their antagonizing effects against the efficacy of
MTX. Thus, we hypothesized that concomitant use of ASA decreases
the MTX-mediated S phase arrest and plays a role in quenching the
G1 to S phase transition to protect cells from MTX-mediated
cytotoxicity. Additionally, the level of cyclin A is low during G1
phase but begins to accumulate at the onset of the S phase and
contributes to the process of the S phase (32), and subsequently, is degraded during
the prometaphase (33). Here, we
showed that the MTX-induced growth inhibition was associated with a
marked S phase arrest as well as a significant accumulation of
cyclin A. Generally, downregulation of cyclin A results in cell
cycle arrest in the S phase. The accumulation of cyclin A suggests
that the MTX-induced S phase arrest was the result of a blockade of
DNA synthesis, instead of the result of a direct regulation of the
cyclin-Cdk pathway. Moreover, ASA may decrease the MTX-mediated
accumulation of cells in the S phase and cyclin A by maintaining
cells in the G1 phase.
Caspase-3 and Bcl-2 proteins play a critical role in
determining the threshold of apoptotic cell death. Here, we
demonstrated that ASA treatment may protect cells from apoptosis
through its ability to reverse the MTX-mediated upregulation of
caspase-3 and downregulation of Bcl-2. The FAS death receptor is
known to be expressed not only in immune cells but also in various
types of tumor cells. Binding of FAS to its ligand, FasL, triggers
a signaling cascade that leads to apoptosis (34). However, recent evidence suggests
that intracellular FAS can also be activated via a FasL-independent
pathway (35). During tumor
development, expression of FasL was found to be increased and was
associated with decreased expression of FAS in solid tumors
(36). Collectively, restoration of
the Fas/FasL pathway is a viable approach for novel therapeutic
strategies (37). In the present
study, MTX treatment increased the FAS protein level that may have
had an effect on the cytotoxicity of MTX. Importantly, while
treatment with ASA alone did not affect the FAS level, ASA
significantly antagonized the MTX-mediated increase in FAS protein.
The data shown in Fig. 5 suggest
that the decreased MTX-mediated apoptosis by ASA was likely through
the downregulation of FAS. Since the FAS/FasL system is important
in inducing cancer cell apoptosis (35), the effect of the downregulation of
FAS by ASA may present a novel mechanism underlying the MTX + ASA
antagonism that warrants further in-depth investigation.
Antifolates are classic antitumor agents that
inhibit key enzymes in DNA synthesis, i.e., DHFR (38,39).
The rhythmic change in MTX efficacy was observed to correspond to
changes in the DHFR activity of cells (3). MTX inhibits cancer cell proliferation
through tight-binding and by depletion of DHFR activity, and it was
thus important to elucidate whether DHFR activity is involved in
the MTX + ASA antagonism. Fig. 6
shows that almost all of the MTX-treated cancer cells were
recovered in terms of viability following folic acid addition.
Moreover, addition of folic acid reversed the MTX-mediated
anticancer effect and the antagonism noted following the MTX + ASA
combination treatment. The ASA-mediated anti-proliferative effect
noted following treatment with ASA alone was not affected. Folic
acid also restored the effects of MTX-mediated DHFR inhibition and
DNA replication, as well as the transition from S phase. The
results suggest that DHFR activity is involved in both the
MTX-mediated cytotoxic effect and the antagonism observed following
combination treatment with MTX + ASA. Several reports have
described that the amplification of DHFR is a common mechanism of
resistance to MTX (40–43). Previous studies have indicated that
DHFR expression is regulated by E2F-1 (38,39).
And E2F has also been found to be expressed in a cell
cycle-dependent manner and is essential for G1/S phase transition.
Our previous results suggest that ASA inhibits E2F-1 expression
(25), indicating that ASA may
control cell cycle progression and thereby may affect MTX efficacy.
As shown in Fig. 5, MTX exposure
upregulated the expression of DHFR protein but not its mRNA
transcript, indicating that the ASA-mediated E2F-1 downregulation
was not involved in the alteration of DHFR in this study. Previous
studies (40,44,45)
have shown that DHFR protein regulates its own transcript
translation through direct binding to its own mRNA; and thereby
constitutes a translational autoregulation loop. Hence, the
adaptive mechanism may allow cells to rapidly respond and to
decrease MTX sensitivity (45).
Binding of MTX to DHFR inhibits DHFR activity as well as the
interaction of the DHFR protein with its mRNA. In the present
study, we also observed the adaptive effect of DHFR translational
upregulation when the lung cancer cells were treated with MTX. In
contrast, ASA did not affect the DHFR translational autoregulation
either with or without MTX, suggesting that the ASA-mediated
attenuation of MTX sensitivity was independent of this DHFR
adaptive mechanism. Taken together, our data revealed that
pharmacologic concentrations of ASA can antagonize the efficacy of
the chemotherapeutic agent MTX. These results are of high clinical
relevance given the widespread use of NSAIDs and COX-1/2
inhibitors. The results also suggest that any agent that causes G1
accumulation may also exert an antagonism against MTX and adversely
influence the treatment outcome of MTX therapy. Further studies are
required to address the precise causes and clinical implications of
the combination of MTX and NSAIDs for RA and cancer therapy.
Acknowledgements
This study was supported by the National Health
Research Institutes (CA-099-PP-09), Department of Health
(DOH100-TD-C-111-004, DOH100-TD-C-111-008) and Wan Fang Hospital
(100swf15), Taiwan, R.O.C.
Abbreviations:
|
ASA
|
aspirin
|
|
NSAIDs
|
nonsteroidal anti-inflammatory
drugs
|
|
MTX
|
methotrexate
|
|
DHFR
|
dihydrofolate reductase
|
References
|
1
|
Fotoohi AK and Albertioni F: Mechanisms of
antifolate resistance and methotrexate efficacy in leukemia cells.
Leuk Lymphoma. 49:410–426. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hider SL, Bruce IN and Thomson W: The
pharmacogenetics of methotrexate. Rheumatology. 46:1520–1524. 2007.
View Article : Google Scholar
|
|
3
|
Yamauchi A, Ichimiya T, Inoue K, Taguchi
Y, Matsunaga N, Koyanagi S, Fukagawa T, Aramaki H, Higuchi S and
Ohdo S: Cell-cycle-dependent pharmacology of methotrexate in HL-60.
J Pharmacol Sci. 99:335–341. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cronstein BN: Low-dose methotrexate: a
mainstay in the treatment of rheumatoid arthritis. Pharmacol Rev.
57:163–172. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tian H and Cronstein BN: Understanding the
mechanisms of action of methotrexate: implications for the
treatment of rheumatoid arthritis. Bull NYU Hosp Jt Dis.
65:168–173. 2007.PubMed/NCBI
|
|
6
|
Weinblatt ME, Coblyn JS, Fox DA, Fraser
PA, Holdsworth DE, Glass DN and Trentham DE: Efficacy of low-dose
methotrexate in rheumatoid arthritis. N Engl J Med. 312:818–822.
1985. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chan ES and Cronstein BN: Molecular action
of methotrexate in inflammatory diseases. Arthritis Res. 4:266–273.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wessels JA, van der Kooij SM, le Cessie S,
Kievit W, Barerra P, Allaart CF, Huizinga TW and Guchelaar HJ: A
clinical pharmacogenetic model to predict the efficacy of
methotrexate monotherapy in recent-onset rheumatoid arthritis.
Arthritis Rheum. 56:1765–1775. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hani N, Casper C, Groth W, Krieg T and
Hunzelmann N: Stevens-Johnson syndrome-like exanthema secondary to
methotrexate histologically simulating acute graft-versus-host
disease. Eur J Dermatol. 10:548–550. 2000.PubMed/NCBI
|
|
10
|
Naidu MU, Ramana GV, Rani PU, Mohan IK,
Suman A and Roy P: Chemotherapy-induced and/or radiation
therapy-induced oral mucositis - complicating the treatment of
cancer. Neoplasia. 6:423–431. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tomoda R, Seto M, Hioki Y, Sonoda J,
Matsumine A, Kusuzaki K and Uchida A: Low-dose methotrexate
inhibits lung metastasis and lengthens survival in rat
osteosarcoma. Clin Exp Metastasis. 22:559–564. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Morison WL, Momtaz K, Parrish JA and
Fitzpatrick TB: Combined methotrexate-PUVA therapy in the treatment
of psoriasis. J Am Acad Dermatol. 6:46–51. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sweeney CJ, Takimoto CH, Latz JE, Baker
SD, Murry DJ, Krull JH, Fife K, Battiato L, Cleverly A, Chaudhary
AK, Chaudhuri T, Sandler A, Mita AC and Rowinsky EK: Two drug
interaction studies evaluating the pharmacokinetics and toxicity of
pemetrexed when coadministered with aspirin or ibuprofen in
patients with advanced cancer. Clin Cancer Res. 12:536–542. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pablos JL: Aspirin antiplatelet therapy
and nonsteroidal antiinflammatory drugs: comment on the 2002 update
of the American College of Rheumatology Guidelines for the
Management of Rheumatoid Arthritis. Arthritis Rheum. 46:31022002.
View Article : Google Scholar
|
|
15
|
Moysich KB, Menezes RJ, Ronsani A, Swede
H, Reid ME, Cummings KM, Falkner KL, Loewen GM and Bepler G:
Regular aspirin use and lung cancer risk. BMC Cancer. 2:312002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Van Dyke AL, Cote ML, Prysak G, Claeys GB,
Wenzlaff AS and Schwartz AG: Regular adult aspirin use decreases
the risk of non-small cell lung cancer among women. Cancer
Epidemiol Biomarkers Prev. 17:148–157. 2008.PubMed/NCBI
|
|
17
|
Cuzick J, Otto F, Baron JA, Brown PH, Burn
J, Greenwald P, Jankowski J, La Vecchia C, Meyskens F, Senn HJ and
Thun M: Aspirin and non-steroidal anti-inflammatory drugs for
cancer prevention: an international consensus statement. Lancet
Oncol. 10:501–507. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kremer JM and Hamilton RA: The effects of
nonsteroidal antiinflammatory drugs on methotrexate (MTX)
pharmacokinetics: impairment of renal clearance of MTX at weekly
maintenance doses but not at 7.5 mg. J Rheumatol. 22:2072–2077.
1995.
|
|
19
|
Stewart CF, Fleming RA, Germain BF,
Seleznick MJ and Evans WE: Aspirin alters methotrexate disposition
in rheumatoid arthritis patients. Arthritis Rheum. 34:1514–1520.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fries JF, Singh G, Lenert L and Furst DE:
Aspirin, hydroxychloroquine, and hepatic enzyme abnormalities with
methotrexate in rheumatoid arthritis. Arthritis Rheum.
33:1611–1619. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chu YW, Yang PC, Yang SC, Shyu YC, Hendrix
MJ, Wu R and Wu CW: Selection of invasive and metastatic
subpopulations from a human lung adenocarcinoma cell line. Am J
Respir Cell Mol Biol. 17:353–360. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Keepers YP, Pizao PE, Peters GJ, van
Ark-Otte J, Winograd B and Pinedo HM: Comparison of the
sulforhodamine B protein and tetrazolium (MTT) assays for in vitro
chemosensitivity testing. Eur J Cancer. 27:897–900. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chou TC: Theoretical basis, experimental
design, and computerized simulation of synergism and antagonism in
drug combination studies. Pharmacol Rev. 58:621–681. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: the combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yan KH, Yao CJ, Chang HY, Lai GM, Cheng AL
and Chuang SE: The synergistic anticancer effect of troglitazone
combined with aspirin causes cell cycle arrest and apoptosis in
human lung cancer cells. Mol Carcinog. 49:235–246. 2010.PubMed/NCBI
|
|
26
|
Rooney TW, Furst DE, Koehnke R and
Burmeister L: Aspirin is not associated with more toxicity than
other nonsteroidal antiinflammatory drugs in patients with
rheumatoid arthritis treated with methotrexate. J Rheumatol.
20:1297–1302. 1993.PubMed/NCBI
|
|
27
|
Kurth T, Hennekens CH, Buring JE and
Gaziano JM: Aspirin, NSAIDs, and COX-2 inhibitors in cardiovascular
disease: possible interactions and implications for treatment of
rheumatoid arthritis. Curr Rheumatol Rep. 6:351–356. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ranganathan P: An update on methotrexate
pharmacogenetics in rheumatoid arthritis. Pharmacogenomics.
9:439–451. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Krajinovic M and Moghrabi A:
Pharmacogenetics of methotrexate. Pharmacogenomics. 5:819–834.
2004. View Article : Google Scholar
|
|
30
|
Bock JM, Menon SG, Goswami PC, Sinclair
LL, Bedford NS, Domann FE and Trask DK: Relative non-steroidal
anti-inflammatory drug (NSAID) antiproliferative activity is
mediated through p21-induced G1 arrest and E2F inhibition. Mol
Carcinog. 46:857–864. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Janssen A, Maier TJ, Schiffmann S, Coste
O, Seegel M, Geisslinger G and Grösch S: Evidence of COX-2
independent induction of apoptosis and cell cycle block in human
colon carcinoma cells after S- or R-ibuprofen treatment. Eur J
Pharmacol. 540:24–33. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Resnitzky D, Hengst L and Reed SI: Cyclin
A-associated kinase activity is rate limiting for entrance into S
phase and is negatively regulated in G1 by p27Kip1. Mol Cell Biol.
15:4347–4352. 1995.PubMed/NCBI
|
|
33
|
Mateo F, Vidal-Laliena M, Canela N, Busino
L, Martinez-Balbas MA, Pagano M, Agell N and Bachs O: Degradation
of cyclin A is regulated by acetylation. Oncogene. 28:2654–2666.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gloire G, Charlier E and Piette J:
Regulation of CD95/APO-1/Fas-induced apoptosis by protein
phosphatases. Biochem Pharmacol. 76:1451–1458. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mollinedo F and Gajate C: Fas/CD95 death
receptor and lipid rafts: new targets for apoptosis-directed cancer
therapy. Drug Resist Updat. 9:51–73. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kim R, Emi M, Tanabe K, Uchida Y and Toge
T: The role of Fas ligand and transforming growth factor β in tumor
progression: molecular mechanisms of immune privilege via
Fas-mediated apoptosis and potential targets for cancer therapy.
Cancer. 100:2281–2291. 2004.
|
|
37
|
O’Brien DI, Nally K, Kelly RG, O’Connor
TM, Shanahan F and O’Connell J: Targeting the Fas/Fas ligand
pathway in cancer. Expert Opin Ther Targets. 9:1031–1044. 2005.
|
|
38
|
Sowers R, Toguchida J, Qin J, Meyers PA,
Healey JH, Huvos A, Banerjee D, Bertino JR and Gorlick R: mRNA
expression levels of E2F transcription factors correlate with
dihydrofolate reductase, reduced folate carrier, and thymidylate
synthase mRNA expression in osteosarcoma. Mol Cancer Ther.
2:535–541. 2003.
|
|
39
|
Slansky JE, Li Y, Kaelin WG and Farnham
PJ: A protein synthesis-dependent increase in E2F1 mRNA correlates
with growth regulation of the dihydrofolate reductase promoter. Mol
Cell Biol. 13:1610–1618. 1993.PubMed/NCBI
|
|
40
|
Skacel N, Menon LG, Mishra PJ, Peters R,
Banerjee D, Bertino JR and Abali EE: Identification of amino acids
required for the functional up-regulation of human dihydrofolate
reductase protein in response to antifolate treatment. J Biol Chem.
280:22721–22731. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mishra PJ, Humeniuk R, Longo-Sorbello GS,
Banerjee D and Bertino JR: A miR-24 microRNA binding-site
polymorphism in dihydrofolate reductase gene leads to methotrexate
resistance. Proc Natl Acad Sci USA. 104:13513–13518. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Snijders AM, Hermsen MA, Baughman J,
Buffart TE, Huey B, Gajduskova P, Roydasgupta R, Tokuyasu T, Meijer
GA, Fridlyand J and Albertson DG: Acquired genomic aberrations
associated with methotrexate resistance vary with background
genomic instability. Genes Chromosomes Cancer. 47:71–83. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bertino JR, Göker E, Gorlick R, Li WW and
Banerjee D: Resistance mechanisms to methotrexate in tumors. Stem
Cells. 14:5–9. 1996. View Article : Google Scholar
|
|
44
|
Ercikan-Abali EA, Banerjee D, Waltham MC,
Skacel N, Scotto KW and Bertino JR: Dihydrofolate reductase protein
inhibits its own translation by binding to dihydrofolate reductase
mRNA sequences within the coding region. Biochemistry.
36:12317–12322. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hsieh YC, Skacel NE, Bansal N, Scotto KW,
Banerjee D, Bertino JR and Abali EE: Species-specific differences
in translational regulation of dihydrofolate reductase. Mol
Pharmacol. 76:723–733. 2009. View Article : Google Scholar : PubMed/NCBI
|