Introduction
Genomic imprinting refers to the epigenetic
modification of a genome without the alteration in the DNA
sequence, and imprinted genes are expressed in a parent-of-origin
dependent manner. Insulin-like growth factor 2 gene (IGF2) is a
paternally expressed imprinted gene, but IGF2 imprinting is lost in
a host of human neoplasm, leading to increased IGF2 expression
(1,2). IGF2 imprinting is regulated by either
an imprinting control region (ICR) or the differentially methylated
domain (DMD) of the IGF2/H19 imprinting domain located on
chromosome 7 (3,4). It acts by regulating interactions
between the H19 and IGF2 promoters and their shared enhancers,
which lie downstream of the H19. Deletion of the ICR/DMD results in
loss of imprinting (LOI) of the IGF2 gene (5). Proper imprinting of IGF2 requires that
the ICR/DMD is methylated on the paternal allele and unmethylated
on the maternal allele. The ICR located between the IGF2 and H19
contains four CTCF binding sites that are differentially methylated
according to their parental origin (6,7). When
unmethylated, such regions are bound by the insulator protein
CCCTC-binding factor (CTCF) (6–8). As an
insulator of transcription, CTCF is a suitable candidate as one of
the putative imprinting factors. When CTCF levels are diminished by
RNA interference (RNAi) in mouse fibroblasts, IGF2 imprinting is
partially lost (9). CTCF binding to
the DMD is critical in the establishment of IGF2 imprinting in the
mouse. Deletion of the locus containing the DMD led to biallelic
expression of IGF2 (5,10). Mutation of each CTCF binding site in
the DMD also altered IGF2 imprinting (11). Using a transgenic RNAi-based
approach to generate oocytes with reduced CTCF expression, Fedoriw
et al found that CTCF protected the ICR from de novo
methylation during oocyte growth and was required for normal
preimplantation development (12).
A number of studies have observed loss of imprinting
(LOI) of IGF2 in both tumor tissues and tumor cell lines. For
example, the LOI of IGF2 was identified in 8 out of 14 different
tumor cell lines, possibly as the result of inactivation or
mutation of CTCF complex (13),
suggesting that the LOI of IGF2 is associated with the loss of
activity of CTCF due to its inactivation or mutation in tumor cells
(known as LOI of IGF2), and that an abnormal tumor epigenotype
could be corrected by in vitro reprogramming.
To obtain sufficient antitumoral effect, it is
critical to deliver therapeutic genes efficiently into target
cancer cells (14,15). Adenovirus (Ad) vectors can infect a
broad range of human cells with high efficiency and achieve high
levels of transgene expression. Moreover, the Ad viral genome is
genetically stable and the inserted foreign genes are generally
maintained without change through successive rounds of viral
replication (16). These features
make Ad vectors attractive in gene therapy. As a therapeutic gene,
we chose the Ad5 early region gene 1A (E1A), whose products are the
first adenoviral proteins produced upon infection and function
primarily as the activation of transcription of the other viral
early gene products (17). The E1A
proteins affect host cell growth that is thought to allow more
efficient production of viral progeny. In addition, E1A may induce
apoptosis (18), and E1A may
enhance sensitivity to chemotherapeutics and radiation. Of note,
normal cells appear to be unaffected by the E1A protein (19).
The present study constructed an adenoviral vector
for tumor cell targeted therapy based on the principle of LOI of
IGF2 in tumor cells. The therapeutic potential of a vector carrying
the E1A gene driven by enhancer-DMD-H19 promoter complex was tested
both in HRT-18 and HT-29 human ileocolon cells (LOI of IGF2),
HCT-116 human ileocolon cells (MOI of IGF2) and MCF-7 human breast
cancer cells (MOI of IGF2) and GES-1 human gastric epithelial cells
(MOI of IGF2). The constructed vector carrying the E1A gene was
found to infect and replicate selectively with high efficiency, and
had an effective antitumor activity in HRT-18 and HT-29 human colon
cells as well as xenografts in nude BALB/c mice in vivo.
Materials and methods
Cell lines and cell culture
Human colon cancer cells (HRT-18 and HT-29) with
loss of imprinting of IGF2 gene (LOI) were obtained from the
American Type Culture Collection (ATCC, Manassas, VA, USA). The
human colon cancer cells (HCT-116), breast cancer cells (MCF-7) and
human gastric epithelial cells (GES-1) which carried maintenance of
imprinting of IGF2 gene (MOI each) were also obtained from the
ATCC. All cells were maintained in DMEM (HyClone, Logan, UT, USA)
supplemented with 10% fetal bovine serum (FBS; Hyclone) and 100
mg/ml penicillin, and 50 mg/ml streptomycin, at 37ºC under
humidified conditions of 95% air and 5% CO2.
Plasmid construction and virus
packaging
The original adenoviral shuttle plasmid used in this
study was pDC312. The mouse H19 enhancer exon 1 (258 bp) and the
mouse H19 enhancer exon2 (360 bp) were amplified by PCR from mouse
genomic DNA, and the mouse DMD exon 1–2 (429 bp), DMD exon 3 (207
bp) and DMD exon 4 (156 bp) were also amplified by PCR from mouse
genomic DNA, and then they were linked to a single fragment by PCR.
Subsequently, the enhancer-DMD (1376 bp) was cloned into the
plasmid pDC312 using restriction endonuclease XbaI and
SalI. The mouse H19 promoter (302 bp) was amplified by PCR
from mouse genomic DNA using the upper primer 5′-GCAAGCTT
CCACCGTTCTATGAAGGGCTTC-3′ (5′-primer for mouse H19 with
SalI) and lower primer 5′-AAGAATTCTCATCAG CGCCCATCTCTAGCC-3′
(5′ primer for mouse H19 with HindIII), cloned into the
downstream of the enhancer-DMD using restriction endonuclease
SalI and HindIII. The E1A segment (1013 bp) was
amplified by PCR from the pDC312 using the upper primer
5′-CCCAAGCTTGGGCCCTATG AGACATATTATCT-3′ (5′ primer for E1A with
EcoRI) and lower primer 5′-CGCGGATCCCGCAATCACAGGTTTAC
ACCTTA-3′ (5′ primer for E1A with BamHI). The enhanced green
fluorescent protein (EGFP) reporter gene from pEGFP-C1 vector
(Clontech, Mountain View, CA, USA) and the E1A gene were inserted,
respectively, into the downstream of the H19 promoter using
restriction endonuclease EcoRI and BamHI to construct
pDC312-enhancer-DMD-H19-EGFP and pDC312-enhancer-DMD-H19-E1A. The
product was confirmed by DNA sequencing. The plasmids carrying the
target gene were transfected into HEK293 cells using liposome
Lipofectamine™ 2000 (Invitrogen-Life Technologies, Carlsbad, CA,
USA) together with the adenoviral vector Ad5. The culture solution
was changed after 6 h, and the cytopathic effect (CPE) was observed
continuously. CPE was found after 7–10 days. When the majority of
the pathologically abnormal cells had come off the bottom of the
culture flask, the abnormal cells and supernatant were collected,
frozen and thawed at −80ºC/37ºC three times and centrifuged; the
supernatant was then collected and, finally, two sets of
adenoviruses were obtained: Ad-EGFP and Ad-E1A.
Analysis of EGFP expression in the
constructed plasmids
All cells were infected with Ad-EGFP (10 plaque
forming units (PFU)/cell), respectively. EGFP expression was
examined at 24, 48 and 72 h after infection using an Olympus
microscope (Axioskop; Carl Zeiss Inc., Oberkochen, Germany) with a
fluorescent filter set (excitation 450–490 nm).
Analysis of the expression of hexon and
E1A mRNA in virus-infected cells by real-time quantitative PCR
The hexon and E1A mRNA expression was determined by
real-time quantitative PCR (RT-qPCR). All cells were infected with
Ad-E1A (10 PFU/cell), respectively. Total RNA was extracted from
the three cell lines with TRIzol (Invitrogen-Life Technologies)
according to the manufacturer’s instructions. Following treatment
with DNase I (Takara Biotechnology) at 37ºC for 30 min, RNA
quantification was performed using spectrophotometry. The RNA (1
μg) was subsequently incubated with 1 μl of Oligo dT primer (50
μM), 1 μl of Random 6 mers (100 μM), 1 μl of PrimeScript™ RT Enzyme
MixI, 4 μl of 5X PrimeScript™ Buffer and RNase Free
dH2O, and first-strand cDNA synthesis was performed in a
total volume of 20 μl. The primers used for hexon, E1A and β-actin
are shown in Table I. The PCR
reactions were performed in a LightCycler apparatus using real-time
PCR Master Mix SYBR-Green I (Yotobo Biotech Co., Ltd., Osaka,
Japan). Thermocycling was carried out in a final volume of 25 μl
containing 1 μl of cDNA sample, 0.5 μl of the up-primer, 0.5 μl
down-primer, 12.5 μl of SYBR-Green Real-time PCR Master Mix and
10.5 μl of dH2O. After 15 sec at 95ºC to denature the
cDNA and to activate Taq DNA polymerase, the cycling conditions
were: 40 cycles consisting of denaturation at 95ºC for 5 sec,
annealing at 60ºC for 5 sec and extension at 72ºC for 30 sec. The
Ct used in the real-time PCR quantification was defined as the PCR
cycle number that crossed an arbitrarily chosen signal threshold in
the log phase of the amplification curve. To verify the fold-change
of E1A gene expression, calculated Ct values were normalized to Ct
values of β-actin amplified from the same sample [ΔCt = Ct (E1A) −
Ct (β-actin)], and the 2−ΔΔCt method was used to
calculate fold-change. Each sample had triplicates and all
reactions were triplicated independently to ensure the
reproducibility of the results.
 | Table IThe primers of hexon, E1A and β-actin
for real-time PCR. |
Table I
The primers of hexon, E1A and β-actin
for real-time PCR.
| Target | Primer |
|---|
| hexon | Sense:
5′-ATGATGCCGCAGTGGTCTTA-3′
Antisense: 5′-GTCAAAGTACGTGGAAGCCAT-3′ |
| E1A | Sense:
5′-CCCAAGCTTGGGCCCTATGAGACATATTATCT-3′
Antisense: 5′-CGCGGATCCCGCAATCACAGGTTTACACCTTA-3′ |
| β-actin | Sense:
5′-CTGGAACGGTGAAGGTGACA-3′
Antisense: 5′-AAGGGACTTCCTGTAACAACGCA-3′ |
Analysis of E1A protein expression by
western blot analysis
The E1A protein expression was evaluated by western
blot analysis. Cells were harvested and lysed by three cycles of
freeze/thaw at −80ºC. Total protein was separated by SDS-PAGE in
12% gels and transferred to a polyvinylidene difluoride (PVDF)
membrane. The immunoblotting was performed as described elsewhere
(20), and 5% skimmed milk powder
(soluble in TBST buffer solution) was used at room temperature
under sealed conditions for 1 h, with mouse anti-E1A primary
antibodies (1:500) and rabbit anti-human β-actin primary antibodies
(1:500) incubated at room temperature for 2 h, and then washed by
vibrating with TBST solution. The proteins were visualized by
ECL.
Analysis of the cytotoxic effect of the
virus by MTT and flow cytometry
The assay was based on the ability of viable cells
to reduce MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl
tetrazolium bromide; Sigma Aldrich, St. Louis, MO, USA] to
insoluble colored formazan crystals. The cells were infected with
adenoviruses increased multiplicity of infection (MOI) from 0 to
100 PFU/cell, and fresh medium was added at 5 h after infection and
every 2 days thereafter. After 48, 72 and 96 h, MTT (5 mg/ml) was
added to each well. After 4 h of incubation at 37ºC, the medium was
removed, and 200 μl of DMSO (Sigma) were added into each well to
dissolve the crystals. The absorbance was measured in a microplate
reader at 490 nm (Bio-Rad Laboratories, Richmond, CA, USA).
Quantitative evaluation of apoptosis was performed by flow
cytometry after double staining with Annexin V fluorescein
isothiocyanate (FITC) apoptosis detection kit, which allowed
discrimination among early apoptotic (single Annexin V positive),
and necrotic cells [double Annexin V/propidium iodide (PI)
positive], which could differentiate cells that had lost membrane
integrity (necrotic cells) from living cells by means of red
staining of their nuclei with PI. Cells (1×106
cells/well) were cultured in 6-well dishes, and then Ad-E1A were
infected at 10 PFU/cell. Cell apoptosis was analyzed at 72 h after
infection. Infection with Ad-EGFP (10 PFU/cell) served as a
negative control.
Treatment of tumor-bearing nude mice with
the adenoviral vectors
Cells were trypsinized to a single cell suspension
and resuspended in 109 cells/100 μl PBS, then
subcutaneously injected into the flank area of adult (8-week-old)
athymic male nude mice (Harlan-Sprague-Dawley, Indianapolis, IN,
USA). Two weeks after injection of HRT-8 or HT-29 cells, the
developed tumors were measured in two dimensions. Then, Ad-E1A was
injected into the left side and Ad-EGFP, serving as a viral vector
control, was injected into the right side. Each adenoviral vector
(a total dosage of 109 PFU/mouse) was injected into a
growing tumor from three directions on 3 successive days, and the
tumor volume was observed for 28 days. Tumor dimensions were
measured, and the tumor volume was calculated according to the
formula (width)2 × length × 0.5.
TUNEL assay
Apoptosis in tumor cells was detected by terminal
deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end labeling
(TUNEL) assay. It was performed using In Situ Cell Death
Detection kit (Roche, Mannheim, Germany) as per the manufacturer’s
protocol. Sections were fixed in 30% formalin and rinsed. The
sections were then incubated in 3% H2O2,
permeabilized with 0.5% Triton X-100, rinsed again and incubated in
the TUNEL reaction mixture. The sections were rinsed and visualized
using Converter-POD with diaminobenzidine (DAB). Harris hematoxylin
was used for counterstaining. The slides were air-dried at room
temperature and cover slips were mounted using Permount. The number
of TUNEL-positive cells was counted in six fields (at ×400
magnification of light microscope) selected at random, and the
apoptosis index for each field was calculated as the percent of
TUNEL-positive cells relative to the total.
Statistical analysis
Experimental data are presented as the means ± SD
(standard deviation) and assessed by the Student’s t-test and
one-way ANOVA at a significance level of P<0.05.
Results
IGF2 imprinting
We measured the expression of IGF2 in selected human
cell lines by RT-PCR, and three polymorphic restriction enzymes
(ApaI, AluI and HhaI) in the last exon for
IGF2 were used to analyze allele-specific expression. As shown in
Table II, human colon cancer cells
(HRT-18 and HT-29) were with loss of imprinting of IGF2 gene (LOI),
and the human colon cancer cells (HCT-116), breast cancer cells
(MCF-7) and human gastric epithelial cells (GES-1) were maintenance
of imprinting of IGF2 gene (MOI each).
| Table IIGenomic imprinting of IGF2 in
selected human cell lines. |
Table II
Genomic imprinting of IGF2 in
selected human cell lines.
| Cell lines | Tumor type | PREs | IGF2
imprinting |
|---|
| HRT-18 | Colon cancer | HhaI | LOI |
| HT-29 | Colon cancer | ApaI | LOI |
| HCT-116 | Colon cancer | ApaI | MOI |
| MCF-7 | Breast cancer | AluI | MOI |
| GES-1 | Human gastric
epithelial cells | ApaI | MOI |
Verification of the recombinant plasmid
and packaging of adenovirus
The recombinant plasmid pDC-312-enhance-DMD was
digested by restriction endonuclease XbaI and SalI to
verify that the recombinant plasmid contained the enhancer-DMD
fragment (1376 bp). The recombinant plasmid
pDC-312-enhancer-DMD-H19 was digested by restriction endonuclease
SalI and HindIII to verify that the recombinant
plasmid contained the H19 fragment (302 bp). The recombinant
plasmid pDC-312-enhancer-DMD-H19-E1A and
pDC-312-enhancer-DMD-H19-EGFP were digested by restriction
endonuclease HindIII and BamHI, to verify that the
recombinant plasmid contained the E1A fragment (1013 bp) and EGFP
fragment (718 bp), respectively. We confirmed that the recombinant
plasmid had been constructed successfully. The plasmids
pDC312-enhancer-DMD-H19-EGFP and pDC312-enhancer-DMD-H19-E1A were
then transfected into HEK293 cells, respectively. After 8 days, the
cells showed significant CPE phenomenon, and we obtained two sets
of adenoviruses, i.e., Ad-EGFP and Ad-E1A.
Analysis of selective expression of
adenoviruses in the different tumor cells
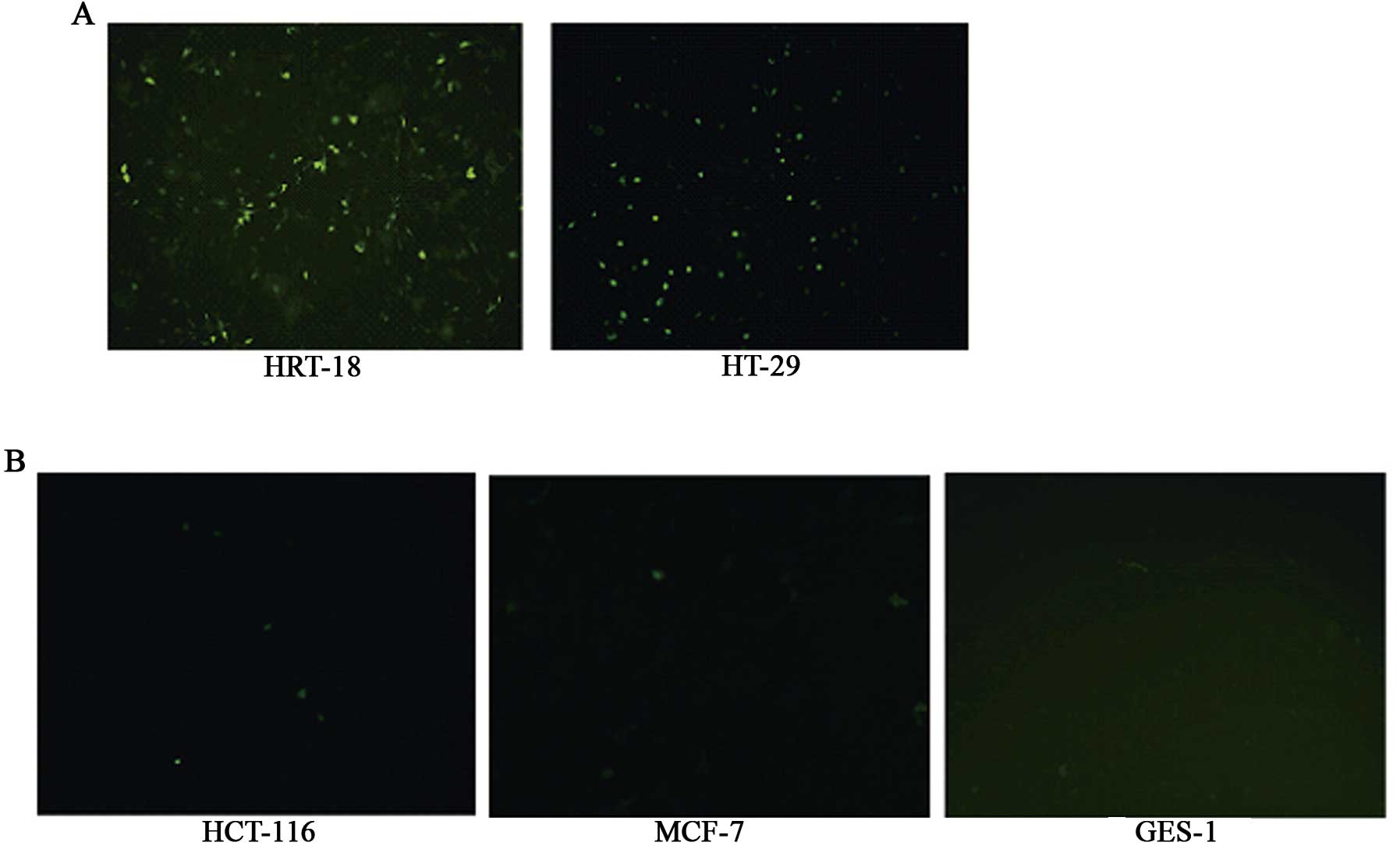
At 24 h after infection with Ad-EGFP (10 PFU/cell),
the expression of EGFP protein was seen to be positive in the
HRT-18 and HT-29 tumor cell lines, but negative in the other three
types of cells (HCT-116, MCF-7 and GES-1). After 48 h, weak
expression could be observed in the other three groups of cells
(HCT-116, MCF-7 and GES-1), and the increase in the amount of the
adenovirus or the infection time did not change the expression
(Fig. 1).
Levels and duration of the hexon gene
expression in the different tumor cells infected with recombinant
adenoviral vectors
The expression of hexon mRNA was determined by
RT-qPCR at various time-points after infection. The present study
demonstrated that hexon expression levels in LOI cell lines (HRT-18
and HT-29) after infecting with Ad-E1A at 6, 12, 24, 48 and 72 h
were significantly higher compared to hexon mRNA levels obtained
with Ad-E1A in MOI cell lines (HCT-116, MCF-7 and GES-1)
(P<0.05; P<0.01) (Fig.
2).
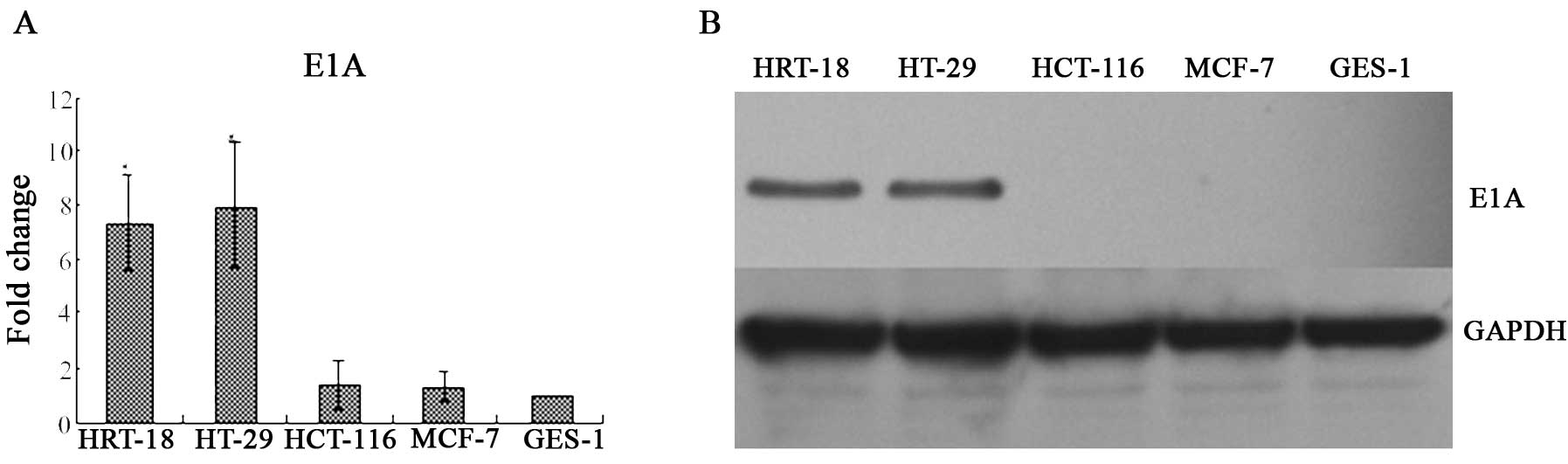
E1A mRNA transcript and protein
expression
The expression of E1A mRNA was determined by RT-qPCR
at 24 h after infection, and the E1A protein expression was
determined by western blot analysis at 48 h after infection. The
present study demonstrated that the expression of E1A mRNA in the
HRT-18 cells was 7.36-fold increased compared with that in the
GES-1 group (P<0.01), and the expression of E1A mRNA in the
HT-29 group was 7.94-fold increased compared with that in the GES-1
group (P<0.01). The expression of E1A mRNA in the HCT-116 group
was 1.34-fold increased compared with that in the GES-1 group
(P>0.05), and the expression of E1A mRNA in MCF-7 was 1.25-fold
increased compared with that in the GES-1 group (P>0.05)
(Fig. 3A). The E1A protein was
positive in the HRT-18 and HT-29 tumor cell lines, and negative in
HCT-116, GES-1 and MCF-7 cells (Fig.
3B).
Cytotoxic effect and apoptosis induced by
the adenoviruses
Cell viability was assessed by MTT assay at 48, 72
and 96 h after infecting by recombinant adenoviral vectors
increased MOI from 0 to 100 PFU/cell. Infecting with Ad-EGFP served
as negative control to evaluate the cytopathic effect of adenoviral
infection itself. The cytotoxicity in HCT-116, MCF-7 and GES-1
cells infected with Ad-E1A (0–10 PFU/cell) 48, 72 and 96 h showed
no significant increase compared with the control group
(P>0.05), but minimal cytotoxicity was observed when infected
with Ad-E1A (50–100 PFU/cell) after 72 and 96 h (P>0.05). The
cell viability in HRT-18 and HT-29 cells infected with Ad-E1A (100
PFU/cell) 48, 72 and 96 h showed a significant decrease compared
with the control group (P<0.05), and when infecting with Ad-E1A
(10–50 PFU/cell) 72 and 96 h also resulted in evident growth
inhibition in the LOI cells (HRT-18 and HT-29) compared with the
control group (P<0.05) (Fig.
4A–C). To assess the influence of the E1A on cell apoptosis, we
analyzed the apoptosis in the three types of cells by flow
cytometry at 72 h after infection. Infection with Ad-EGFP served as
negative control to evaluate the cytopathic effect of adenoviral
infection itself. The apoptosis rate in the HRT-18 and HT-29 cells
(~20%) infected with Ad-E1A (10 PFU/cell) was higher than that in
the control group (~6%) (P<0.01), but the percentage of
apoptosis in HCT-116, MCF-7 and GES-1 cells infected with Ad-E1A
(10 PFU/cell) showed no significant increase compared with the
control group (P>0.05) (Fig.
4D).
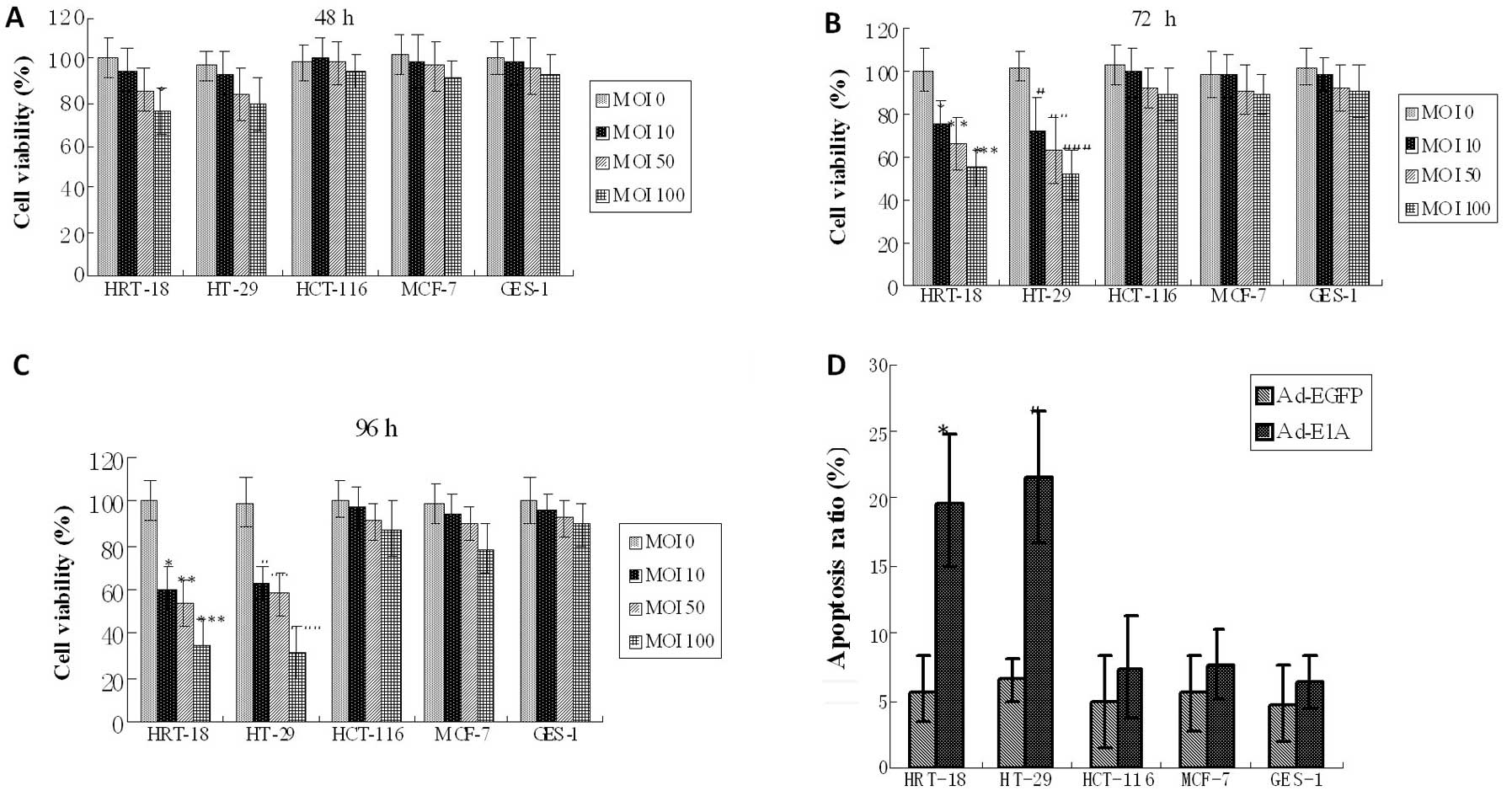 | Figure 4In vitro cytotoxic effect of
adenoviral vectors carrying the E1A gene. (A–C) Cell viability was
determined by MTT assay 48, 72 and 96 h after infecting with
increased MOI from 0 to 100 PFU/cell in five cell lines. The cell
viability in HCT-116, MCF-7 and GES-1 cells infected with Ad-E1A
(0–10 PFU/cell) showed no significant increase compared with the
control group (P>0.05), but minimal cytotoxicity was seen when
infected with Ad-E1A (50–100 PFU/cell) at 48, 72 and 96 h
(P>0.05). The cell viability in LOI cells (HRT-18 and HT-29)
infected with Ad-E1A (100 PFU/cell) showed a significant decrease
compared with the control group at 48 h (*P<0.05;
#P<0.05). The cell viability in LOI cells infected
with Ad-E1A (10–100 PFU/cell) showed a significant decrease
compared with the control group at 72 h (*P<0.05,
**P<0.05, ***P<0.01;
#P<0.05, ##P<0.05,
###P<0.01). The cell viability in LOI cells infected
with Ad-E1A (10–100 PFU/cell) showed a significant decrease
compared with the control group at 96 h (*P<0.01,
**P<0.01, ***P<0.01;
#P<0.01, ##P<0.01,
###P<0.01). Graphs are representative of 3 separate
experiments. (D) The apoptosis of cells was investigated using flow
cytometry analysis 72 h after infecting with Ad-E1A or Ad-EGFP (10
PFU/cell). The apoptotic ratio in MOI cell lines (HCT-116, MCF-7
and GES-1) infected with Ad-E1A showed no significant increase
compared with the control group (P>0.05), but the apoptotic
ratio in LOI cell lines (HRT-18 and HT-29) infected with Ad-E1A
showed a significant increase compared with the control group
(*P<0.01, #P<0.01). Graphs are
representative of 3 separate experiments. |
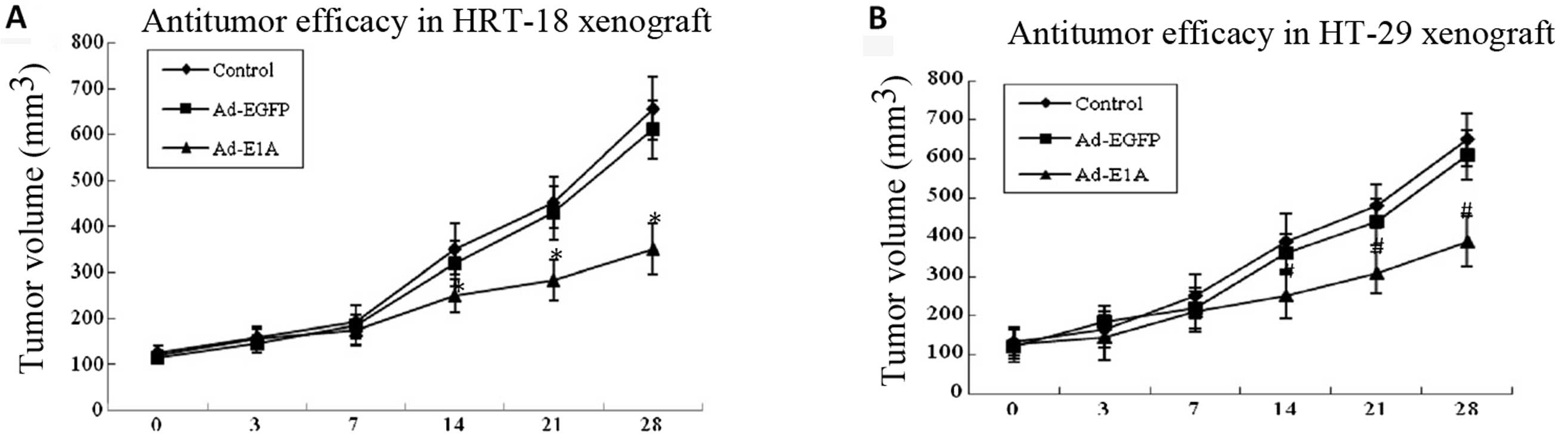
In vivo tumor growth inhibition by
adenoviruses
We further confirmed the antitumor effect by
injecting the indicated adenoviruses (a total dosage of
109 PFU/mouse) into nude mice carrying HRT-18 and HT-29
tumor xenografts, respectively. The tumor growth was found to be
similar and steady for all groups at the beginning, but after ~14
days the suppression of tumor growth was found to be strong and
significantly superior in the Ad-E1A-treated groups, with the tumor
inhibition rate of 30% in HRT-18 tumor xenografts compared to the
buffer-control group (P<0.01) and 35% in HT-29 tumor xenografts
compared to the buffer-control group (P<0.01). By comparison, no
significant antitumor effect was observed in the Ad-EGFP group
compared to the buffer-control group in HRT-18 and HT-29 tumor
xenografts. (P>0.05) (Fig.
5).
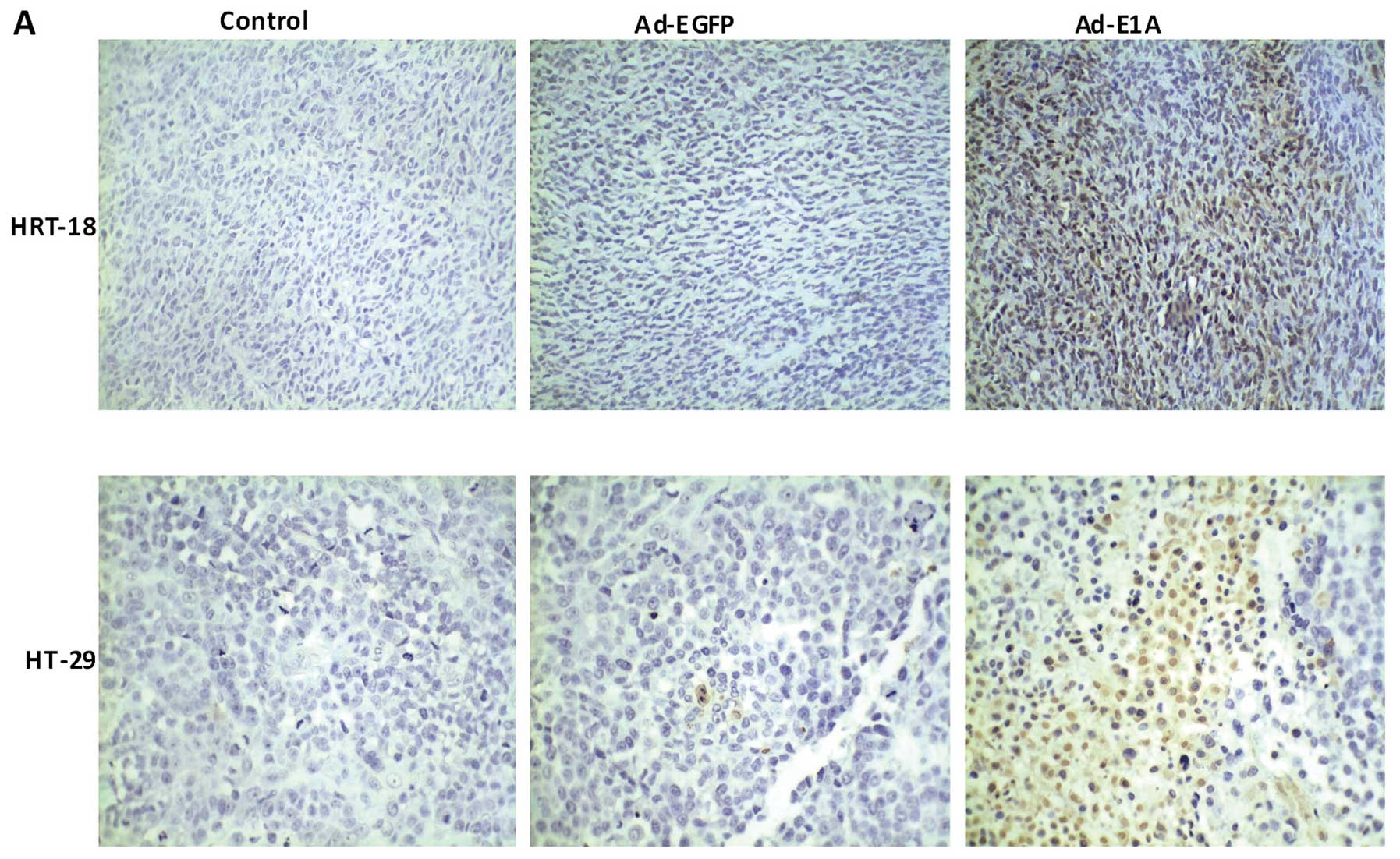
Immunohistology by TUNEL assay
In order to further elucidate the mechanisms of the
suppression of growth by Ad-E1A, the TUNEL assay was applied. Tumor
apoptosis in HRT-18 and HT-29 tumor treatment nude mouse model was
confirmed after treatment with Ad-E1A or Ad-EGFP or PBS by TUNEL
assay. Nuclei of TUNEL-positive cells were stained brown and their
shapes were condensed or segmented, whereas those of TUNEL-negative
cells were stained blue by hematoxylin. Fig. 6A indicates a marked increase in
apoptotic bodies in the Ad-E1A-treated mice tumor compared to
Ad-EGFP-treated or PBS-treated groups. Apoptosis index was measured
by the percentage of TUNEL-positive cells vs. total number of
cells. The apoptotic indexes in HRT-18 of PBS, Ad-EGFP and Ad-E1A
were 2±1, 4±2 and 34±9, respectively. The apoptotic indexes in
HT-29 of PBS, Ad-EGFP and Ad-E1A were 3±2, 5±4 and 45±12,
respectively. As mentioned above, the apoptotic index of HRT-18 and
HT-29 tumors from mice treated with Ad-E1A was significantly higher
than that from other groups (P<0.01) (Fig. 6B).
Discussion
Genomic imprinting is essential for the normal
growth and development of organisms (21). IGF2 is a paternally expressed
imprinted gene, but IGF2 imprinting is lost in a host of human
neoplasm, resulting in biallelic IGF2 expression and abnormally
high IGF2 production. Earlier studies have shown that LOI of IGF2
is associated with somatic overgrowth and embryonal tumors and has
been linked to more than 20 types of cancer in humans, including
Wilms’ tumor and colorectal, lung, breast and prostate cancer
(22). The current model of
imprinting regulation of IGF2 is that binding of CTCF to the
unmethylated maternal ICR/DMD protects it from de novo
methylation and prevents downstream enhancers from activating IGF2,
and CTCF is unable to bind the methylated paternal ICR/DMD that
results in the expression of IGF2. Although it is now well
established that the ICR/DMD acts as a CTCF-dependent
insulator/enhancer blocker, the mechanism of insulation remains
incomplete. Li et al(23)
confirmed that the inactivation or mutation of the CTCF complex was
closely related to the LOI of IGF2 in tumor cells by chromatin
immunoprecipitation (CHIP) and chromosome conformation capture (3C)
technique (23). Hu et al
demonstrated that IGF2 aberrant epigenotype can be corrected by
transferring nuclei from human tumor cells (LOI cells) into
enucleated mouse and human fibroblasts (MOI cells) (13,24).
The results demonstrate that an abnormal tumor epigenotype can be
corrected by in vitro reprogramming, and suggest that LOI is
associated with the inactivation or mutation of the CTCF complex;
therefore, we designed our experiment based on the following
mechanisms that in the MOI cells, the active CTCF can bind to the
DMD, blocking the activity of enhancer and inhibiting the
expression of the downstream genes, but that the inactive CTCF in
the LOI cells cannot bind to the DMD, leading to increased
expression of downstream genes as a result of the effect of the
promoter activated by the enhancers. This is due to the fact that
the enhancers physically interact with the IGF2 promoters on the
paternal chromosome. However, interactions on the maternal
chromosome remain unclear. Kurukuti et al(25) found maternal-specific silencing of
IGF2 when the ICR/DMD interacts with a matrix attachment region and
a differentially methylated region at the IGF2 locus to generate a
tight loop around the IGF2 gene, thereby physically impeding IGF2
expression. By contrast, Yoon et al demonstrated that the
ICR/DMD forms a transcriptionally unproductive association with
enhancers and the inactive IGF2 promoters on the maternal
chromosome, which leads to silencing of IGF2 (26).
In the present study, we examined the effects of
enhancer-DMD-H19 promoter-driven E1A expression on the growth of
tumor cells based on loss of imprinting of IGF2 gene. We
constructed the expression vector to express E1A protein only seen
in the cells that were loss of imprinting of IGF2 (inactive CTCF).
Earlier studies from our group and others showed that normal cells
were maintenance of IGF2 imprinting (active CTCF), we speculated
that there was no cytotoxicity of E1A in normal cells. Firstly, the
use of our expression system was identified by EGFP reporter
assays, and data demonstrated that green fluorescence was positive
in HRT-18 and HT-29 tumor cell lines that was loss of imprinting of
IGF2, but only weak expression observed in HCT-116 cells, MCF-7
cells and GES-1 cells that were maintenance of imprinting of IGF2
regardless of the multiplicity of infection or the prolonged
infection time. In the present study, expression of hexon genes was
observed in the five infected cell lines at various time-points,
suggesting that the recombinant adenovirus could infect LOI tumor
cells effectively and gradually declined with time.
The E1A has been found to have an anticancer effect,
and it is a tumor-suppressing gene commonly used in gene therapy.
In the present study, the five types of the cells of different gene
imprinting systems were infected with Ad-E1A and the cell viability
was assessed by MTT. The results showed that there was no
detectable cytotoxicity in HCT-116, MCF-7 and GES-1 cells when
infected with Ad-E1A, and minimal cytotoxicity was seen when
co-infected with Ad-E1A (50–100 PFU/cell) at 72 and 96 h. The
results showed that E1A was very weak in inducing apoptosis in MOI
of IGF2 cell lines (HCT-116, MCF-7 and GES-1), whereas it
significantly induced apoptosis in LOI of IGF2 cell lines (HRT-18
and HT-29). Thus, we could observe imprinting-dependent induction
of apoptosis in tumor cells infected with the E1A expression
vectors. We also used the HRT-18 and HT-29 cell lines to establish
a xenograft mouse model. The results showed a significant
inhibition of tumor growth in Ad-E1A-treated groups compared to the
control group. The apoptosis in HRT-18 animal model and HT-29
animal model were also confirmed using the immunohistochemical
staining. The results showed that Ad-E1A increased the apoptotic
index and suggested that Ad-E1A resulted in an increased apoptosis
of tumor cells, thereby leading to the tumor growth inhibition,
indicating that the E1A expression vector had a high therapeutic
potential and was a promising candidate for colon cancer therapy in
humans.
In conclusion, an IGF2 imprinting-based gene therapy
vector has been developed that is effective in inhibiting the
growth of human colon cancer cells in vitro and in
vivo. This study provides, to the best of our knowledge, the
first evidence of the use of this system for cancer gene therapy
in vitro and in vivo. Since our data focused only on
colon cancer therapy, further studies are required to explore the
use of these vectors to gene therapy for other types of cancer that
are loss of IGF2 imprinting, such as hepatoma, lung cancer, breast
cancer, leiomyosarcoma, osteosarcoma, leukemia and Wilms’
tumor.
Acknowledgements
This study was supported by grants from the National
Nature Science Foundation of China (no. 81172141), the Nanjing
Science and Technology Committee project (no. 201108025), the
Nanjing Medical Technology Development Project (no. ZKX11025) to
S.K.W, and Nanjing Medical Science and technique Development
Foundation to Y.Q.P (no. QRX11255) and B.S.H (no. QRX11255), and a
NIH grant (1R43 CA103553-01) and The Department of Defence Grant
(W81XWH-04-1-0597) to J.F.H, and the Research Service of the
Department of Veterans Affairs.
References
|
1
|
Rainier S, Johnson LA, Dobry CJ, Ping AJ,
Grundy PE and Feinberg AP: Relaxation of imprinted genes in human
cancer. Nature. 362:747–749. 1993. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Feinberg AP: Genomic imprinting and gene
activation in cancer. Nat Genet. 4:110–113. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Reik W, Constancia M, Dean W, Davies K,
Bowden L, Murrell A, Feil R, Walter J and Kelsey G: Igf2 imprinting
in development and disease. Int J Dev Biol. 4:145–150. 2000.
|
|
4
|
Arney KL: H19 and Igf2-enhancing the
confusion? Trends Genet. 19:17–23. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thorvaldsen JL, Duran KL and Bartolomei
MS: Deletion of the H19 differentially methylated domain results in
loss of imprinted expression of H19 and Igf2. Genes Dev.
12:3693–3702. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hark AT, Schoenherr CJ, Katz DJ, Ingram
RS, Levorse JM and Tilghman SM: CTCF mediates methylation-sensitive
enhancer-blocking activity at the H19/Igf2 locus. Nature.
405:486–489. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bell AC and Felsenfeld G: Methylation of a
CTCF-dependent boundary controls imprinted expression of the Igf2
gene. Nature. 405:482–485. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kanduri C, Pant V, Loukinov D, et al:
Functional association of CTCF with the insulator upstream of the
H19 gene is parent of origin-specific and methylation-sensitive.
Curr Biol. 10:853–856. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ling JQ, Li T, Hu JF, et al: CTCF mediates
interchromosomal colocalization between Igf2/H19 and Wsb1/Nf1.
Science. 312:269–272. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Leighton PA, Saam JR, Ingram RS, Stewart
CL and Tilghman SM: An enhancer deletion affects both H19 and Igf2
expression. Genes Dev. 9:2079–2089. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schoenherr CJ, Levorse JM and Tilghman SM:
CTCF maintains differential methylation at the Igf2/H19 locus. Nat
Genet. 33:66–69. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fedoriw AM, Stein P, Svoboda P, Schultz RM
and Bartolomei MS: Transgenic RNAi reveals essential function for
CTCF in H19 gene imprinting. Science. 303:238–240. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen HL, Li T, Qiu XW, et al: Correction
of aberrant imprinting of IGF2 in human tumors by nuclear
transfer-induced epigenetic reprogramming. EMBO J. 25:5329–5338.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vile RG, Russell SJ and Lemoine NR: Cancer
gene therapy: hard lessons and new courses. Gene Ther. 7:2–8. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gómez-Navarro J, Curiel DT and Douglas JT:
Gene therapy for cancer. Eur J Cancer. 35:2039–2057. 1999.
|
|
16
|
Robbins PD and Ghivizzani SC: Viral
vectors for gene therapy. Pharmacol Ther. 80:35–47. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Branton PE, Bayley ST and Graham FL:
Transformation by human adenoviruses. Biochim Biophys Acta.
780:67–94. 1985.PubMed/NCBI
|
|
18
|
Radke JR, Siddiqui ZK, Miura TA, Routes JM
and Cook JL: E1A oncogene enhancement of caspase-2-mediated
mitochondrial injury sensitizes cells to macrophage nitric
oxide-induced apoptosis. J Immunol. 180:8272–8279. 2008. View Article : Google Scholar
|
|
19
|
Sánchez-Prieto R, Quintanilla M, Cano A,
et al: Carcinoma cell lines become sensitive to DNA-damaging agents
by the expression of the adenovirus E1A gene. Oncogene.
13:1083–1092. 1996.PubMed/NCBI
|
|
20
|
Adler V, Yin Z, Fuchs SY, et al:
Regulation of JNK signaling by GSTp. EMBO J. 18:1321–1334. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Szabó PE and Mann JR: Biallelic expression
of imprinted genes in the mouse germ line: implications for
erasure, establishment, and mechanisms of genomic imprinting. Genes
Dev. 9:1857–1868. 1995.PubMed/NCBI
|
|
22
|
Kaneda A and Feinberg AP: Loss of
imprinting of IGF2: a common epigenetic modifier of intestinal
tumor risk. Cancer Res. 65:11236–11240. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li T, Hu JF, Qiu X, et al: CTCF regulates
allelic expression of Igf2 by orchestrating a promoter-polycomb
repressive complex 2 intrachromosomal loop. Mol Cell Biol.
28:6473–6482. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hu JF, Vu TH and Hoffman AR:
Promoter-specific modulation of insulin-like growth factor II
genomic imprinting by inhibitors of DNA methylation. J Biol Chem.
271:18253–18262. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kurukuti S, Tiwari VK, Tavoosidana G, et
al: CTCF binding at the H19 imprinting control region mediates
maternally inherited higher-order chromatin conformation to
restrict enhancer access to Igf2. Proc Natl Acad Sci USA.
103:10684–10689. 2006. View Article : Google Scholar
|
|
26
|
Yoon YS, Jeong S, Rong Q, Park KY, Chung
JH and Pfeifer K: Analysis of the H19ICR insulator. Mol Cell Biol.
27:3499–3510. 2007. View Article : Google Scholar : PubMed/NCBI
|