Introduction
Gastric cancer is a lethal malignant tumor which
accounts for 10% of all types of invasive cancer in human beings,
particularly in Asia (1); it is
also the second leading cause of cancer-related mortality (2,3). Due
to the aggressiveness of gastric cancer biology and the limitation
of current treatment with advanced disorders, it is critical to
elucidate the mechanisms of gastric cancer growth and to identify
novel targets for the urgently needed adjuvant therapies.
Obesity, a worldwide epidemic (4), is associated with an increased risk of
various malignant tumors such as colon cancer (5) and breast cancer (6). A marked decline in distal gastric
cancer was noted, and the incidence of proximal gastric cancer has
increased, which may be associated with obesity (7). A meta-analysis demonstrated that the
body mass index is closely related to the risk of gastric cancer,
particularly in cardia gastric cancer (8).
Adipokines and other growth factors secreted in the
context of obesity may enhance cancer cell survival and solid tumor
growth (9). The new adipokine,
nicotinamide phosphoribosyltransferase (Nampt) was originally
described as pre-B-cell colony-enhancing factor (PBEF) promoting
early B cell proliferation (10),
and attracted considerable interest after it was reported to
function as an insulin-mimetic adipokine termed visfatin secreted
by visceral fat (11). Nampt has
two different forms: intracellular Nampt (iNampt) and extracellular
Nampt (eNampt, also known as visfatin) (12). iNampt functioning as an enzyme
involved in the NAD+ salvage pathway was significantly
overexpressed in the genesis of numerous types of cancer, such as
colorectal (13), breast (14) and gastric cancer (15). The expression level of visfatin
increases with the increase of obesity. Visfatin contributes to a
general pro-inflammatory state in the periphery, and may well prove
to be an important mechanistic link in the network of factors
affecting obesity-associated tumor growth. Visfatin also acted as
an NAD biosynthetic enzyme similar to iNampt (16).
Silent mating-type information regulation 2
homologue 1 (Sirt1) functions as an NAD+ dependent
histone deacetylase (17) and plays
an important role in cell survival under genotoxic and oxidative
stress by deacetylating key cell cycle proteins and apoptosis
regulatory molecules, such as P53 (18,19).
Previous studies have demonstrated that Sirt1 is upregulated in
numerous types of human and mouse malignancies including human
colon cancer (20) and mouse
pulmonary adenocarcinoma (21),
which may be involved in carcinogenesis through its antiapoptotic
activity (22). Upregulated Sirt1
inactivates P53 by deacetylation (21), and this allows cells with damaged
DNA to proliferate and subsequently prompts tumor development
(23). c-MYC is the key molecule in
a transcription factor network that regulates cellular
proliferation, apoptosis and cell differentiation (24,25).
Deregulation of c-MYC activity has been implicated in the
carcinogenesis of the majority of cancer cases, and its inhibition
represents a possible alternative to current cancer therapies
(26). A recent study revealed the
existence of the positive feedback loop among the c-MYC
oncoprotein, the iNampt enzyme and the Sirt1 deacetylase (27). Thus, obesity may correlate with
gastric cancer growth via a nampt/sirt1/c-myc positive
feedback loop.
Currently, limited fundamental studies have been
performed regarding the mechanisms by which obesity affects gastric
cancer growth. We recently characterized a murine in vivo
gastric cancer model in the context of obesity, and found that
diet-induced obese mice with high serum visfatin, insulin and
glucose levels were insulin-resistant and glucose-intolerant.
Furthermore, subcutaneous injected tumors grew faster and larger in
obese mice than in lean mice. Tumor weights showed a significantly
positive correlation with mouse body weights. In addition, the
histological analysis demonstrated that adipocytes within tumors
from obese mice were significantly larger than those from lean mice
(28). Using this novel in
vivo mouse model, we sought to investigate the underlying
mechanism of gastric cancer growth under conditions of obesity.
Materials and methods
Cell culture
The cancer cell line used in this study, murine
forestomach carcinoma cell line (MFC), was established in the
Chinese Academy of Medical Sciences (29), and purchased from the Type Culture
Collection of the Chinese Academy of Sciences, Shanghai, China. MFC
was cultured in RPMI-1640 medium (Cellgro, Herndon, VA, USA)
containing 10% (v/v) heat-inactivated fetal bovine serum (Valley
Biomedical, Winchester, VA, USA), penicillin (100 U/ml) and
streptomycin (100 mg/ml). Cells were maintained in a 37°C
humidified incubator supplying 5% CO2.
Diet-induced obesity model
Three- to five-week-old male C57BL/6j mice (Slac
Laboratory Animal, Shanghai, China) were weaned on a high-fat diet
(35.5% fat, 36.3% carbohydrate, 20.0% protein) and normal chow
(5.4% fat, 51.0% carbohydrate, 22.9% protein) (30), respectively, for 12 weeks. Then,
24/24 mice on the normal fat diet were referred to as ‘lean’, 24/36
animals consuming the high fat diet and chosen by the criterion
that the body weight exceeded the mean plus 2-fold standard
deviation (SD) of the lean mice body weight were referred to as
‘obese’, and the other 12 mice on the high fat diet were referred
to as ‘non-obese’.
In vivo gastric cancer model
To detect the impact of obesity on tumor growth, 12
obese, 12 lean and 12 non-obese mice were injected subcutaneously
with 2.0×106 MFC cells into the right flank and
monitored daily to check for the presence of palpable tumors.
Another 12 obese and 12 lean mice which had no difference with
relative injected mice in the body weight were injected
subcutaneously with 0.9% normal saline into the right flank as a
control group. Then, all mice were maintained on a normal or a high
fat diet for another 2 weeks, respectively. Once tumors became
palpable, tumor volume was calculated by measuring the length,
width of the tumor with calipers as 1/2 (length ×
width2).
All experiments were performed with the approval of
the Xi'an Jiaotong University Institutional Animal Care and Use
Committee following the principles of laboratory animal care (NIH
publication no. 85-23, revised 1985). All surgery was performed
under sodium pentobarbital anesthesia and all efforts were made to
minimize suffering.
Immunohistochemical staining
Tumors were collected and prepared for
formalin-fixed, paraffin embedded tumor sections. The tumor
sections were dewaxed and dehydrated. Rehydration, antigen
retrieval in citrate buffer, endogenous peroxidase activity was
blocked for 10 min using 3.0% hydrogen peroxide, the sections were
blocked with 10% goat plasma for 30 min, then separately incubated
with the primary antibodies directed against iNampt, Sirt1 and
c-MYC at 4°C overnight. The primary antibodies were detected using
biotinylated secondary antibodies (Zhongshan Goldenbridge
Biotechnology Co. Ltd., China) following the manufacturer's
recommendations. The staining of the sections was performed using
the HRP-streptavidin conjugates for iNampt, Sirt1 and c-MYC (SP
method).
The staining results for iNampt, Sirt1 and c-MYC
were semi-quantitatively expressed by an immunohistochemical score
combined with the percentage of MFC cells showing specific
immunoreactivity. Staining intensity was expressed as four grades:
0, none; 1, weak; 2, moderate; and 3, strong. The percentage of
positive MFC cells was expressed as: 0, <5%; 1, 6–25%; 2,
26–50%; 3, 51–75%; and 4, >75%. The staining intensity and
average percentage of positive MFC cells were assayed for 10
independent high power fields (x400). The total score was
calculated by multiplying the staining intensity and the percentage
of positive MFC cells. Sections with a total score of >1 were
defined as exhibiting positive staining for the above proteins. All
histological analyses were performed by three independent
observers. The primary antibodies recognizing iNampt, Sirt1 and
c-MYC and were purchased from Santa Cruz Biotechnology (Santa Cruz,
CA, USA).
Serum assays
Mice were fasted overnight and sacrificed following
anesthesia. Blood drawn from the retro-orbital venous plexus was
preserved for analysis of metabolites. Enzyme-linked immunosorbent
assay (ELISA) was used to determine serum concentration of insulin
(Crystal Chem, Inc., Downers Grove, IL, USA) and visfatin (Linco
Research, Inc., St. Charles, MO, USA) following the manufacturer's
protocol. Serum glucose was determined by colorimetric assay.
Cellular proliferation assay
Cellular proliferation was evaluated by the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. MTT and dimethyl sulfoxide (DMSO) were obtained from Sigma
Chemical. After exposure to RPMI-1640 or 5% sera of obese or lean
mice, MFC cells were incubated with 0.5% MTT solution at 37°C for
another 4 h. Subsequently, 150 μl of DMSO was added to solubilize
the MTT tetrazolium crystal. Finally, the optical density was
determined at 490 nm by a Benchmark Plus microplate reader
(Bio-Rad, Hercules, CA, USA). All experiments were repeated in
triplicate.
Cell migration assay
To detect the invasiveness of MFC cells in response
to exposure to sera from obese or lean mice, we measured the
ability of cell migration. Briefly, 5×104 MFC cells
suspended in serum-free RPMI-1640 were added to the top wells of
migration chambers. The lower chambers were filled with 5% sera
obtained from obese or lean mice, or serum-free RPMI-1640, and the
top chambers were placed in 24-well plates and incubated for 24 h
at 37°C. After removal of the cells from the upper surface of the
filters, the remaining cells on the lower surface were counted
randomly in 10 different low-power fields (original magnification,
×40) using a microscope.
Cellular apoptosis analysis
To determine phosphatidylserine externalization (on
the surface of cell membrane), an indicator of early apoptosis,
flow cytometry was performed with propidium iodide (PI) and
fluorescein isothiocyanate (FITC)-labeled Annexin V (Joincare
Biosciences, Zhuhai, China) (31).
After exposure to RPMI-1640 or 5% sera of obese or lean mice for 24
h, the remaining intact MFC cells were collected and 500 μl of 1X
binding buffer, 10 μl of PI staining solution and 5 μl of
FITC-labeled Annexin V were added to them, and mixed well
incubating at room temperature for 10 min in the dark. The cells
were then analyzed for cell apoptosis on a FACScan (BD Biosciences,
USA).
Cell cycle analysis
After exposure to RPMI-1640 or 5% sera of obese or
lean mice for 24 h, MFC cells were harvested and resuspended at
1–5×105 cells/ml with PBS. Aliquot 1 ml cells were added
into 3 ml cold (−20°C) absolute ethanol and fixed overnight at
−20°C, then washed with PBS. Following centrifugation at 1500 rpm
for 5 min, 1 ml PI and 50 μl RNase A stock solution were added to
the cell pellet and mixed gently. After incubation at 25°C for 30
min in the dark, the cells were analyzed for cell cycle on a
FACScan (BD Biosciences).
RNA expression studies
After exposure to RPMI-1640 or 5% sera of obese or
lean mice for 24 h, total RNA was extracted from the cultured cells
with the TRIzol reagent (Invitrogen, San Diego, CA, USA), then
reverse transcribed to cDNA with High Capacity 1st Strand Synthesis
kit (Takara Biochemicals, Japan). The cycling program was based on
the manufacturer's instructions. Expression of nampt, sirt1
and c-myc mRNA was quantified by RT-PCR (Takara
Biochemicals). Transcript levels were normalized to β-actin.
Briefly, the reaction was carried out using an Icycler (Bio-Rad)
with the following thermal profile: a pre-heating step at 95°C for
10 min followed by 30 repeats of 94.0°C for 30 sec and 55.0°C for
30 sec, then 72°C for 1 min. The primer sequences were generated
using NCBI primer-BLAST. Murine β-actin was amplified using forward
primer 5′-TTCTTTGCAGCTCCTTCGTTGCCG-3′ and reverse primer
5′-TGGATGGCTACGTACATGGCTGGG-3′ (NM 007393.3), which yielded a
product of 467 bp. Murine nampt was amplified using forward
primer 5′-TCGGTTCTGGTGGCGCTTTGCTAC-3′ and reverse primer
5′-AAGTTCCCCGCTGGTGTCCTATGT-3′ (NM 021524.2), which yielded a
product of 181 bp. Murine sirt1 was amplified using forward
primer 5′-TTGTGAAGCTGTTCGTGGAG-3′ and reverse primer
5′-GCGTGGAGGTTTTTCAGTA-3′ (NM 001159590.1), which yielded a product
of 412 bp. Murine c-myc was amplified using forward primer
5′-CAGCGACTCTGAAGAAGAGCAAG-3′ and reverse primer
5′-GGGTTTGCCTCTTCTCCACAG-3′ (NM 001177354.1), which yielded a
product of 71 bp. For validation, each experiment was performed in
triplicate.
Protein extraction and western blot
analysis
We examined the expression of iNampt, Sirt1 and
c-MYC in the cultured cells with exposure to RPMI-1640 or 5% sera
of obese mice or lean animals to detect whether obesity may be
responsible for the growth of MFC cells. After treatment for 24 h,
cells were lysed in RIPA buffer on ice. Protein concentration was
measured via the BCA method (Pierce), bromophenol blue and NuPage
Reducing Agent (Invitrogen) were added to the lysate, and the final
mixture was heated. Equal amounts of total protein were run on a
10% SDS-PAGE and were electrotransferred onto nitrocellulose
membranes (Millipore, Bedford, MA, USA) using a Bio-Rad Mini
PROTEAN 3 System according to the standard protocol. The
nitrocellulose membranes were then blocked in TBST with 5% non-fat
dry milk at 37°C for 2 h. Subsequently, the membranes were
incubated with a 1:200 dilution of the primary antibodies for
iNampt, Sirt1 and c-MYC, and a 1:2,000 dilution of anti-β-actin at
4°C overnight. An antibody against rabbit IgG was used as the
secondary antibody. The membranes were developed with enhanced
chemiluminescence (Pierce) by an enhanced chemiluminescence
detection system (Amersham Biosciences, Piscataway, NJ, USA). The
primary antibodies recognizing iNampt, Sirt1, c-MYC and β-actin
were purchased from Santa Cruz Biotechnology.
Statistical analysis
Values are expressed as the mean ± SD. Statistical
differences were estimated by one-way analysis of variance (ANOVA)
followed by Dunnett's test or the Spearman rank test. P-value
<0.05 was considered to indicate statistically significant
differences. Analysis of the data and plotting of the figures were
performed with the aid of software (SPSS version 13.0).
Results
Metabolic changes in obese mice and their
effects on tumor growth
Obese, non-obese and lean mice were selected and
used as recipients of MFC cells to determine the influence of
obesity on gastric cancer growth. At the time of transplantation,
the lean mice (body weight, 28.5±1.2 g, ~26.5–30.5 g) were
significantly lighter than the obese mice (35.4±2.8 g, ~32.0–40.5g)
(P<0.05), and had no difference with non-obese mice (28.7±2.3 g,
~26.0–31.0 g) (P>0.05).
No mouse died and no metastasis was detected during
the experimental time frame. The tumors became palpable 4 days
after injection and tumor growth was detected in 83.3% (10/12) of
the lean mice, in 75% (9/12) of the non-obese mice and in 100%
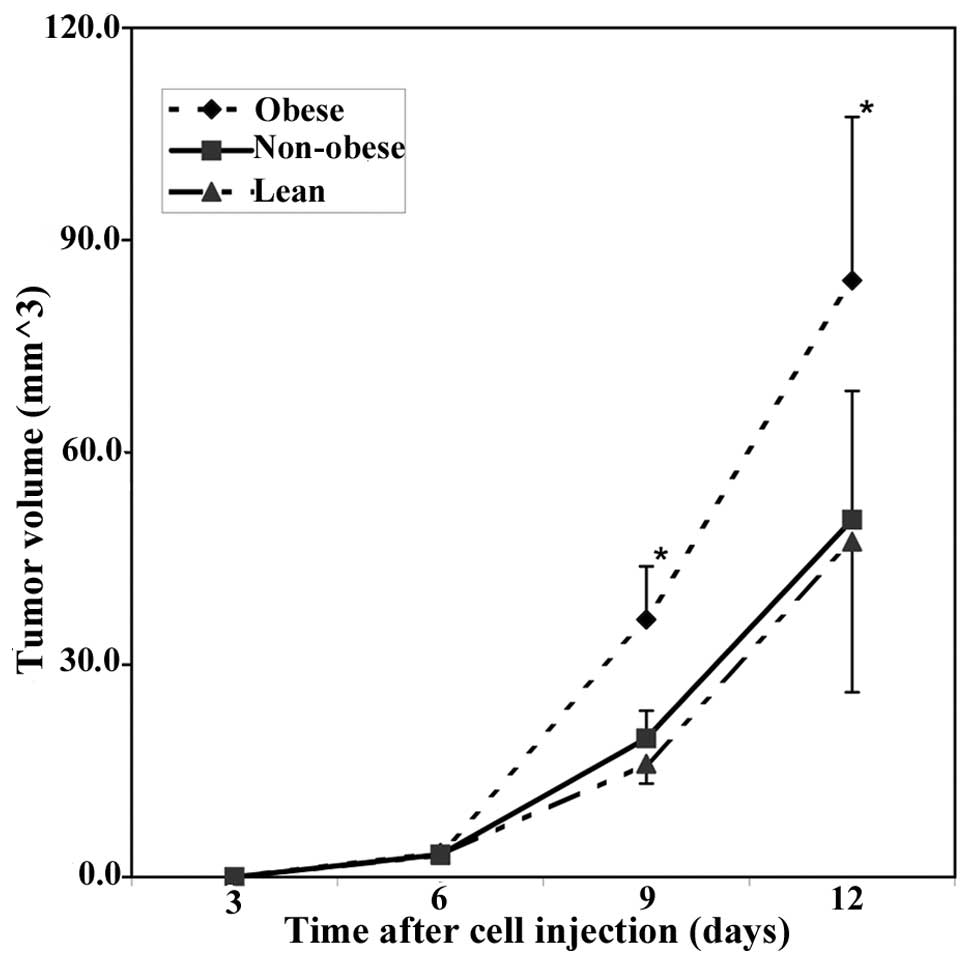
(12/12) of the obese animals. Tumors grew faster in the obese mice
than in the lean and non-obese animals within 2 weeks (Fig. 1). Then, all mice were sacrificed and
the tumors were collected. The tumor weights were: lean, 77.2±14.9
mg (P<0.01 vs. obese and P>0.05 vs. non-obese); non-obese,
83.4±15.3 mg (P<0.01 vs. obese); obese, 134.2±17.3 mg, and
showed a significantly positive correlation with the body weight of
the mice (r=0.75, P<0.05).
There was no difference between injected mice and
relative control group animals in the body weight, serum glucose,
insulin and visfatin concentrations. The obese mice with insulin
resistance and glucose intolerance (28) were hyperglycemic, hyperinsulinemic,
and had high serum visfatin concentration (Table I). The tumor weights correlated
significantly with serum visfatin concentration (r=0.47, P<0.05)
and insulin level (r=0.37, P<0.05), but did not correlate with
serum glucose concentration (r=0.22, P>0.05). This indicated
that obesity may promote murine gastric cancer growth by endocrine
mechanisms.
 | Table ISerum metabolic changes in mice. |
Table I
Serum metabolic changes in mice.
| Groups |
|---|
|
|
|---|
| Parameters | Obese | Non-obese | Lean |
|---|
| Visfatin
(ng/ml) | 44.3±3.6a | 38.9±2.7b | 39.6±3.4 |
| Insulin
(ng/ml) | 0.51±0.07a | 0.41±0.03b | 0.40±0.04 |
| Glucose
(mmol/l) | 11.9±1.6a | 8.1±2.0b | 7.8±1.6 |
Expression of iNampt, Sirt1 and c-MYC in
the tumors and their correlation
The mechanisms by which obesity affects gastric
cancer growth remain unclear. We considered the possibility that
obesity prompts gastric cancer growth by the new pro-survival
signal, nampt/sirt1/c-myc positive feedback loop. Therefore,
we investigated the expression of these related genes nampt,
sirt1 and c-myc in the subcutaneous tumors by
immunohistochemical staining which might be altered in the context
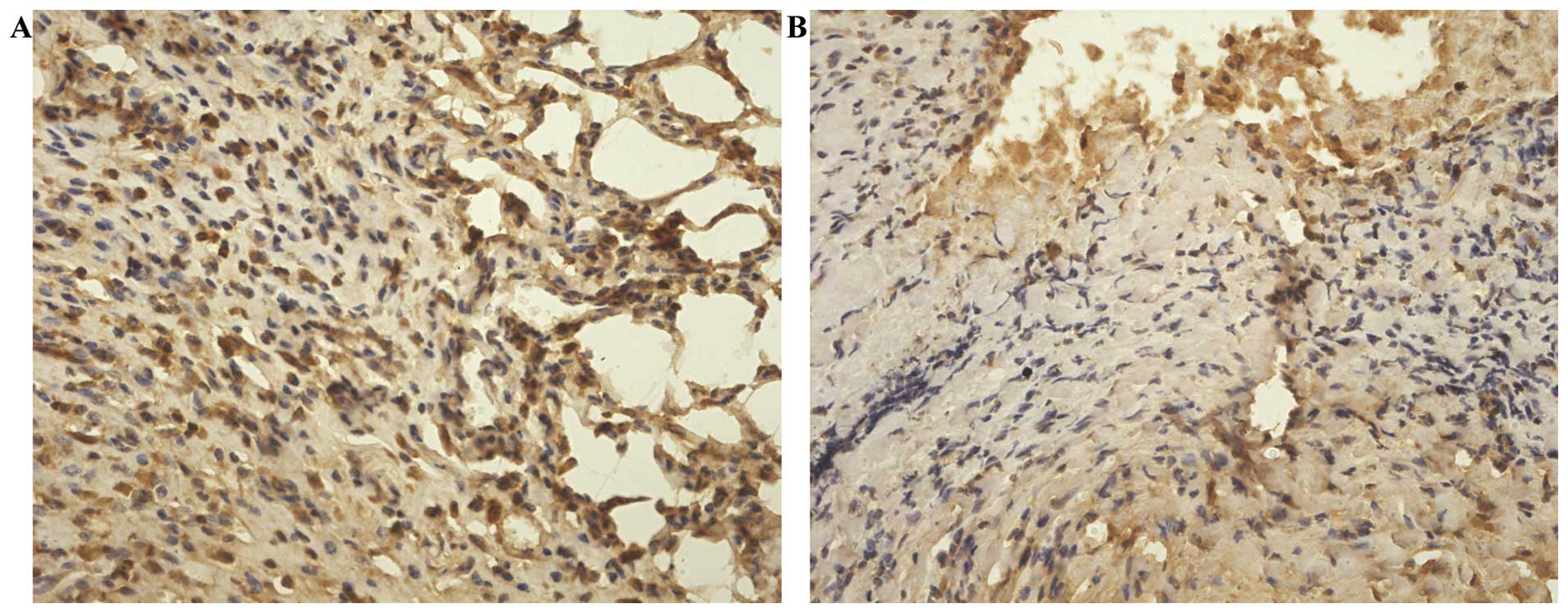
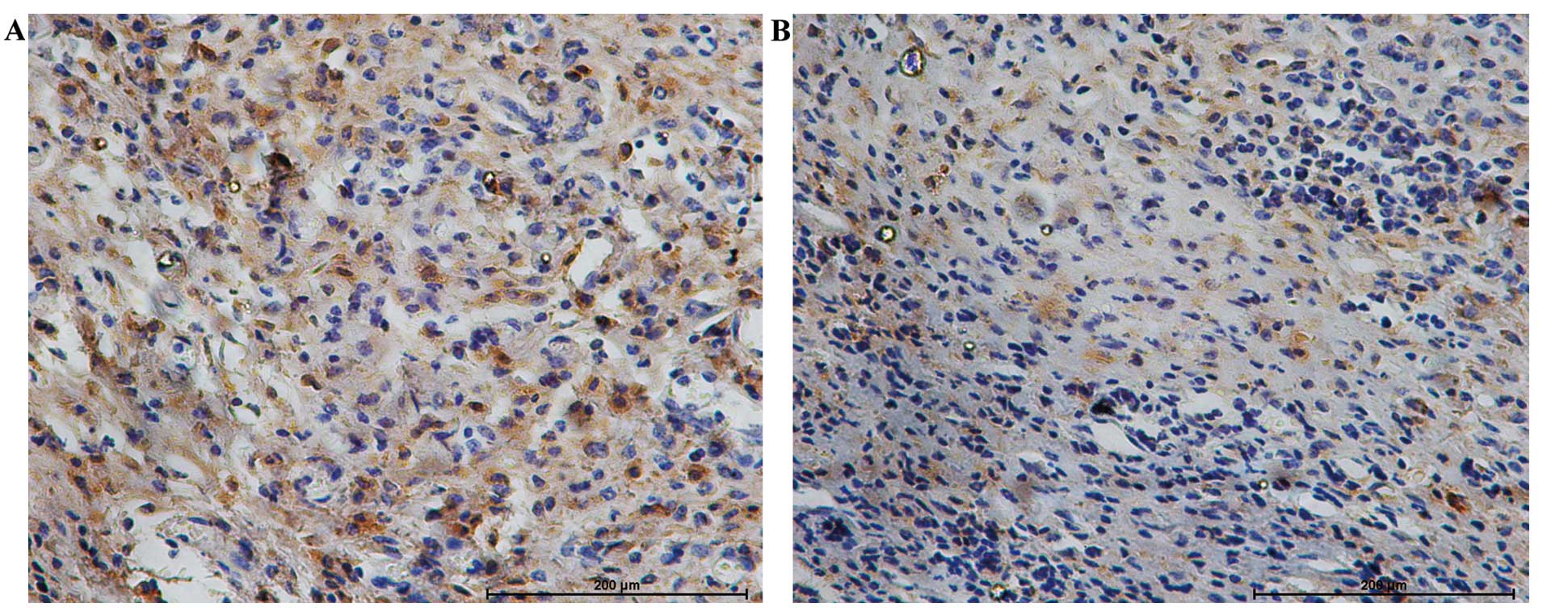
of obesity. The protein expression of iNampt, Sirt1 and c-MYC was
assessed semi-qualitatively by immunohistochemical analysis. iNampt
protein expressed in the cytoplasm and nucleus, Sirt1 and c-MYC
proteins localized mainly in the nucleus were significantly
elevated in the tumors from obese mice than in those from lean mice
(P<0.01) (Figs. 2–4). Additionally, the level of iNampt
protein was positively correlated with that of c-MYC (r=0.54,
P=0.01) and Sirt1 (r=0.71, P<0.001) in the tumors, and Sirt1
protein expression was positively correlated with c-MYC (r=0.43,
P=0.02<0.05), as assessed by the Spearman rank test. Of note,
iNampt protein expression in these tumors was positively correlated
with serum visfatin (r=0.76, P<0.001) and insulin
concentrations(r=0.39, P=0.02<0.05), but not with fasting
glucose level (r=0.12, P>0.05).
Effects of obesity on tumor cell
migration and proliferation
Individual cell migration is an important
characteristic of invasive tumor cells, therefore we examined the
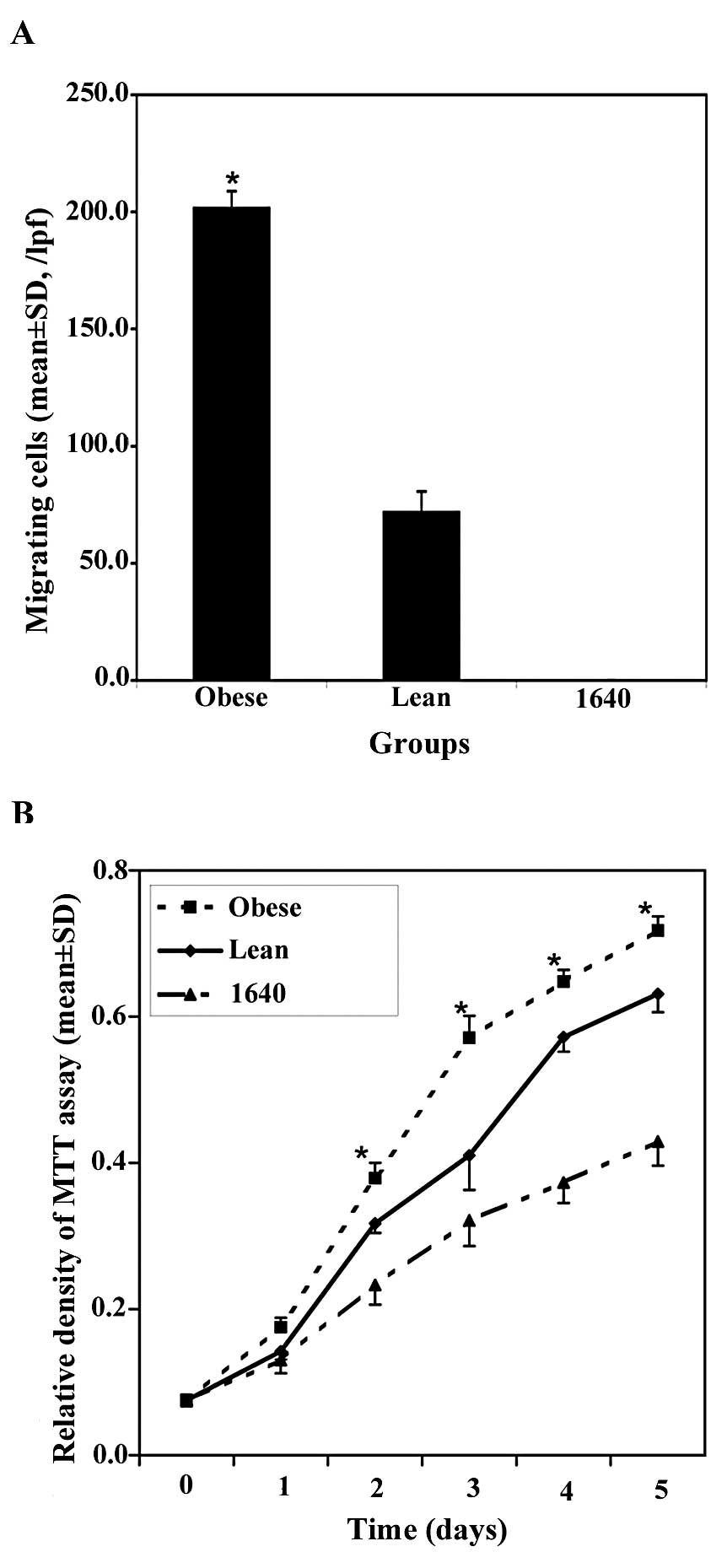
effects of obesity on modulating cell migration. The migrating cell
quantity of the obesity group was significantly larger than that of
the lean group in the low-power fields (original magnification,
×40) (201.60±7.21 vs. 71.80±8.88/lpf, P<0.01, Fig. 5A), and no cell migration existed in
the RPMI-1640 group. It was apparent that obesity induced the
ability of MFC cells traversing the filter to a highly significant
degree.
In this study, we also used MTT assay to assess MFC
cell proliferation. These groups were as follows: obesity, lean,
and RPMI-1640 as the control group. After treatment, cell
proliferation by MTT assay was followed over a course of 5 days.
The results of cell growth curve assay showed that obesity prompted
MFC cell growth significantly faster compared to the lean group
(Fig. 5B), and obesity also
promoted MFC proliferation in the subcutaneous tumors by DNA
incorporation of 5-bromodeoxyuridine (5-Brdu) in our previous study
(28).
Effects of obesity on tumor cell cycle
and apoptosis
To further explore how obesity promoted gastric
cancer cell survival and growth, we assessed MFC cell cycle and
apoptotic status using FACS analysis after treatment for 24 h. The
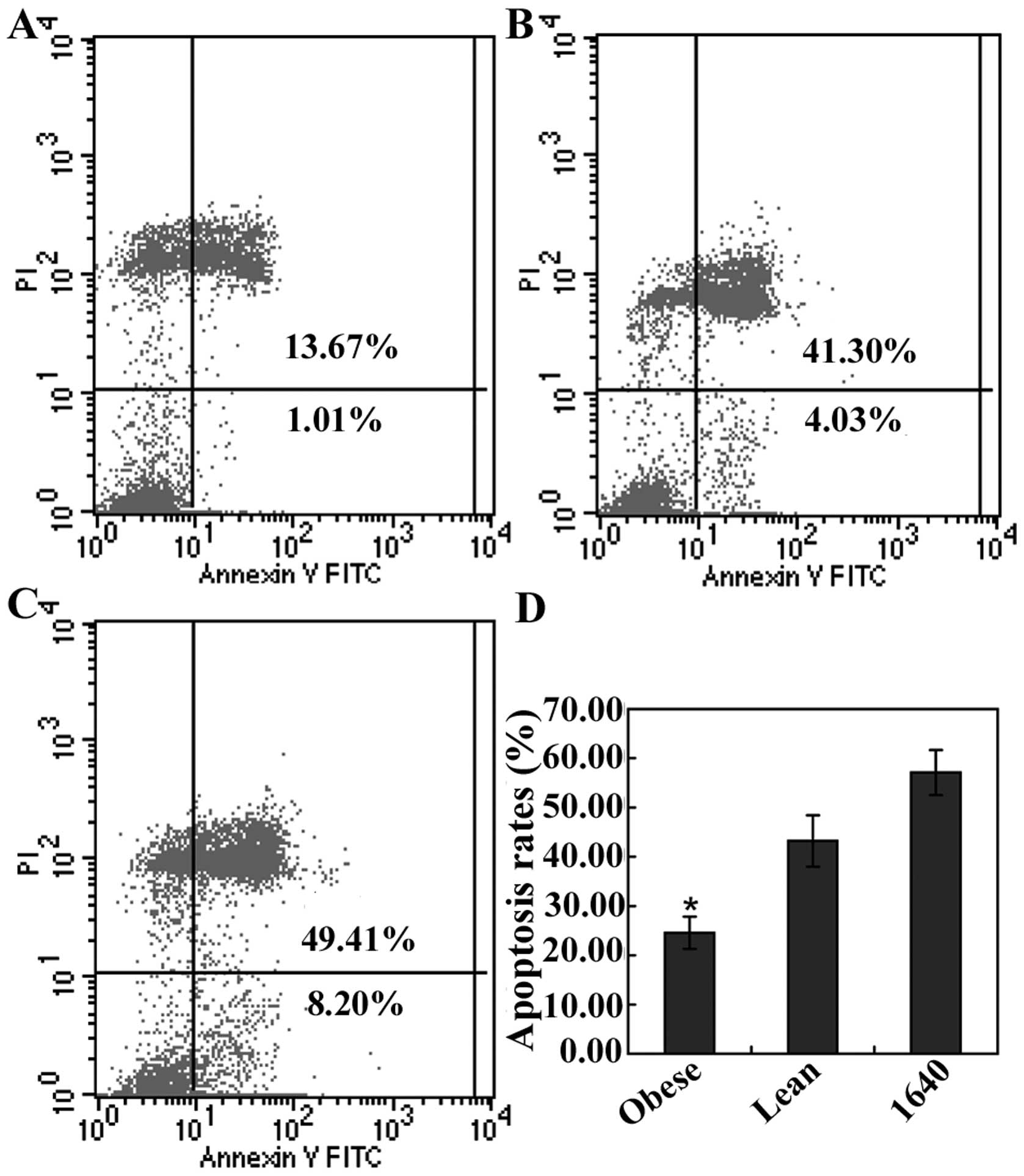
apoptosis assay results indicated that the average rate of
apoptosis in the Annexin V-positive (apoptosis portion) area in
obesity, lean and RPMI-1640 group was 24.59±3.26%, 43.21±5.21% and
57.10±4.55%, respectively, and there was a statistically
significant difference between the obesity and the lean group
(P<0.05, Fig. 6). It appeared
that obesity had an important influence on prompting MFC cell
survival and decreasing the cell apoptosis, and this was verified
in the subcutaneous tumors by TUNEL assay in our previous study
(28).
The data of cell cycle analysis showed that obesity
played a key role in cell cycle progression, and obesity
significantly increased S phase (P<0.05) and decreased G0/G1
phase (P<0.05); however, it had no influence on G2/M phase
(Table II). This demonstrated that
obesity accelerated cell cycle progression.
| Table IICell cycle in MFC cells over various
culture conditions for 24 h. |
Table II
Cell cycle in MFC cells over various
culture conditions for 24 h.
| Group | G0/G1 (mean ± SD,
%) | S (mean ± SD,
%) | G2/M (mean ± SD,
%) |
|---|
| Obese | 70.88±1.53a | 6.40±0.84a | 22.72±1.03 |
| Lean | 78.34±2.18 | 0.18±0.19 | 21.47±2.06 |
| RPMI-1640 | 77.60±2.49 | 0.25±0.27 | 22.15±2.34 |
Obesity promotes the growth of gastric
cancer by nampt/sirt1/c-myc positive feedback loop
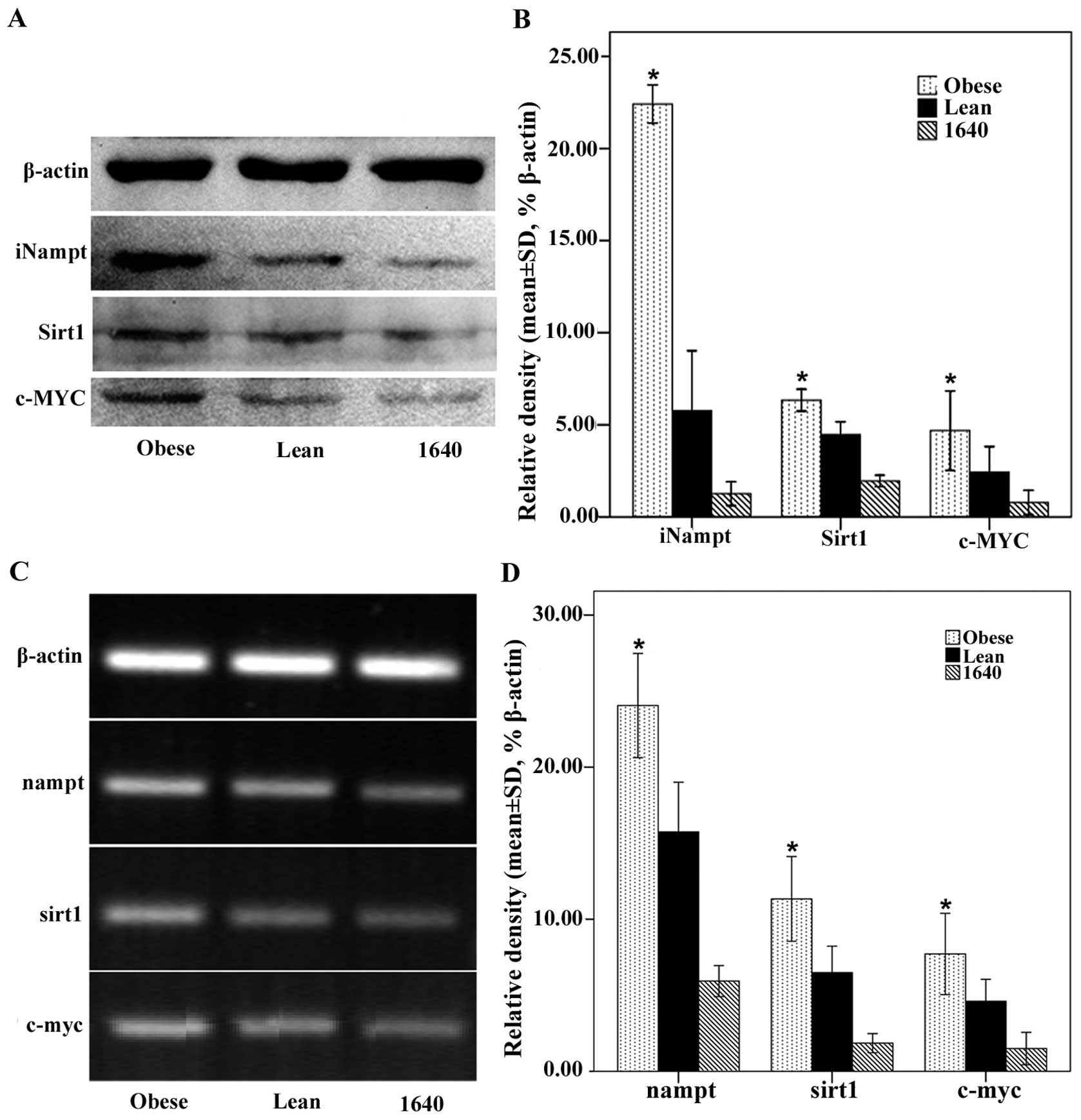
For validation, we also investigated the expression
of these related genes nampt, sirt1 and c-myc in the
cultured MFC cells which might be altered in the context of
obesity. Obesity significantly upregulated iNampt, Sirt1 and c-MYC
protein expression of MFC cells (Fig.
7A and B), and these results were verified at the mRNA level by
RT-PCR (Fig. 7C and D). This showed
that obesity could potentiate MFC cell migration and proliferation,
decrease MFC cellular apoptosis, and accelerate cell cycle
progression including endocrine mechanisms.
Discussion
Elucidating the mechanisms linking obesity and
gastric cancer is complicated due to the numerous biological
effects of obesity. Therefore, we developed a gastric cancer in
vivo model and cell culture in vitro utilizing the sera
of mice to explore this correlation. The results demonstrated that
MFC cells survived longer and grew larger in obese C57BL/6j mice
compared to non-obese and lean animals within 2 weeks, which
strongly indicated a positive correlation between obesity and tumor
growth. Additionally, the effect of obesity was not affected by the
high fat diet. Our previous histological data revealed that
injected tumors were surrounded by adipocytes and fat depots were
also detected within the tumors, which suggested that the adipocyte
microenvironment might be a protective niche for MFC cell survival
and growth, although the microvessel density was not significantly
different between the lean and the obese groups (28). This is consistent with the
hypothesis that adipose tissue plays a protective role in the
microenvironment of breast cancer (32), colon cancer (33) and multiple myeloma (34).
This study also showed that obesity potentiated MFC
cell migration and proliferation, decreased MFC cellular apoptosis,
and accelerated cell cycle progression through endocrine
mechanisms. Obesity increased the expression levels of pro-survival
signals, which may shift the apoptotic balance of MFC toward
survival. The main targets of this obesity-mediated effect are
probably the nampt/sirt1/c-myc positive feedback loop. Their
expression in MFC cells was upregulated at the mRNA and protein
levels in the context of obesity which could prompt gastric cancer
survival and growth. However, it is unclear which specific adipose
depots are relevant to MFC cell growth.
White adipose tissue, considered to be an inert
tissue as an energy store, drew more attention for secreting a
great number of adipokines in the past years. Adipokines secreted
mainly by adipocytes are small, biologically active factors which
could play an important role in stimulating tumor development.
Adipokines including resistin, leptin and adiponectin are
implicated in cell growth, proliferation, cell cycle control and
angiogenesis (35).
The new adipokine, visfatin, may facilitate tumor
proliferation and metastasis in various types of cancer including
breast (36,37) and prostate cancer (38). In addition, it was reported that
circulating visfatin concentrations correlated with tumor
progression in malignant astrocytomas (39), gastric cancer (40), and colorectal cancer (41). This study demonstrated directly the
correlation between gastric cancer growth and visfatin. Visfatin,
also acting as an NAD biosynthetic enzyme similar to iNampt,
catalyzes the transfer of a phosphoribosyl group from
5-phosphoribosyl-1-pyrophosphate to nicotinamide, forming
nicotinamide mononucleotide (NMN) and pyrophosphate (16). NMN is then converted to NAD by
nicotinamide mononucleotide adenylyltransferase (Nmnat) (42,43)
through the NAD+ salvage pathway.
We previously demonstrated that iNampt was
overexpressed in gastric cancer, and the specific iNampt inhibitor
FK866 suppressed gastric cancer cell proliferation and growth
(15). As such, visfatin enriched
in obesity may promote gastric cancer development by functioning as
an NAD biosynthetic enzyme similar to iNampt in the NAD+
salvage pathway to increase NAD+ content, which directly
affects the ability of Sirt1 or by other ways, then activates
c-MYC, iNampt and Sirt1 to form a positive feedback loop. Insulin
may also influence obesity-mediated tumor development through
insulin receptor A isoform (IR-A), IR-B, and heterodimeric
receptors including IGF-IR and IR (44), then stimulating downstream
activation of AKT and MAPK and prompting cancer cell proliferation
(30). The novel findings of this
study that serum visfatin and insulin concentrations were
positively correlated with the subcutaneous tumor growth and the
expression of iNampt protein in the tumors was consistent with the
notion that adipokines and other growth factors secreted in the
context of obesity may enhance cancer cell survival and solid tumor
growth.
In conclusion, this study clearly revealed that
obesity potentiated transplanted tumor growth in mice, promoted
gastric cancer cell migration, proliferation and survival, and
increased cell cycling in vitro. These were correlated with
the elevated expression levels of nampt, sirt1 and
c-myc.
Acknowledgements
The authors thank Dr Hai-Tao Shi, from the GI
Medicine Department of the Second Affiliated Hospital and the
Institution of Genetic Disease Research of Xi'an Jiaotong
University, for his technical assistance. This study was supported
by a grant from the National Natural Science Foundation of China
(no. 81172357).
References
|
1
|
Brenner H, Rothenbacher D and Arndt V:
Epidemiology of stomach cancer. Methods Mol Biol. 472:467–477.
2009. View Article : Google Scholar
|
|
2
|
David MR: The epidemiology of gastric
cancer. Gastric Cancer. 5:5–11. 2002. View Article : Google Scholar
|
|
3
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
4
|
Flegal KM, Carroll MD, Kit BK and Ogden
CL: Prevalence of obesity and trends in the distribution of body
mass index among US adults, 1999–2010. JAMA. 307:491–497. 2012.
|
|
5
|
El-Serag HB: Obesity and disease of the
esophagus and colon. Gastroenterol Clin North Am. 34:63–82. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Whiteman MK, Hillis SD, Curtis KM,
McDonald JA, Wingo PA and Marchbanks PA: Body mass and mortality
after breast cancer diagnosis. Cancer Epidemiol Biomarkers Prev.
14:2009–2014. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Crew KD and Neugut AI: Epidemiology of
gastric cancer. World J Gastroenterol. 12:354–362. 2006.
|
|
8
|
Yang P, Zhou Y, Chen B, Wan HW, Jia GQ,
Bai HL and Wu XT: Overweight, obesity and gastric cancer risk:
results from a meta-analysis of cohort studies. Eur J Cancer.
45:2867–2873. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang Y, Daquinag A, Traktuev DO, et al:
White adipose tissue cells are recruited by experimental tumors and
promote cancer progression in mouse models. Cancer Res.
69:5259–5266. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Samal B, Sun Y, Stearns G, Xie C, Suggs S
and McNiece I: Cloning and characterization of the cDNA encoding a
novel human pre-B-cell colony-enhancing factor. Mol Cell Biol.
14:1431–1437. 1994.PubMed/NCBI
|
|
11
|
Fukuhara A, Matsuda M, Nishizawa M, et al:
Visfatin: a protein secreted by visceral fat that mimics the
effects of insulin. Science. 307:426–430. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bi TQ and Che XM: Nampt/PBEF/visfatin and
cancer. Cancer Biol Ther. 10:119–125. 2010. View Article : Google Scholar
|
|
13
|
Van Beijnum JR, Moerkerk PT, Gerbers AJ,
De Bruïne AP, Arends JW, Hoogenboom HR and Hufton SE: Target
validation for genomics using peptide-specific phage antibodies: a
study of five gene products overexpressed in colorectal cancer. Int
J Cancer. 101:118–127. 2002.PubMed/NCBI
|
|
14
|
Folgueira MA, Carraro DM, Brentani H, et
al: Gene expression profile associated with response to
doxorubicin-based therapy in breast cancer. Clin Cancer Res.
11:7434–7443. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bi TQ, Che XM, Liao XH, Zhang DJ, Long HL,
Li HJ and Zhao W: Overexpression of Nampt in gastric cancer and
chemopotentiating effects of the Nampt inhibitor FK866 in
combination with fluorouracil. Oncol Rep. 26:1251–1257.
2011.PubMed/NCBI
|
|
16
|
Revollo JR, Grimm AA and Imai S: The
regulation of nicotinamide adenine dinucleotide biosynthesis by
Nampt/PBEF/visfatin in mammals. Curr Opin Gastroenterol.
23:164–170. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Voelter-Mahlknecht S and Mahlknecht U:
Cloning, chromosomal characterization and mapping of the
NAD-dependent histone deacetylases gene sirtuin 1. Int J Mol Med.
17:59–67. 2006.PubMed/NCBI
|
|
18
|
Luo J, Nikolaev AY, Imai S, et al:
Negative control of p53 by Sir2alpha promotes cell survival under
stress. Cell. 107:137–148. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vaziri H, Dessain SK, Ng Eaton E, et al:
hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell.
107:149–159. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kuzmichev A, Margueron R, Vaquero A, et
al: Composition and histone substrates of polycomb repressive group
complexes change during cellular differentiation. Proc Natl Acad
Sci USA. 102:1859–1864. 2005.PubMed/NCBI
|
|
21
|
Chen WY, Wang DH, Yen RC, Luo J, Gu W and
Baylin SB: Tumor suppressor HIC1 directly regulates SIRT1 to
modulate p53-dependent DNA-damage responses. Cell. 123:437–448.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Saunders LR and Verdin E: Sirtuins:
critical regulators at the crossroads between cancer and aging.
Oncogene. 26:5489–5504. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lim CS: Human SIRT1: a potential biomarker
for tumorigenesis? Cell Biol Int. 31:636–637. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lüscher B and Vervoorts J: Regulation of
gene transcription by the oncoprotein MYC. Gene. 494:145–160.
2012.PubMed/NCBI
|
|
25
|
Eilers M and Eisenman RN: Myc's broad
reach. Genes Dev. 22:2755–2766. 2008.
|
|
26
|
Soucek L, Whitfield J, Martins CP, et al:
Modelling Myc inhibition as a cancer therapy. Nature. 455:679–683.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Menssen A, Hydbring P, Kapelle K, et al:
The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1,
and the SIRT1 deacetylase form a positive feedback loop. Proc Natl
Acad Sci USA. 109:E187–E196. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li HJ, Che XM, Zhao W, He SC, Zhang ZL and
Chen R: Diet-induced obesity potentiates the growth of gastric
cancer in mice. Exp Ther Med. 4:615–620. 2012.PubMed/NCBI
|
|
29
|
Qian SS, Gao J, Wang JX, Liu Y and Dong
HY: Establishment of a mouse forestomach carcinoma cell line (MFC)
with spontaneous hematogenous metastasis and preliminary study of
its biological characteristics. Zhonghua Zhong Liu Za Zhi.
9:261–264. 1987.(In Chinese).
|
|
30
|
Yakar S, Nunez NP, Pennisi P, et al:
Increased tumor growth in mice with diet-induced obesity: impact of
ovarian hormones. Endocrinology. 147:5826–5834. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vermes I, Haanen C, Steffens-Nakken H and
Reutelingsperger C: A novel assay for apoptosis. Flow cytometric
detection of phosphatidylserine expression on early apoptotic cells
using fluorescein labelled Annexin V. J Immunol Methods. 184:39–51.
1995. View Article : Google Scholar
|
|
32
|
Iyengar P, Combs TP, Shah SJ, et al:
Adipocyte-secreted factors synergistically promote mammary
tumorigenesis through induction of anti-apoptotic transcriptional
programs and proto-oncogene stabilization. Oncogene. 22:6408–6423.
2003. View Article : Google Scholar
|
|
33
|
Amemori S, Ootani A, Aoki S, et al:
Adipocytes and preadipocytes promote the proliferation of colon
cancer cells in vitro. Am J Physiol Gastrointest Liver Physiol.
292:G923–G929. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Caers J, Deleu S, Belaid Z, et al:
Neighboring adipocytes participate in the bone marrow
microenvironment of multiple myeloma cells. Leukemia. 21:1580–1584.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tilg H and Moschen AR: Adipocytokines:
mediators linking adipose tissue, inflammation and immunity. Nat
Rev Immunol. 6:772–783. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kim SR, Park HJ, Bae YH, et al: Curcumin
down-regulates visfatin expression and inhibits breast cancer cell
invasion. Endocrinology. 153:554–563. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kim JG, Kim EO, Jeong BR, et al: Visfatin
stimulates proliferation of MCF-7 human breast cancer cells. Mol
Cells. 30:341–345. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Patel ST, Mistry T, Brown JE, Digby JE,
Adya R, Desai KM and Randeva HS: A novel role for the adipokine
visfatin/pre-B cell colony-enhancing factor 1 in prostate
carcinogenesis. Peptides. 31:51–57. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Reddy PS, Umesh S, Thota B, et al:
PBEF1/NAmPRTase/Visfatin: a potential malignant
astrocytoma/glioblastoma serum marker with prognostic value. Cancer
Biol Ther. 7:663–668. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nakajima TE, Yamada Y, Hamano T, et al:
Adipocytokine levels in gastric cancer patients: resistin and
visfatin as biomarkers of gastric cancer. J Gastroenterol.
44:685–690. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nakajima TE, Yamada Y, Hamano T, et al:
Adipocytokines as new promising markers of colorectal tumors:
adiponectin for colorectal adenoma, and resistin and visfatin for
colorectal cancer. Cancer Sci. 101:1286–1291. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Garten A, Petzold S, Körner A, Imai S and
Kiess W: Nampt: linking NAD biology, metabolism and cancer. Trends
Endocrinol Metab. 20:130–138. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pilz S, Mangge H, Obermayer-Pietsch B and
März W: Visfatin/pre-B-cell colony-enhancing factor: a protein with
various suggested functions. J Endocrinol Invest. 30:138–144. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sciacca L, Costantino A, Pandini G, et al:
Insulin receptor activation by IGF-II in breast cancers: evidence
for a new autocrine/paracrine mechanism. Oncogene. 18:2471–2479.
1998. View Article : Google Scholar : PubMed/NCBI
|