Introduction
Various genetic alterations such as point mutations,
insertions/deletions (indels) and gene fusions directly participate
in human carcinogenesis. Some of such changes confer growth
advantage to cancer cells, and targeting their encoded proteins is
one of the most effective means to treat cancer. Activating
mutations in EGFR(1) or the
gene fusion between EML4 and ALK(2) are, for instance, identified in subsets
of non-small cell lung cancer (NSCLC), and reagents targeting EGFR
or ALK are proved clinically effective against NSCLC positive for
the corresponding genetic lesions (3,4).
Peripheral T-cell lymphoma (PTCL) is a subdivision
of non-Hodgkin’s malignant lymphoma (5). In contrast to the improved prognosis
for individuals with B-cell lymphoma with recent treatment
modalities, most subtypes of PTCL still have a 5-year survival rate
of only <30% (6). Molecular
mechanism for PTCL carcinogenesis is mainly enigmatic, except for
the facts that i) a part of PTCL is associated with infection with
Epstein-Barr virus (EBV) or human T-lymphotropic virus type I
(HTLV-I), and ii) activating ALK fusions are found in the
anaplastic large cell lymphoma (ALCL) subtype. Given the fact that
an ALK inhibitor is effective in the treatment of ALCL (7), identification of essential growth
drivers in other subsets of PTCL is urgently needed.
While exome sequencing of cancer specimens is used
for the detection of somatic mutations in the cancer genome, such
approach fails to detect gene fusions, because gene fusions usually
take place at intronic regions. To simultaneously detect point
mutations/indels/gene fusions in a single experiment, we previously
reported the ‘cDNA capture system’ that conducts massive
resequencing on purified cDNAs for cancer-related genes (8). Herein we applied such technology to
the cDNAs isolated from a PTCL specimen, and discovered a STK10
amino acid substitution that turned out to exert anti-apoptotic
effects. While this amino acid change was recently deposited as a
single nucleotide polymorphism (SNP) in the 1000 genome database
(http://www.1000genomes.org), we also
confirmed that other nonsynonymous, somatic mutations within
STK10 confer marked anti-apoptotic activity. These results
suggest that STK10 may contribute to carcinogenesis, either through
polymorphism or somatic mutations, by suppressing apoptotic
signaling in cancer.
Materials and methods
Cell lines and specimens
PTCL specimens were collected in Fukushima Medical
University and The Cancer Institute. A human embryonic kidney 293T
(HEK293) cell line was obtained from American Type Culture
Collection (ATCC: Manassas, VA, USA), and maintained in Dulbecco’s
modified Eagle’s medium-F12 medium (Invitrogen, Carlsbad, CA, USA)
supplemented with 10% fetal bovine serum (FBS) and 2 mM L-glutamine
(both from Invitrogen). An IL-2-dependent mouse cytotoxic T-cell
line, CTLL-2, and a human T-cell line, Jurkat, were both obtained
from ATCC, and maintained in RPMI1640 medium (Invitrogen)
supplemented with 10% FBS. Mouse recombinant IL-2 (Peprotech, Rocky
Hill, NJ, USA) was added to the culture medium of CTLL-2 at the
concentration of 2 ng/ml. Total RNA was extracted from cell lines
and a PTCL specimen with an RNeasy mini kit (Qiagen, Valencia, CA,
USA), and was subjected to reverse transcriptase (RT) with an
oligo-dT primer. Written informed consent was obtained from the
subjects who provided cancer specimens, and the study was approved
by the human ethics committee of the University of Tokyo, Jichi
Medical University, Fukushima Medical University, and The Cancer
Institute of the Japanese Foundation for Cancer Research.
Resequencing of target-captured cDNA in a
PTCL specimen
Resequencing with a custom cDNA-capture system was
performed as previously described (8). In brief, RNA probes of 120 bases were
designed to interrogate cDNAs of 906 human protein-coding genes,
and were synthesized by Agilent Technologies (Santa Clara, CA,
USA). Hybridization of cDNA fragments isolated from a PTCL specimen
to the RNA probes was performed according to the protocols
recommended for the SureSelect Target Enrichment system (Agilent
Technologies). Purified cDNA fragments were then subjected to deep
sequencing with a Genome Analyzer IIx (GAIIx; Illumina, San Diego,
CA, USA) for 76 bases from both ends by the paired-end sequencing
system.
Plasmid construction
The full-length cDNA for the wild-type or the R634H
mutant of STK10 was amplified by PCR from cDNA of a PTCL specimen.
The cDNAs for other mutant forms of STK10 were further generated by
a polymerase chain reaction (PCR)-based mutagenesis approach. The
cDNA for PLK1, IKK-α, IKK-β or IKK-γ was amplified by PCR from our
cancer cell lines.
Luciferase-based reporter assay
HEK293 cells were transfected with the expression
plasmids, a promoter-less Renilla luciferase plasmid
pGL-4.70 (Promega, Madison, WI, USA), and firefly luciferase-based
reporter plasmid for Fos (pFL700) (9), Myc (pHXL) (10), NF-κB (Stratagene, La Jolla, CA,
USA), Bcl-xL (11), Notch
(pGa981-6) (12), Wnt (TOP-flash,
Upstate Biotechnology, Lake Placid, NY, USA) or Rho (pSRE.L)
(13) signaling pathway. Luciferase
activities were determined with the Dual-Luciferase Reporter Assay
System (Promega), and the firefly luciferase activities were
normalized to the Renilla luciferase activities.
Quantitative real-time RT-PCR
Quantitative real-time RT-PCR was performed with
QuantiTect SYBR Green PCR kit (Qiagen) and an Applied Biosystems
7900HT Fast Real-Time PCR System. The PCR conditions include the
first incubation at 50°C for 2 min and then 95°C for 15 min,
followed by 60 cycles of 94°C for 15 sec and 60°C for 30 sec and
72°C for 1 min. Relative expression levels of target mRNAs to
GAPDH were normalized to those in the mock-transfected
cells. The primer sequences used for the PCR reactions were:
5′-TTCATTCCTGGCAAGTGGATCATT-3′ and 5′-ATGGCAGCATCATTGTTCTCATCA-3′
for TLR2, 5′-TGACAACCTTCTGGTTGGTAGGGA-3′ and
5′-CCAAGGTCATGGT TGTCCAAAGAC-3′ for BCL2, 5′-AAGAATCACCAGCA
GCAAGTGTCC-3′ and 5′-TTGGGTTGTGGAGTGAGTGTTCAA-3′ for CCL2,
and 5′-CCAGGTGGTCTCCTCTGACTTCAA-3′ and
5′-CACCCTGTTGCTGTAGCCAAATTC-3′ for GAPDH.
Antibodies, immunoprecipitation and
immunoblotting
Antibodies used in this study were: anti-FLAG M2
(Sigma-Aldrich, St. Louis, MO, USA), anti-PLK1 (Santa Cruz
Biotechnology, Santa Cruz, CA, USA), anti-phospho-PLK1 (BD
Pharmingen, San Jose, CA, USA), anti-ACTB (Cell Signaling
Technology, Danvers, MA, USA), anti-mouse IgG and anti-rabbit IgG
(both from GE Healthcare, Piscataway, NJ, USA). HEK293 cells were
transfected with the appropriate expression vectors, and then lysed
in lysis buffer [1% NP-40, 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1
mM NaF, 1 mM Na3VO4, 1 mM
phenylmethylsulfonyl fluoride and aprotinin]. Target proteins were
purified by incubation of the cell lysates with appropriate
antibodies and Protein G Sepharose Fast Flow (Sigma-Aldrich) for 3
h at 4°C. Immunoblot analyses were visualized with SuperSignal West
Femto Maximum Sensitivity Substrate (Thermo Scientific, Waltham,
MA, USA).
In vitro kinase assay
Immunoprecipitates were washed with kinase buffer
[50 mM NaCl, 50 mM Tris-HCl (pH 7.4), 10 mM MgCl2, 10 mM
MnCl2 and 0.1 mM Na3VO4], and then
incubated in kinase buffer containing [γ-32P]ATP
(Perkin-Elmer, Boston, MA, USA) and histone H2A protein (New
England BioLabs, Ipswich, MA, USA) for 30 min at room
temperature.
Dexamethasone-induced apoptosis
assay
CTLL-2 cells were infected with retrovirus
expressing STK10 and the blasticidin-resistant gene, and
cultured under the presence of 10 μg/ml blasticidin (InvivoGen, San
Diego, CA, USA). Blasticidin-resistant cells were then treated with
1 μM dexamethasone (Sigma-Aldrich), and cell number was determined
every 24 h with CellTiter-Glo Luminescent Cell Viability Assay
(Promega). At 72 h after treatment with dexamethasone, CTLL-2 cells
were collected and subjected to apoptosis assay with flow cytometry
(FACSCanto II; BD Biosciences, San Jose, CA, USA) using Annexin
V/propidium iodide (PI) staining kit (eBioscience, San Diego, CA,
USA).
Results
Identification of STK10(R634H) in a PTCL
specimen
To identify transforming genes in PTCL, cDNAs for
cancer-related genes (n=906) (8)
were isolated from a PTCL specimen obtained from a 25 year-old male
negative for EBV and HTLV-I infection, and subjected to deep
sequencing with GAIIx. We thus obtained a total of 69,552,278
independent, high-quality reads that mapped to 868 cDNAs (95.8%).
Screening of missense mutations, insertions, deletions and fusion
genes through our in-house computational pipeline (8) revealed a total of 19 nonsynonymous
mutations but no indels or gene fusions, which were further
confirmed by Sanger sequencing (Table
I). It should be noted, however, that 9 of the 19 nonsynonymous
mutations were recently registered as single nucleotide
polymorphisms (SNPs) in the 1000 genome project (http://www.1000genomes.org) after our initial
analysis.
 | Table INonsynonymous mutations detected in a
PTCL specimen. |
Table I
Nonsynonymous mutations detected in a
PTCL specimen.
| Gene symbol | GenBank accession
no. | Nucleotide
change | Read coverage | Mismatch reads
(%) | Amino acid
change |
|---|
| AXLa | NM_001699 | c.1506G>T | ×156 | 59.0 | W439L |
| CDC7 | NM_003503 | c.1732C>G | ×317 | 47.0 | H523D |
| CDC7 | NM_003503 | c.1751A>G | ×298 | 48.3 | N529S |
| CREBBP | NM_001079846 | c.2478G>T | ×470 | 54.7 | Q758H |
| E2F1 | NM_005225 | c.1399G>A | ×126 | 30.2 | G420D |
| EIF2AK4 | NM_001013703 | c.3268G>A | ×239 | 46.0 | R1073H |
| FERMT3 | NM_031471 | c.2080G>T | ×48 | 56.3 | R644L |
| FOXO3a | NM_201559 | c.2013G>C | ×1001 | 41.3 | G566A |
| LRRK2a | NM_198578 | c.4314G>A | ×166 | 43.4 | R1398H |
| MAPK7a | NM_139033 | c.2475G>A | ×287 | 61.7 | G697R |
| PDGFRBa | NM_002609 | c.3441G>A | ×129 | 35.7 | R991H |
| PTCH1 | NM_000264 | c.4423C>T | ×60 | 63.3 | P1412L |
|
PTCH2a | NM_003738 | c.2975C>T | ×502 | 61.6 | T988M |
|
RPS6KA2a | NM_021135 | c.1202G>A | ×293 | 41.0 | R328Q |
|
RPS6KB2a | NM_003952 | c.882C>T | ×446 | 54.0 | P267L |
| SOS2 | NM_006939 | c.668A>T | ×338 | 49.1 | E190D |
| STK4 | NM_006282 | c.962G>A | ×7195 | 49.1 | R291Q |
|
STK10a | NM_005990 | c.2201G>A | ×462 | 58.7 | R634H |
| TRIM33 | NM_015906 | c.1049C>T | ×599 | 44.9 | A322V |
One of the 19 nonsynonymous mutations was a
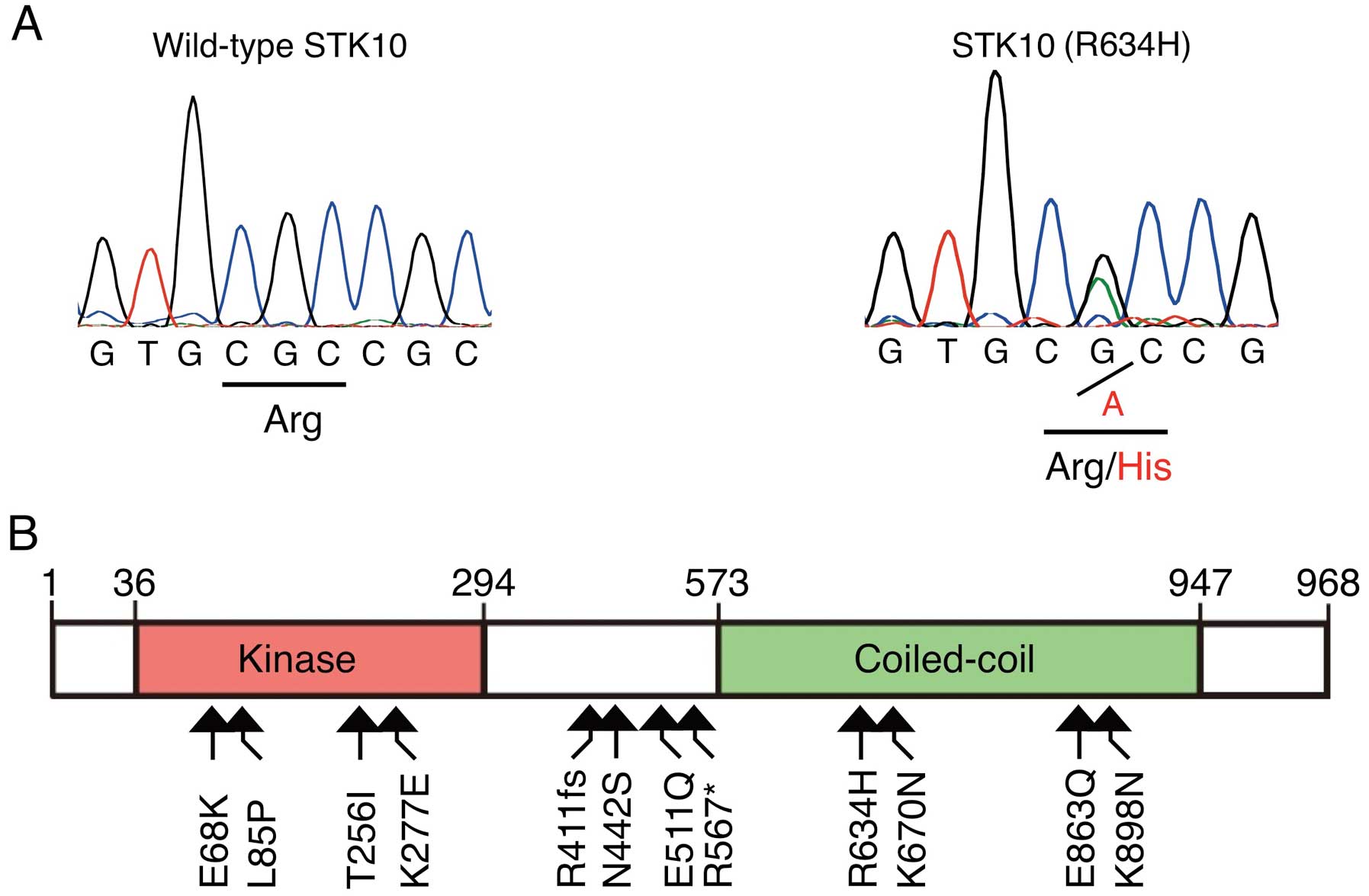
nucleotide substitution of G-to-A at position 2201 of human
STK10 cDNA (GenBank accession number, NM_005990) that was
later deposited as an SNP (rs115974403). Sequencing of this
position in genomic DNA of the same specimen further confirmed this
substitution, which results in replacement of an arginine residue
at amino acid position 634 with a histidine residue (R634H)
(Fig. 1A). We also searched STK10
mutations among our human cancer specimens (n=76), cancer cell
lines (n=56) and the COSMIC database for cancer genome mutations
(Release v58, http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/),
revealing additional 11 nonsynonymous mutations; E68K in a CML cell
line K562, L85P, T256I, R411-frameshift (R411fs), K670N and K898N
in various specimens of ovarian cancer, K277E in a testicular
cancer specimen, N442S in a pancreatic cancer cell line Pa21C,
E511Q in a specimen of upper aerodigestive tract cancer, R567* in
Jurkat, and E863Q in a lung cancer specimen (Fig. 1B).
Activation of NF-κB signaling by STK10
mutants
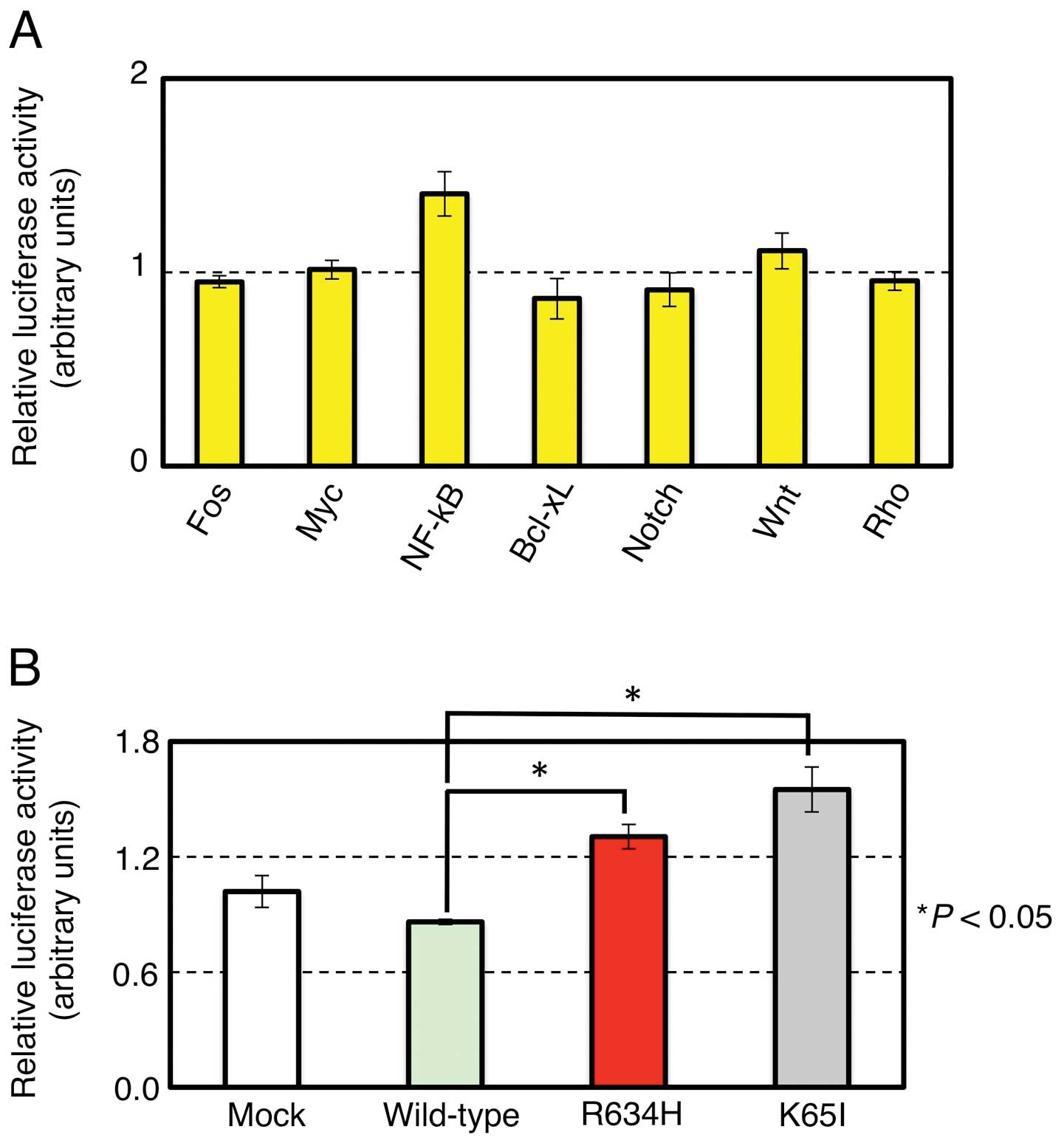
To examine whether STK10(R634H) affects
intracellular signaling pathway related to Fos, Myc, NF-κB, Bcl-xL,
Notch, Wnt or Rho, we examined the reporter activity for each
pathway under the expression of the wild-type or the R634H mutant
of STK10. As shown in Fig. 2A, the
R634H mutant significantly elevated the NF-κB reporter activity
compared to the wild-type (P<0.05, Student’s t-test), but did
not affect the other signaling systems. To further investigate the
role of STK10 in the NF-κB signaling, we generated cDNA for a
kinase-dead mutant (K65I) of STK10, and examined its effects on the
NF-κB signaling. Of note, the wild-type STK10 rather suppressed the
NF-κB pathway, but the R634H mutation abolished such effect,
suggesting that STK10 is a negative regulator for intracellular
NF-κB activity (Fig. 2B). This
hypothesis was further reinforced by the fact that the
kinase-inactive K65I mutant markedly elevates the NF-κB reporter
signaling. We further asked the effects of other STK10 mutants on
NF-κB, and revealed that the L85P and K277E mutants found in the
COSMIC database clearly induced NF-κB (Fig. 2C). Noteworthy, these mutations were
somatically acquired in ovarian and testicular cancers,
respectively. The other STK10 mutations did not affect NF-κB
signaling (Fig. 2C and data not
shown).
To further confirm the effects of STK10 mutants on
the NF-κB activation, expression levels of its target genes were
quantified among cells expressing the wild-type or mutant forms of
STK10 (Fig. 2D). Consistent with
the results of luciferase assay, L85P, K277E, R634H and K65I
mutants each induced the expression of NF-κB target genes including
TLR2, BCL2 and CCL2, compared to the wild-form. These
results indicate that polymorphism as well as somatic mutations of
STK10 may regulate NF-κB signaling in various human cancers.
It is not clear yet how STK10 regulates the NF-κB
pathway. When HEK293 cells were stimulated with tumor necrosis
factor (TNF)-α, K65I amino acid change cancelled the STK10-mediated
NF-κB suppression (Fig. 2E).
However, such regulation was not observed on the IKK complex-driven
NF-κB activation, indicating that STK10 may interact with the NF-κB
pathway between TNF receptors and the IKK complex.
Kinase activities of STK10 mutants
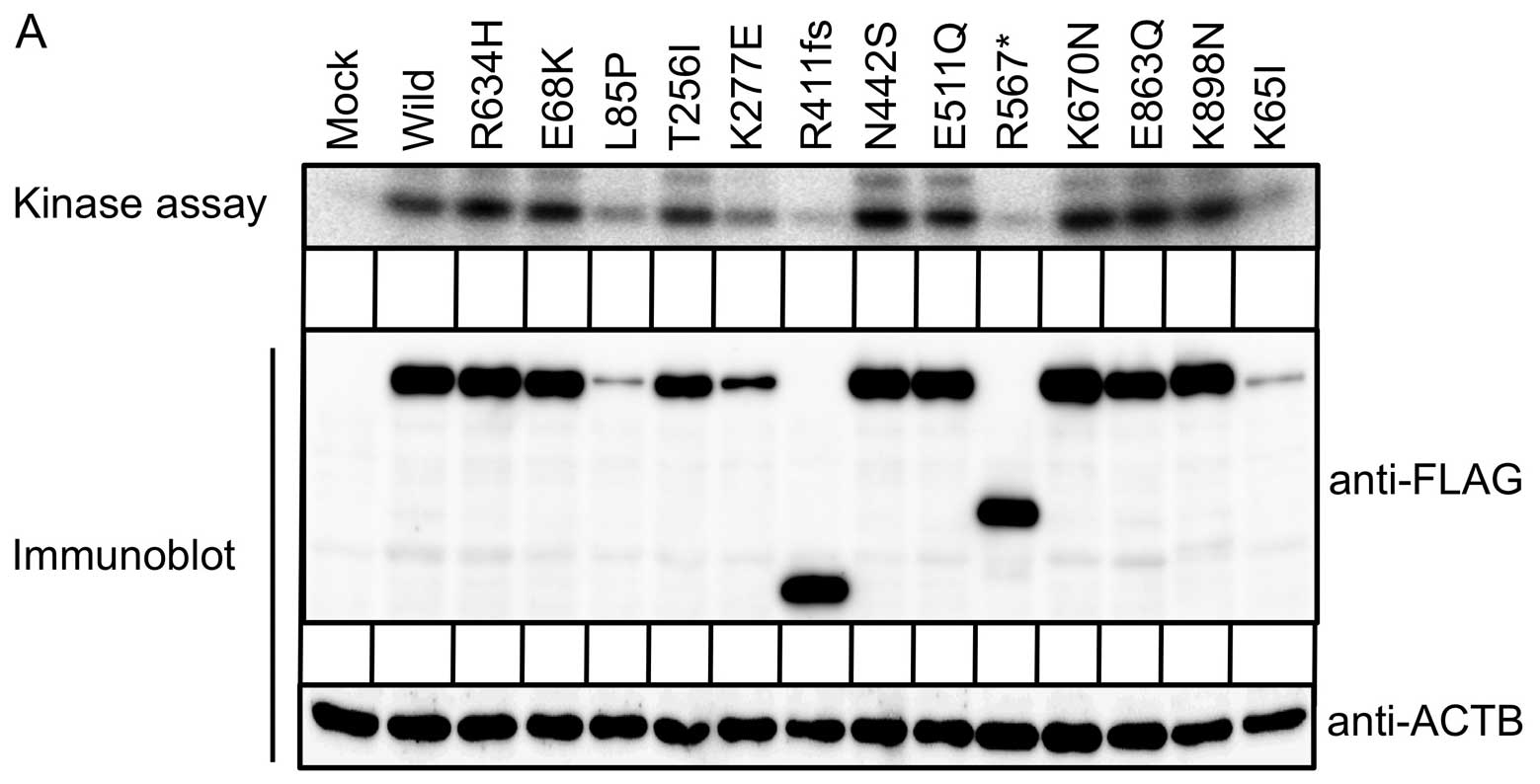
To examine if the STK10 mutations directly affect
its enzymatic activity, FLAG-tagged wild-type or mutant forms of
STK10 was expressed in HEK293 cells, and subjected to
immunoprecipitation with antibodies to FLAG, followed by an in
vitro kinase assay with histone H2A as an exogenous substrate.
As shown in Fig. 3A, while the
wild-type STK10 clearly phosphorylates histone H2A, the L85P, K277E
and K65I mutants remarkably attenuate the substrate
phosphorylation. We could not, however, observe a notable
difference between the wild-type and the R634H mutant in their
ability of H2A phosphorylation, suggesting that R634H mutation may
regulate the NF-κB signaling not through suppressing its enzymatic
activity but through affecting its interaction to other
proteins.
Of note, the R411fs and R567* mutants did not
phosphorylate histone H2A, indicating the coiled-coil domain and/or
carboxyl terminal end of STK10 (Fig.
1B) is indispensable for histone phosphorylation. Immunoblot
analysis with antibodies to FLAG revealed that protein amounts of
the L85P, K277E and K65I mutants were markedly reduced, which may
account for the decreased phosphorylation of histone H2A.
Since STK10 is also known to phosphorylate PLK1
(14), we further investigated the
phosphorylation level of PLK1 when co-expressed with the wild-type
or mutant forms of SKT10 (Fig. 3B).
Consistent with the result of the in vitro kinase assay,
phosphorylation of PLK1 was reduced under the presence of the L85P,
K277E or K65I mutant. Again, R634H mutation did not significantly
affect the PLK1 phosphorylation. Strikingly, however, both of
R411fs and R567* mutants could phosphorylate PLK1 equally to, or
greater than, the wild-type. The carboxyl-terminal half of STK10
may, thus, be dispensable for the phosphorylation of PLK1 in
vivo.
Anti-apoptotic function of STK10
mutants
NF-κB plays pivotal roles in inflammatory and
anti-apoptotic responses in cells, and constitutive NF-κB
activation has been recognized as a hallmark of several lymphoid
malignancies (15). Given the
NF-κB-activating potential of STK10 mutants, we then asked whether
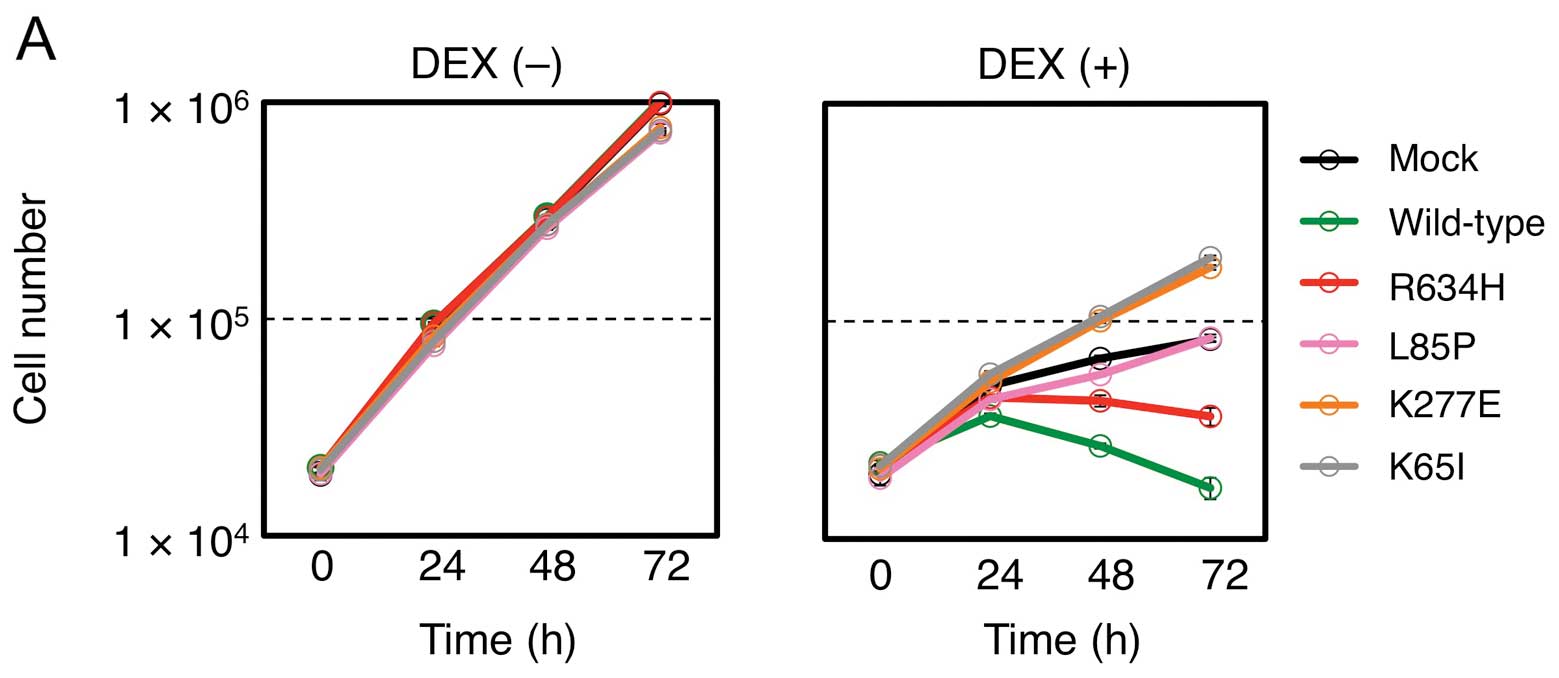
such mutants also regulate cell apoptosis. Glucocorticoids are
known to be a potent apoptosis-inducer in lymphocytes, and have
been used in therapeutic regimens for lymphoid malignancies
(15). As shown in Fig. 4A, treatment of a mouse T-cell line,
CTLL-2, with dexamethasone inhibited cell proliferation, and,
importantly, introduction of the wild-type STK10 markedly augmented
such effect, again confirming its tumor-suppressive role. In
contrast, introduction of NF-κB-activating mutants restored cell
growth, in a parallel manner to their NF-κB-activating
potential.
To determine whether the growth-inhibitory effect
thus observed was related to apoptosis, we conducted the Annexin
V/PI staining assay (Fig. 4B).
Although an increase in the Annexin V-positive fraction was
apparent in dexamethasone-treated, wild-type STK10-expressing
CTLL-2 cells, introduction of the L85P, K277E, R634H or K65 mutant
of STK10 significantly suppressed apoptosis. Thus, a part of
polymorphism/somatic mutations within STK10 not only activate the
NF-κB pathway, but directly exert anti-apoptotic function.
Multiplex deep sequencing of STK10 in
PTCL specimens
As it was demonstrated that some STK10 mutations
induce NF-κB activation and render resistance to
dexamethasone-induced apoptosis, we further searched for
STK10 mutations in genomic DNAs of 92 PTCL specimens. It
should be noted, however, that PTCL specimens frequently contain
numerous normal cells in addition to neoplastic cells, which makes
it difficult to determine the presence or absence of STK10
mutations with conventional Sanger sequencing. We therefore chose a
deep sequencing approach with the GAIIx system (16), leading to the isolation of one
specimen carrying STK10(R634H) (data not shown).
Discussion
STK10 is a member of the STE20 family of
serine/threonine kinases that are involved in a variety of
intracellular functions such as cell proliferation, regulation of
apoptosis, rearrangement of the cytoskeleton and cell motility
(17). While the Lok kinase, a
mouse homolog of STK10, was shown to be highly expressed in
lymphocytes (18), expression of
STK10 may be detected in other tissues as well, including brain,
colon, thymus, kidney, liver, small intestine and lung (14).
In this study, we identified STK10(R634H) in a PTCL
specimen, and demonstrated that the R634H mutation induces NF-κB
activation. We also demonstrated, for the first time, that the
wild-type STK10 is a negative regulator for NF-κB, and may be a
tumor suppressor since it strongly augments dexamethasone-driven
apoptosis in T-lymphocytes. Given the fact that STK10(R634H)
attenuates cell apoptosis, such polymorphism may predispose
lymphomagenesis. Indeed, through a mutation search of additional
PTCL specimens (n=92), we identified another PTCL case positive for
the R634H polymorphism. While the cohort size is still small,
frequency (2/92=2.2%) of this polymorphism in PTCL is significantly
over-represented compared to that (1/2385=0.04%) in the 1000 genome
database (P=4.0×10−3, Fisher’s exact test).
Noteworthy, some of the somatic mutations within
STK10 found in epithelial tumors have more profound effects on the
NF-κB regulation and cell apoptosis than R634H. Thus, dysfunction
of STK10 through somatic mutations is likely a novel, driver event
in human carcinogenesis. Restoration of STK10 function or/and
suppression of NF-κB in the tumors positive for STK10 mutations can
be a novel strategy for raising effective molecular targeted
therapies against human cancer.
Acknowledgements
This study was supported in part by grants for
Third-Term Comprehensive Control Research for Cancer and for
Research on Human Genome Tailor-made from the Ministry of Health,
Labor and Welfare of Japan, by Grants-in-Aid from the Japan Society
for the Promotion of Science, from The Yasuda Medical Foundation,
from The Sagawa Foundation for Promotion of Cancer Research, and
from The Mitsubishi Foundation.
References
|
1
|
Lynch TJ, Bell DW, Sordella R, et al:
Activating mutations in the epidermal growth factor receptor
underlying responsiveness of non-small-cell lung cancer to
gefitinib. N Engl J Med. 350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Soda M, Choi YL, Enomoto M, et al:
Identification of the transforming EML4-ALK fusion gene in
non-small-cell lung cancer. Nature. 448:561–566. 2007.PubMed/NCBI
|
|
3
|
Mok TS, Wu YL, Thongprasert S, et al:
Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N
Engl J Med. 361:947–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kwak EL, Bang YJ, Camidge DR, et al:
Anaplastic lymphoma kinase inhibition in non-small-cell lung
cancer. N Engl J Med. 363:1693–1703. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Foss FM, Zinzani PL, Vose JM, Gascoyne RD,
Rosen ST and Tobinai K: Peripheral T-cell lymphoma. Blood.
117:6756–6767. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vose J, Armitage J and Weisenburger D:
International peripheral T-cell and natural killer/T-cell lymphoma
study: pathology findings and clinical outcomes. J Clin Oncol.
26:4124–4130. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gambacorti-Passerini C, Messa C and
Pogliani EM: Crizotinib in anaplastic large-cell lymphoma. N Engl J
Med. 364:775–776. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ueno T, Yamashita Y, Soda M, et al:
High-throughput resequencing of target-captured cDNA in cancer
cells. Cancer Sci. 103:131–135. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hu Q, Milfay D and Williams LT: Binding of
NCK to SOS and activation of ras-dependent gene expression. Mol
Cell Biol. 15:1169–1174. 1995.PubMed/NCBI
|
|
10
|
Takeshita T, Arita T, Higuchi M, et al:
STAM, signal transducing adaptor molecule, is associated with Janus
kinase and involved in signaling for cell growth and c-myc
induction. Immunity. 6:449–457. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Grillot DAM, Gonzalez-Garcia M, Ekhterae
D, et al: Genomic organization, promoter region analysis, and
chromosome localization of the mouse bcl-x gene. J Immunol.
158:4750–4757. 1997.PubMed/NCBI
|
|
12
|
Kurooka H, Kuroda K and Honjo T: Roles of
the ankyrin repeats and C-terminal region of the mouse notch1
intracellular region. Nucleic Acids Res. 26:5448–5455. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hill CS, Wynne J and Treisman R: The Rho
family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional
activation by SRF. Cell. 81:1159–1170. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Walter SA, Cutler RE Jr, Martinez R,
Gishizky M and Hill RJ: Stk10, a new member of the polo-like kinase
kinase family highly expressed in hematopoietic tissue. J Biol
Chem. 278:18221–18228. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jost PJ and Ruland J: Aberrant NF-kappaB
signaling in lymphoma: mechanisms, consequences, and therapeutic
implications. Blood. 109:2700–2707. 2007.PubMed/NCBI
|
|
16
|
Choi YL, Soda M, Ueno T, et al: Oncogenic
MAP2K1 mutations in human epithelial tumors. Carcinogenesis.
33:956–961. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dan I, Watanabe NM and Kusumi A: The Ste20
group kinases as regulators of MAP kinase cascades. Trends Cell
Biol. 11:220–230. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kuramochi S, Moriguchi T, Kuida K, Endo J,
Semba K, Nishida E and Karasuyama H: LOK is a novel mouse
STE20-like protein kinase that is expressed predominantly in
lymphocytes. J Biol Chem. 272:22679–22684. 1997. View Article : Google Scholar : PubMed/NCBI
|