Introduction
The most commonly used source of mesenchymal stem
cells (MSCs) is bone marrow aspirate, but its disadvantages
restrict clinical and laboratory practice, such as pain, morbidity
and low cell number upon harvest. Recent studies have shown that
MSCs can be obtained from trabecular bone fragments obtained during
total hip/knee replacements, which can avoid the disadvantages of
using bone marrow as a source. Human trabecular bone-derived cell
populations are pluripotent stem cells, which have multilineage
potential and can give rise not only to osteoblasts, but also to
adipocytes, when subjected to appropriate treatment protocols
(1). Sottile et al(2) compared the characteristics of
mesenchymal cell cultures established either from trabecular bone
or from bone marrow, and the results showed that both of the cell
cultures actually had similar characteristics to bone
marrow-derived MSCs, differentiating into osteoblasts, chondrocytes
and adipocytes under appropriate differentiating conditions.
Therefore, trabecular bone tissue is a good source of adult MSCs
for in vitro investigation.
The fate and commitment of MSCs are regulated by
microenvironmental conditions, such as injury, inflammation and
tumors. For example, under conditions of chronic inflammation, MSCs
may contribute to adverse manifestations, such as the accumulation
of fat deposits in bone and muscles, impaired healing and fibrosis
after severe injury, or altered hematopoiesis and autoimmunity
(3). On the other hand, MSCs also
exert anti-inflammatory effects that are important in maintaining
homeostatic balance. To date, MSCs have been used as a treatment
modality for several inflammation-related diseases, such as
inflammatory bowel disease (IBD) (4), graft vs. host disease (5), rheumatoid arthritis (6) and multiple sclerosis (7). In addition, MSCs secrete large amounts
of inflammatory cytokines which regulate the inflammatory process.
Thus, these findings indicate a complex, functional interaction
between MSCs and the inflammatory microenvironment.
T lymphocytes play a central role in the initiation
and maintenance of inflammatory processes. Accumulation of T cells
at inflammatory sites is one of the characteristic features of
chronic inflammatory diseases (8).
During the development of inflammation, T cells are activated by
inflammatory messengers, and ‘activated’ T cells are a sign of
ongoing inflammation. T cell activation depends on signals
delivered from antigen-presenting cells (APCs) through triggering
of their T cell receptor (TCR) complex and a co-stimulatory
receptor such as CD28 (9). The T
cell activation pathway is triggered when a T cell encounters its
cognate antigen, such as plant mitogen phytohemagglutinin (PHA),
which has a marked selectivity for T lymphocytes. Following
short-term pre-incubation with PHA, T cell activity is maintained.
T cells activated with PHA express Ia-like antigens (which play a
role in the stimulation of T lymphocytes by autologous PHA-T
lymphocytes) and acquire the ability to stimulate autologous T
lymphocytes in mixed lymphocyte reaction (10). Therefore, T cell activation using
PHA stimulation is a good pathway with which to curb
inflammation.
To better understand the effects of T cell
activation and inflammation on the adipogenic and osteogenic
differentiation of MSCs, PHA-activated T cells were used for
co-culturing with MSCs in the present study. The inflammatory gene
expression was detected by quantitative RT-PCR, and the adipo- and
osteo-specific proteins were determined by western blotting.
TGF-β/Smad signaling was described by western blotting; and ALP
activity was detected by pNPP method; and the
adipo-/osteo-differentiation of MSCs was further verified using
histological and immunohistochemical staining.
Materials and methods
All the procedures were approved by the Ethics
Committee of the Union Hospital and the Tongji Medical College.
Informed consent was obtained from the patients included in the
study.
Isolation and culture of MSCs
Human trabecular bone-derived MSCs were isolated
from patients undergoing total hip replacement. One piece of bone
was collected from the removed femoral neck under sterilization.
The bone was rinsed several times (3–5 times) with PBS to remove
excess blood. Subsequently, the bone was placed onto a sterile
Petri dish and cancellous bone was broken into 2–3 mm2 ×
1 mm fragments (the cortical bone was discarded). Subsequently, the
bone fragments were placed into 15-ml centrifuge tubes in
collagenase solution (3 mg/ml), and the tubes were incubated on a
rotator at 37°C for 3 h. After 3 h of digestion, the same volume of
complete culture medium (CM) was added to neutralize the reaction,
and the supernatant was filtered through a 70-μm cell strainer.
Finally, we obtained MSC solution, the cell density was adjusted
and cells were seeded in T-75 cell culture flasks. The flask was
placed in a humidified incubator at 37°C with 5% CO2.
After a 24-h culture, the culture medium was refreshed; the culture
medium was replaced every 2–3 days.
In vitro osteogenic, adipogenic and
chondrogenic differentiation of MSCs
MSCs of passage 3–5 were used in the experiments.
MSCs were plated in a 24-well plate in coverslips at a density of
1.5×104 cells/ml. For osteogenic differentiation, after
becoming confluent, the cells were incubated in an osteogenic
medium (OS) containing dexamethasone, ascorbate-phosphate and
β-glycerolphosphate in complete medium. OS was replaced every 2–3
days for 3 weeks. The cells were fixed using 4% paraformaldehyde,
and subjected to Alizarin Red S staining and immunostaining (mouse
anti-human osteocalcin monoclonal antibody, osteogenic marker). For
adipogenic differentiation, MSCs were seeded at a density of
7.4×104 cells/ml. The subconfluent cells were cultured
in adipogenic differentiation medium, containing hydrocortisone,
isobutylmethylxanthine and indomethacin in complete medium. After
7–21 days of stimulation, the cells were fixed and then detected
using Oil Red staining and immunostaining (goat anti-mouse FABP-4
polyclonal antibody, adipogenic marker). As previously described, a
pellet culture system was used for chondrogenic differentiation. A
total of 2.5×105 cells were transferred into a 15-ml
tube and pelleted by spin-down. The pellet was cultured in 0.5 ml
of chondrogenic differentiation medium, containing dexamethasone,
ascorbate-phosphate, proline, pyruvate, TGF-β3, insulin,
transferrin and selenious acid in complete medium. The chondrogenic
culture medium was changed every 2–3 days for 3 weeks. The pellets
were then fixed and subjected to frozen sections. Immunostaining
method was used to detect the expression of aggrecan protein (goat
anti-human aggrecan polyclonal antibody, chondrogenic marker).
T cell isolation and activation
Peripheral blood mononuclear cells (PBMCs) were
collected from healthy donors and separated by Ficoll-Hypaque
density gradient centrifugation. The T cells were then isolated and
purified from PBMCs by magnetic-activated cell sorting using the
CD3 MicroBead-based isolation kit according to the manufacturer’s
instructions (MACS; Miltenyi Biotec). To achieve higher purities,
the positively selected CD3+ cell fractions were
separated again over a new, freshly prepared LS column. The
viability of CD3+ cell fractions was measured by trypan
blue, with cells generally being >95%. For activation,
CD3+ cell fractions (T cells) were resuspended in α-MEM
medium containing PHA (2.5 μg/ml), 10% FCS, penicillin (100 U/ml),
streptomycin (100 μg/ml), and L-glutamine (2 mM) and incubated at
37°C for 2 days in tissue culture tubes with filtered caps.
MTS assay for cell proliferation
MTS assay was used to detect the cell proliferation
of MSCs after treatment with different concentrations of
PHA-activated T cells. MSCs were placed into each well of 96-well
plates at a density of 6×103 cells/ml. At 24 h after
plating for attachment, cells were incubated with different
concentrations of activated T cells (6×10 to 6×105
cells/ml). On day 4, suspended cells were removed by gentle washing
with phosphate-buffered saline (PBS). The number of adherent cells
remaining in each well was then quantified using a coupled
enzymatic assay, which resulted in the conversion of a tetrazolium
salt into a red formazan product (MTS assay). Recording of the
absorbance at 490 nm in the MTS assay was carried out.
MSC differentiation under T cell
activation and inflammation
MSCs were co-cultured with activated T cells to
mimic the inflammatory microenvironment. In the experiment, the
cells were divided into the control group and inflammatory group
(inflammatory). In the inflammatory group, MSCs were seeded onto
plates at a concentration of 1.2×104 cells/ml in CM,
containing 10% FCS, 100 μg PSN (penicillin, streptomycin and
neomycin) and 100 μM L-ascorbic acid phosphate in α-MEM. Activated
T cells were then added at a concentration of 0.6×105
cells/ml, after the cultures became 90% confluent. In the control
groups, MSCs were cultured without T cell exposure. The cultures
were refreshed with complete medium (CM) every 2–3 days for the
following measurements.
RNA isolation and quantitative
RT-PCR
To investigate the effects of the inflammatory
microenvironment induced by activated T cells, quantitative RT-PCR
was used to detect the expression of inflammatory genes and
osteo-/adipo-specific genes. After a 1-day co-culture with
activated T cells, MSCs were collected in TRIzol reagent
(Invitrogen, Carlsbad, CA, USA), followed by RNA isolation
according to the manufacturer’s instructions. The RNA samples were
subjected to cDNA synthesis, followed by quantitative PCR assays.
The reverse transcriptase reaction was carried out using
ThermoScript™ reverse transcription reagents (Roche Applied
Science). PCRs were performed according to the real-time PCR
machine manufacturer’s instructions (MJ Research, Inc., Watertown,
MA, USA), which allow real-time quantitative detection of the PCR
product by measuring the increase in SYBR-Green fluorescence caused
by binding of SYBR-Green (Bio-Rad Laboratories, Québec, Canada) to
double-stranded DNA. To amplify specific gene products, the
following primers were used: IL-6 (forward, CCTCGACGGCATCTCAGCCC
and reverse, TGCCCAGTGGACAGGTTTCTGAC); IL-10 (forward,
CAAGGCCGTGGAGCAGGTGAA and reverse, GGTTTCTCAAGGGGCTGGGTCA); INFγ
(forward, TCG CCAGCAGCTAAAACAGGGA and reverse, GCTGCCTA
GTTGGCCCCTGA); PPAR-γ (forward, GCTGTTATGGGT GAAACTCT and reverse,
ATGGAATGTCTTCGTAATGT); RUNX2 (forward, GTGCCTAGGCGCATTTCA and
reverse: GCTCTTCTTACTGAGAGTGGAAGG); and 18S rRNA (forward,
CCGCAGCTAGGAATAATGGAATA and reverse, TCTAGCGGCGCAATACGAAT), which
served as an internal control. Amplification was performed using a
profile at 94°C for 1 min (denaturation), 60°C for 30 sec
(annealing), 72°C for 45 sec (elongation) for a total of 38 cycles,
followed at the end by 72°C for 5 min (extension). Negative
controls without RT were carried out in parallel for every PCR
reaction to exclude amplification of contaminating DNA.
Western blot analysis
Under the inflammatory microenvironment induced by
activated T cells, cell lysates of MSCs were collect at 0, 1, 3, 6,
12 and 18 h and stored at −20°C for western blot analysis. The
protein concentrations were measured using the Bio-Rad protein
assay, and equalized protein samples were used for the
electrophoresis. The proteins were then electro-transferred to 0.45
μm-pore-diameter polyvinylidene difluoride (PVDF) membranes
(Invitrogen). PVDF membranes were blocked in blocking agent (5%
skim milk, 0.1% Tween-20 in basic buffer) overnight at 4°C. After
blocking of non-specific immunoglobulin (IgG) binding, the
membranes were incubated in primary antibodies at a 1:400 dilution
for 2 h at 4°C under rocking. The membranes were then incubated for
2 h at 4°C with a 1:2,000 dilution of goat secondary antibodies in
antibody diluents. Finally, the ECL-Plus western blotting system
was used, and immunoreactive bands were revealed and quantified
using ImageQuant LAS 400 software (GE Healthcare Life Sciences).
The primary antibodies were anti-PPAR-γ, anti-RUNX2, anti-TGF-β1,
anti-Smad3 and anti-P-Smad3 produced in rabbit (Sigma-Aldrich).
Immunohistochemical staining
After 2–3 weeks of culture, the cells were fixed and
saved for immunostaining, to assess the possible adipogenic or
osteogenic differentiation of MSCs. The culture medium was
aspirated and the cells were washed twice PBS. The cells were fixed
with 4% paraformaldehyde for 20 min at room temperature. After
fixation, the cultures were washed with 1% BSA in PBS and blocked
with 0.3% Triton X-100, 1% BSA, 10% normal donkey serum in PBS for
45 min. Subsequently, the primary antibody was added and incubated
overnight at 4°C. After washing, the secondary antibody was added
and incubation was carried out in the dark for 60 min at room
temperature. The primary antibodies used in the experiment were
goat anti-human FABP4 and mouse anti-human osteocalcin antibodies
(R&D Systems); and the secondary antibodies were
fluorochrome-conjugated donkey anti-goat and donkey anti-mouse
(R&D Systems). Staining was examined using fluorescence
microscopy immediately.
Oil Red O staining and Alizarin Red
staining
To determine whether the inflammatory
microenvironment induced by activated T cells affects the
differentiation of MSCs, the adipogenic and osteogenic
differentiation of the cells was evaluated after 2–3 weeks of
culture. Briefly, the cultures were washed with PBS, 2.5%
glutaraldehyde was added (0.5 ml/well) (24-well plate), and the
reaction was carried out at room temperature for 20 min. For
adipogenic differentiation, Oil Red O solution was added to the
fixed cells; for osteogenic differentiation, 2% Alizarin Red
solution was added. The plate was kept at 37°C in an incubator for
10–20 min. The staining was monitored under a microscope every 2–5
min and images were captured under the microscope immediately for
analysis.
In vitro osteogenic differentiation and
pNPP assay
For osteogenic differentiation, the cells were
cultured in OS, containing dexamethasone, ascorbate-phosphate and
β-glycerolphosphate in CM. After 2–3 weeks of culture, the cells
were subjected to pNPP alkaline phosphatase assay to detect
alkaline phosphatase (ALP) activity. Assay buffer 1X was prepared
and the cells were gently washed twice with the buffer. Then, 300
μl of 1X assay buffer in Triton X-100 per well (6-well plate) was
added to the cells, and the adherent cells were scraped off and
transferred into a microcentrifuge tube. The cell suspension was
incubated at 4°C for 10 min under agitation. After centrifugation,
the supernatant was collected for alkaline phosphatase assay,
according to the instructions for the SensoLyte® pNPP
alkaline phosphatase assay kit. Meanwhile, the concentrations of
the proteins were determined using Bio-Rad protein assay. The
results of ALP activity was determined using the standard curve and
normalized by the total protein content. ALP activity = ALP
(μg/ml)/protein (mg/ml).
Statistical analysis
All experiments were repeated three times, and the
results are expressed as the means ± SEM. Data were analyzed by
ANOVA or t-test, and P<0.05 was considered to indicate a
statistically significant result.
Results
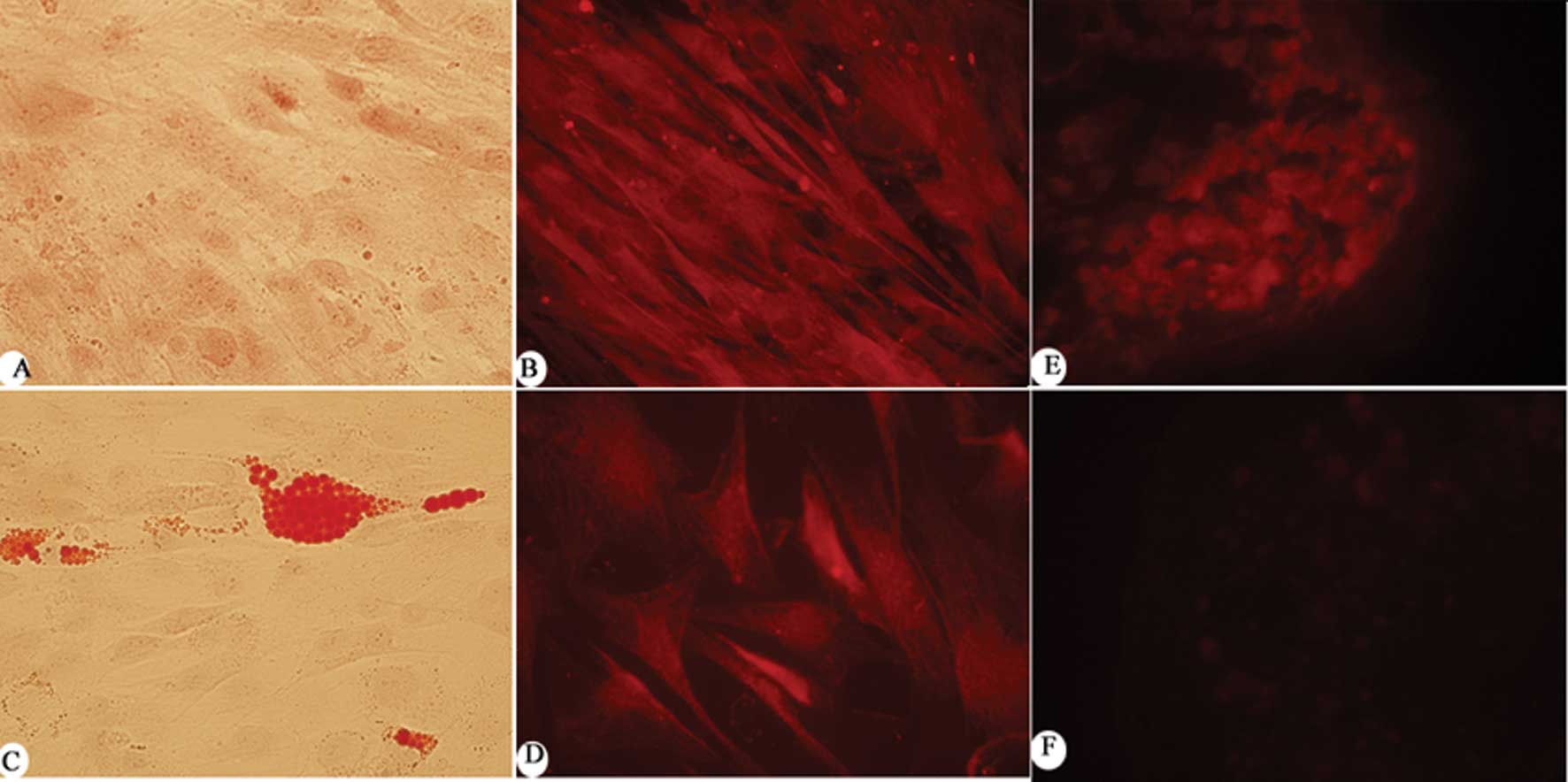
Characterization of MSCs isolated from
human trabecular bone
One of the defining characteristics of MSCs is their
multilineage differentiation potential. Under certain inductive
conditions, MSCs are able to acquire the characteristics of cells
derived from the embryonic mesoderm, such as osteoblasts,
chondrocytes and adipocytes. In order to define the characteristics
of the trabecular bone-derived MSCs, cell type-specific
histological and immunochemical staining were used. To identify
adipogenic and osteogenic differentiation in vitro, the
cultures were stained with Oil Red O and Alizarin Red S,
respectively. Trabecular bone-derived MSC differentiation also was
verified by analyzing the gene expression of adipogenic,
chondrogenic and osteogenic markers, such as osteocalcin, FABP-4
and aggrecan (Fig. 1). The results
showed that the MSCs were successfully differentiated into
adipocytes, osteocytes and chondrocytes.
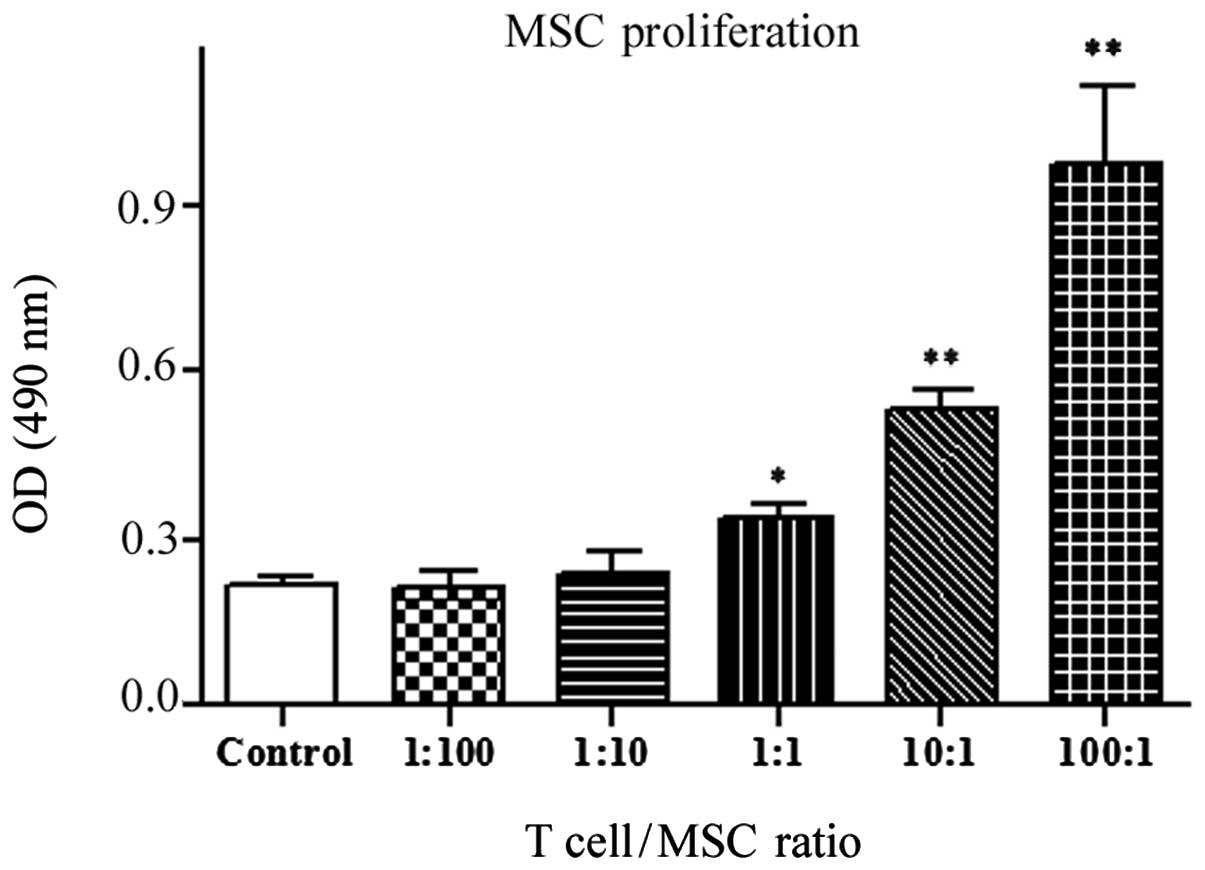
MSC proliferation with PHA-activated T
cell supplements
MTS is a common and useful method for monitoring
cell proliferation, for the detection of viable cells. To quantify
the proliferation of trabecular bone-derived MSCs following
exposure to activated T cells of different concentrations, an MTS
assay was performed (Fig. 2). The
results showed that lower concentrations (ratios of 1:100 and 1:10)
of T cells had no effect on the proliferation of MSCs. However,
high concentrations of T cells significantly increased the
proliferation of MSCs, particularly at ratios of 10:1 and 100:1
(P<0.01).
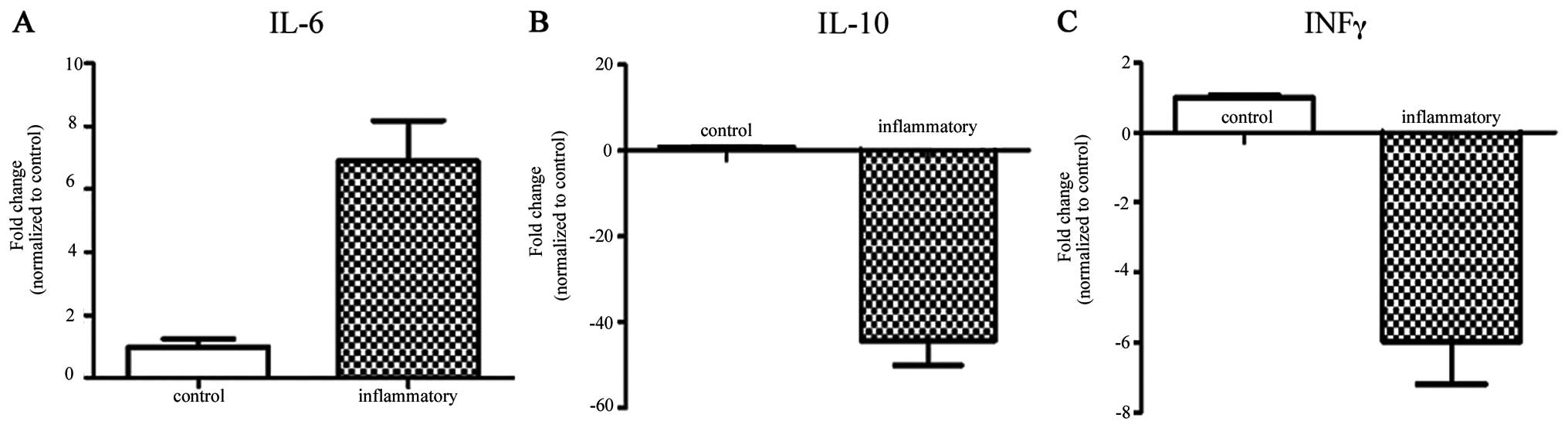
Expression of inflammatory factors
induced by T-cell exposure
T cells induce the expression of genes mostly
related to inflammatory cytokines, which are thought to play an
important role in the process of chronic inflammation. The present
study was undertaken to detect the gene expression profile of MSCs
exposed to activated T cells using qRT-PCR. The results showed that
the expression of the proinflammatory gene IL-6 was significantly
upregulated (6.89-fold compared with the control) and acted to
promote excessive inflammation. On the other hand, the expression
of anti-inflammatory genes, such as IL-10 (−44.24-fold) and INFγ
(−5.96-fold), was significantly downregulated when compared to the
control group (Fig. 3). Therefore,
T-cell exposure increased the expression of proinflammatory genes
but inhibited the expression of anti-inflammatory genes and led to
an excessive inflammatory microenvironment.
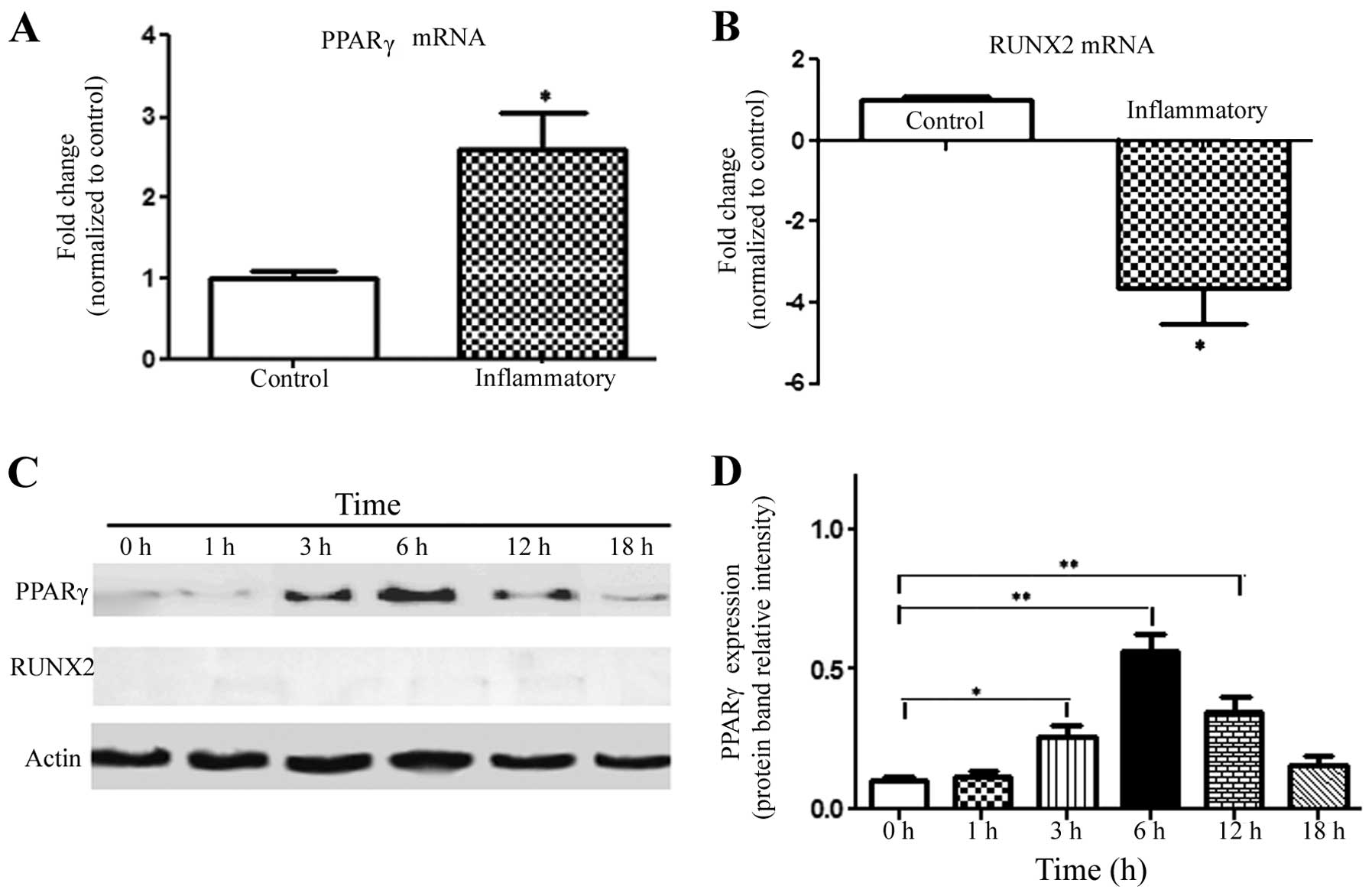
Adipo-/osteo-specific gene expression is
influenced by activated T cells and inflammation
In order to investigate the effects of an
inflammatory microenvironment induced by activated T cells on the
adipogenic and osteogenic differentiation of MSCs, qRT-PCR and
western blotting were performed. The results of qRT-PCR showed that
the adipo-specific gene expression of PPARγ was significantly
upregulated (2.58-fold compared with the control); while the
osteogenic-specific gene expression of RUNX2 was significantly
downregulated (−3.63-fold) (Fig. 4A and
B). Western blot analysis revealed that the expression level of
PPARγ was upregulated in MSCs after inflammatory stimulation with
activated T cells, reaching a peak value at 6 h, which was
subsequently decreased (Fig. 4C).
In contrast, MSCs expressed barely detectable levels of RUNX2
protein for the duration of the experiment; thus, we could not
compare the expression levels of RUNX2 protein between the groups.
However, the results still indicated that the alterations resulted
in increased adipogenic differentiation and decreased osteogenic
differentiation of MSCs at an early stage.
Adipogenic differentiation of MSCs is
regulated by activated T cells
MSCs were cultured in CM supplied with activated T
cells. After a 2-week culture, the cultures were fixed; half of
them were subjected to Oil Red O staining, and the other half were
used for the detection of the expression of FABP4 using
fluorescence immunohistochemistry. The fluorescence staining showed
a very weak diffuse fluorescence pattern in the control group
(Fig. 5A) but strong FABP4
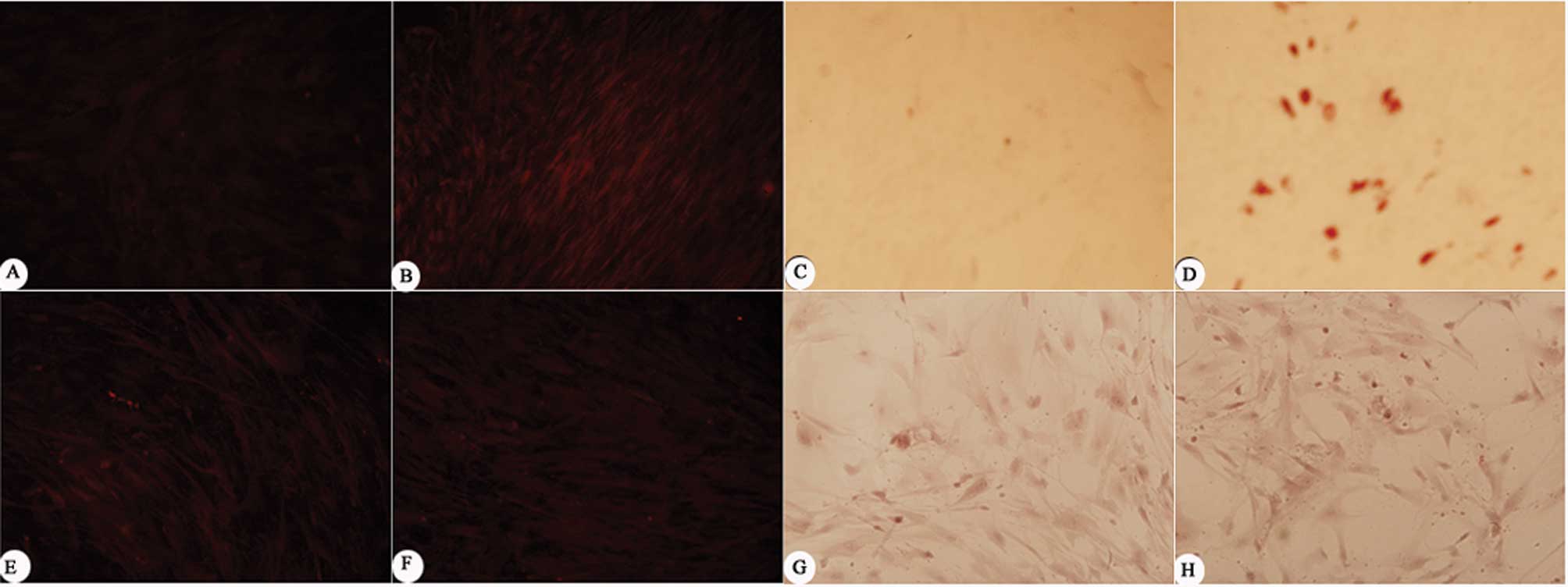
expression of MSCs in the inflammatory group (Fig. 5B). The results of Oil Red O staining
showed accumulated intracellular lipid droplets in the inflammatory
group (Fig. 5D), while no lipid
droplet was found in the control group (Fig. 5C). Both types of staining indicated
that an inflammatory microenvironment induced by activated T cells
promoted the adipogenic differentiation of MSCs.
Osteogenic differentiation of MSCs in the
presence of activated T cells
In order to investigate the effect of activated T
cells and inflammation on the osteogenic differentiation of MSCs,
fluorescence immunohistochemical staining and Alizarin Red staining
were used. The fluorescence staining showed that MSCs expressed
lower levels of osteocalcin in both the inflammatory group and the
control group. Alizarin Red staining showed similar results; only
mild mineralization was observed in both of the groups (Fig. 5E–H). These results confirmed the
finding that the inflammatory microenvironment induced by activated
T cells had no significant effect on the osteogenic differentiation
of MSCs.
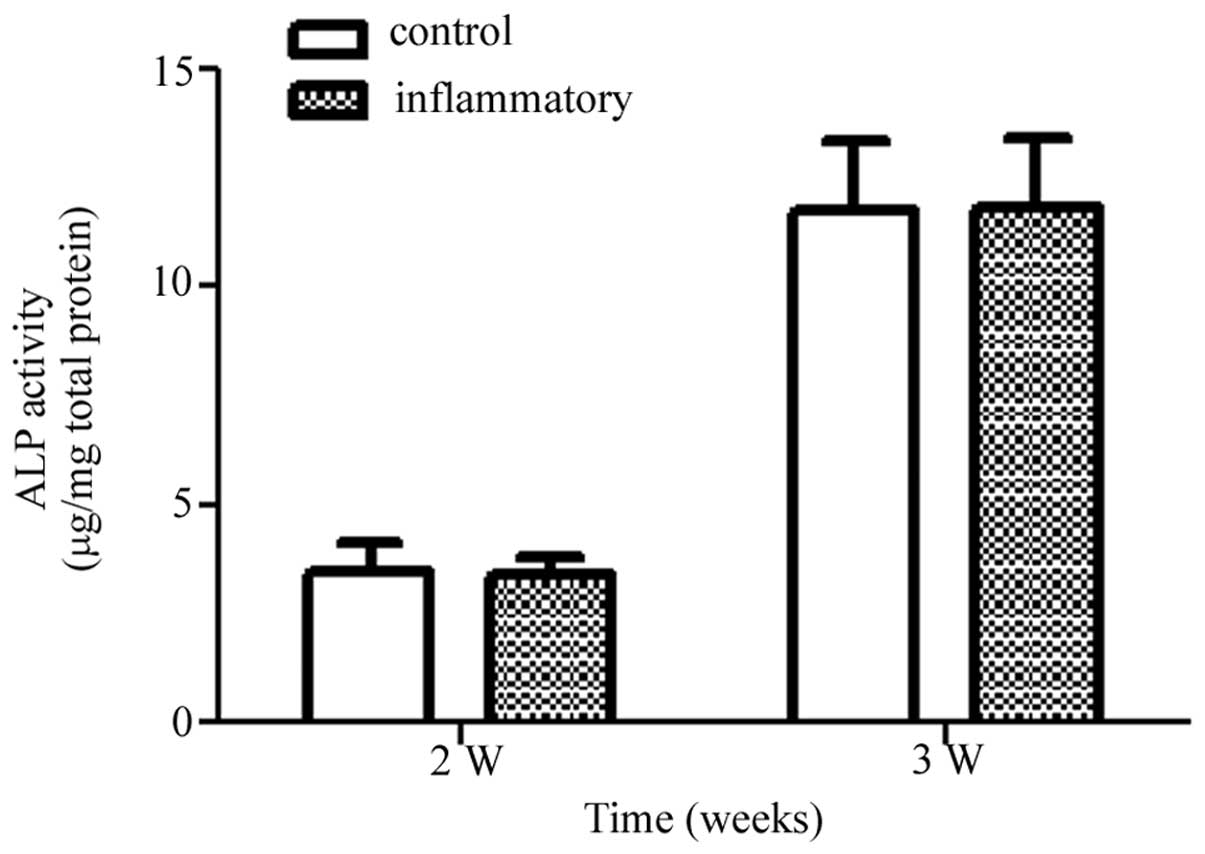
ALP activity as determined by pNPP
method
The activity of ALP was detected to evaluate the
effect of activated T cells on the osteogenic differentiation of
MSCs in OS medium by pNPP method. The ALP levels expressed by MSCs
increased with time in both the control and inflammatory groups in
the presence of OS medium. However, at the time-points studied
(week 2 and 3), no significant difference was noted in ALP activity
due to the inflammatory microenvironment induced by the activated T
cells, when compared to the control group (Fig. 6). Therefore, the results confirmed
that the activated T cells had no obvious effects on the osteogenic
differentiation of MSCs at the late stage.
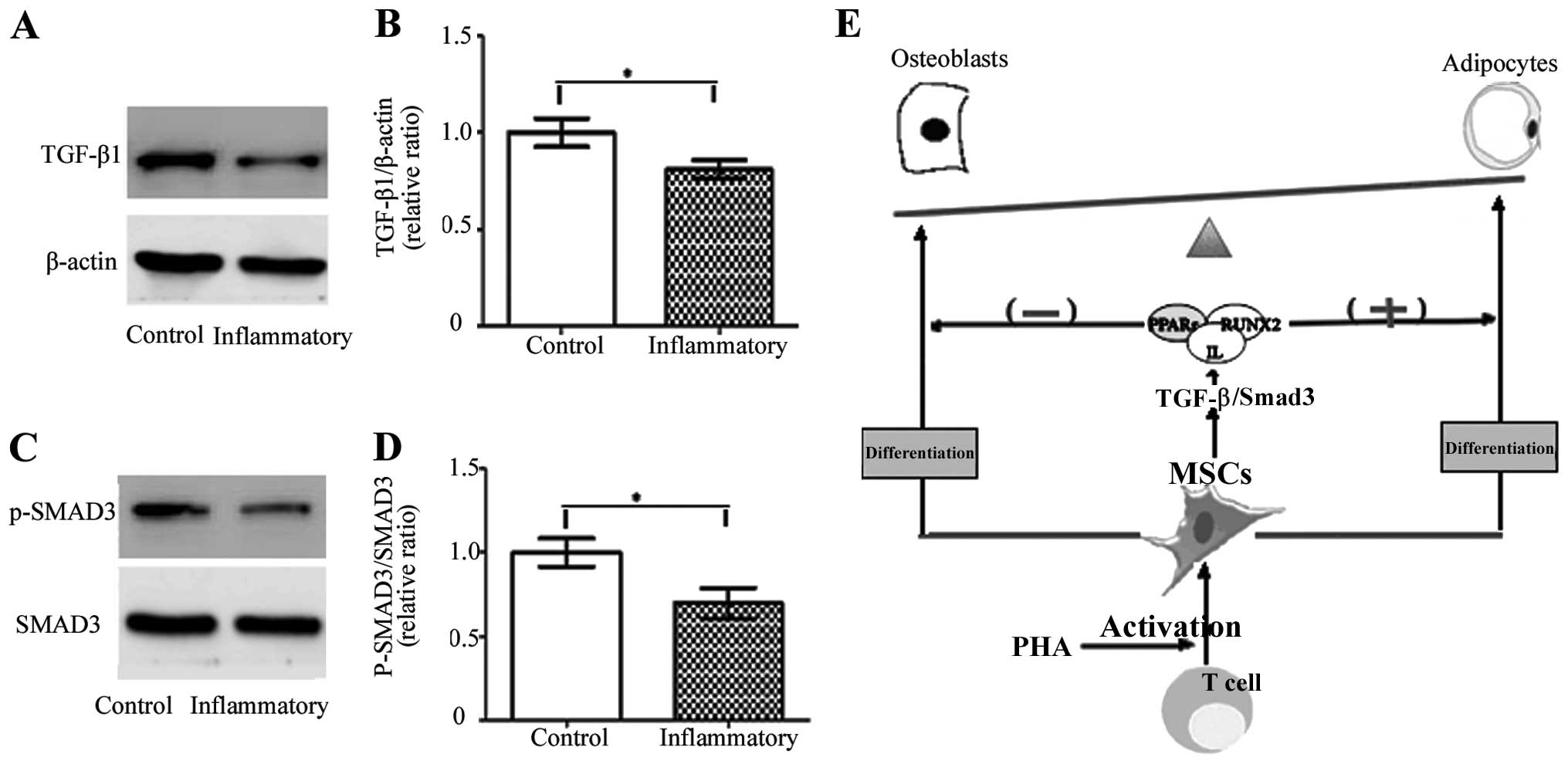
TGF-β/Smad3 signaling
To evaluate the effect of T cell treatment on the
TGF-β/smad pathway, the TGF-β1 level was detected by western
blotting. The expression of the TGF-β1 gene in MSCs decreased
significantly after inflammatory stimulation with activated T cells
at 6 h, compared to the control group. In addition, the gene
expression of Smad3 and the phosphorylation levels of the MSC
culture in the absence or presence of T cells were investigated by
western blotting. At this time-point, the phosphorylation level of
Smad3 protein declined in the presence of the T cells, when
compared to control group. Therefore, T cell treatment inhibited
the expression of TGF-β1, resulting in the weakening of the
TGF-β/Smad3 pathway and enhancing the adipogenic differentiation of
MSCs (Fig. 7).
Discussion
Mesenchymal stem cells (MSCs) have chemotactic
ability and appear to migrate to sites of inflammation (11), where they appear to play an active
role in tissue remodeling. MSCs are generally isolated from an
aspirate of bone marrow; in addition, MSCs have been isolated from
bone marrow, periosteum, trabecular bone, adipose tissue, synovium,
skeletal muscle and deciduous teeth (12). Trabecular bone-derived mesenchymal
stem cells (MSCs) are multipotent cells, which can differentiate
into a number of different types of cells, including osteocytes,
adipocytes, chondrocytes, myocytes and neurocytes (1,2).
Differentiation of MSCs into different lineages of cells is
strictly regulated by various instructive signals, and alteration
or malfunction of this regulation results in pathological
consequences, such as osteoporosis or a high bone mass phenotype
(13,14). In the present study, histological
and immunohistochemistry methods were used to confirm the
multipotential characteristics of the trabecular bone-derived
cells. Oil Red O staining and anti-FABP-4 staining showed
trabecular bone-derived MSCs had adipogenic differentiation
potential; Alizarin Red S staining and anti-osteocalcin staining
showed osteogenic differentiation capability; and chondrogenesis of
MSCs was evidenced by anti-aggrecan fluorescent staining.
Therefore, the results showed that MSCs derived from trabecular
bone were multipotent stem cells; that is, they can give rise to
diverse cell types.
Another property of MSCs is the ability to reduce
the proliferation of lymphocytes of various types (15,16).
For example, BM-MSCs have been reported to impair the proliferation
of activated T cells (17). In
contrast, activated T cells do affect the proliferation of in
vitro cultured MSCs, but this depends on the concentration of
the cells. In the present study, we examined the effects of various
concentrations of activated T cells on the proliferation of MSCs by
MST assay. The results showed that activated T cells had no effect
on the proliferation of MSCs at low concentrations, but higher
concentrations of activated T cells significantly increased the
proliferation of MSCs, particularly at the ratios of 10:1 and 100:1
(T cells/MSCs; P<0.01). Therefore, in order to exclude effects
of an increase in the proliferation of precursors on the
differential outcome of MSCs; in subsequent experiments, T cells at
a lower concentration were added to the cultures after MSCs reached
90% confluency.
Furthermore, one of the undisputed features of MSCs
is their ability to produce a wide variety of chemokines and
cytokine receptors, including those for tumor necrosis factor α
(TNF-α), interleukin (IL)-10, IL-6 and interferon γ (INFγ). The
cytokine IL-10, which is produced by various immune cells, in
particular monocytes/macrophages and T cell subsets, has a crucial
role in limiting the inflammatory response caused by activated T
cells (18). MSCs are also known to
produce high levels of IL-10 (19).
What is more, MSCs constitutively expressed mRNA of IL-6, which
acts as a predominantly pro-inflammatory cytokine. IL6 is produced
by undifferentiated dividing MSCs, and the secretory pathway of
MSCs is mediated through the activation of p38 MAPK (20). In addition, excessive release of
INFγ is associated with the pathogenesis of chronic inflammatory
and autoimmune diseases. Previous research showed that INFγ
possesses both pro-inflammatory and anti-inflammatory effects
depending on the pathophysiological conditions. INFγ is
pro-inflammatory in mouse models of diabetes, thyroiditis and
myasthenia gravis, but anti-inflammatory in models such as
experimental autoimmune encephalomyelitis, uveitis and
collagen-induced arthritis (21).
Both MSCs and T cells express inflammatory
cytokines, thus ELISA methods cannot be used directly to measure
the expression levels of cytokines secreted by MSCs. Alternatively,
the detection of mRNA expression was used. The expression of
inflammatory genes was regulated in MSCs exposed to PHA-activated T
cells. According to the results, IL-6 (pro-inflammatory gene) was
significantly upregulated, while IL-10 and INFγ (anti-inflammatory
genes) were significantly downregulated. This indicated that the
inflammatory microenvironment induced by activated T cells could
increase the expression of proinflammatory genes but inhibited the
expression of anti-inflammatory genes, leading to an excessive
inflammatory microenvironment. Actually, these inflammatory factors
not only attribute to inflammatory response but also play an
important role in the differentiation of MSCs. Previous studies
have shown that MSCs cultured in OS differentiated along the
osteogenic lineage and downregulated protein and mRNA levels of
IL-6 (22,23). Our results are consistent with the
findings of the adipogenic differentiation of MSCs following
upregulated mRNA levels of IL-6.
The balance between peroxisome
proliferator-activated receptor-γ (PPARγ) and Runt-related
transcription factor 2 (RUNX2) leads to MSC differentiating into
adipocytes or osteoblasts, respectively. PPARγ is a member of the
nuclear receptor superfamily of ligand-activated transcriptional
factors, which act as a key regulator of adipogenesis. PPARγ is
commonly termed the master regulator of adipogenesis; no factor has
yet been identified that can induce normal adipogenesis in the
absence of PPARγ (24). In
contrast, RUNX2 promotes osteogenesis but inhibits adipogenesis of
MSCs. These master regulators of different lineages are expressed
at low levels in undifferentiated cells, maintaining the
differentiation potential of MSCs (25,26).
In the experiment, expression of PPARγ and RUNX2 genes was detected
by qRT-PCR and western blot analyses. The results showed that
PHA-activated T cells significantly upregulated the expression of
PPARγ (adipo-specific gene) in MSCs at both the RNA and protein
levels. However, the expression of RUNX2 was altered at the RNA
level but not at the protein level. qRT-PCR showed that the
expression of RUNX2 was significantly downregulated in MSCs treated
with activated T cells. Whether treated or not with activated T
cells and inflammation, RUNX2 protein expressed by MSCs remained at
or below the limits of detection for the duration of the
experiment. Thus, it was difficult to compare the expression levels
between the groups. According to the results, activated T cells and
inflammation promoted adipogenesis in MSCs and it may play a role
in the early stage of MSC osteogenesis.
To carry out the specialized functions of T cell
activation and inflammation, the adipogenic differentiation of MSCs
was further verified using histological and immunohistochemical
staining. Fatty acid binding protein 4 (FABP4), also called aP2
(adipocyte protein 2), has a high affinity for a variety of fatty
acids and facilitates their storage, trafficking and solubilization
(27). FABP4 has been used as a
marker (specific gene) to follow the differentiation of adipocytes.
The expression of FABP4 can be induced by fatty acids, likely
through changes in PPARγ expression or activity, both at the
transcriptional and post-transcriptional level (28,29).
In accordance with its stimulatory effect on PPARγ expression, we
found that PHA-activated T cells upregulated the expression of
FABP4 in MSCs, as detected by fluorescence immunohistochemistry. In
addition, the increase in FABP4 expression by activated T cells was
consistent with the observed increase in Oil Red O staining.
Therefore, these studies confirmed once again that PHA-activated T
cells promoted MSC adipogenesis, by increasing the expression of
key adipogenic genes.
Whether or not T cell activation and inflammation
have an effect at later stages of osteogenic differentiation from
MSCs was investigated. The osteogenic differentiation of MSCs was
evaluated by the expression of osteocalcin, mineralization and
alkaline phosphatase (ALP) activity. According to the results, it
had no obvious effect on osteogenic differentiation from MSCs.
Osteocalcin (bone gla-protein), secreted by osteoblasts, is
generally regarded as an osteoblast-specific marker (30). The cells showed low-level expression
of osteocalcin and mild mineralization in both groups, as evidenced
by Alizarin Red S staining and fluorescence immunohistochemical
staining. The findings were consistent with the protein expression
of RUNX2. No significance was observed between the groups. Alkaline
phosphatase (ALP) activity is another marker for osteoblast
differentiation (31). Under
osteogenic conditions (OS), pNPP assay revealed that activated T
cells had no significant effect on ALP activity on the 14th and
21st day of osteogenic differentiation. Totally, these results
indicated that PHA-activated T cells promote adipogenesis without
affecting the osteogenesis of MSCs.
Growth factors such as transforming growth factor-β1
(TGF-β1) are known to inhibit adipocyte differentiation in
vitro. MSCs can produce significant levels of transforming
growth factor-β1 (TGF-β1) which can be further enhanced by
anti-inflammatory cytokines (32).
Moreover, Smad proteins play a key role in the regulation of TGF-β
signaling and Smad3 is the central intracellular mediator of TGFβ
signaling. After activation of Smads, the effectors of TGFβ
signaling result in Smad translocation from the cytoplasm into the
nucleus where they act as transcriptional comodulators to regulate
target gene expression (33). The
TGF-β/Smad3 signaling pathway was found to play a key role in
adipogenesis, which inhibited adipogenesis independent from the Wnt
and β-catenin pathway (34). TGF-β
inhibits adipocyte differentiation. Previous results indicate that
endogenous TGF-β signaling regulates the rate of adipogenesis, and
that Smad3 has distinct functions in this endogenous control of
differentiation (35). TGF-β
targets the transcription factor cascade upstream of PPARγ, and the
inhibition of adipogenesis by TGF-β was accompanied by reduced mRNA
and protein levels of PPARγ. In the present study, western blot
analysis confirmed that T cell treatment inhibited the protein
expression of TGF-β1 and the phosphorylation of Smad3, resulting in
a weakening of the TGF-β/Smad pathway which enhanced the adipogenic
differentiation of MSCs (Fig.
7).
In conclusion, we demonstrated the effects of T cell
activation and inflammation on osteoblast and adipocyte
differentiation of MSCs. The results showed that PHA-activated T
cells upregulated the expression of adipocyte-specific genes and
led to adipogenic differentiation, possibly due to gene expression
of PPARγ which plays a more decisive role in the differentiation of
MSCs exposed to activated T cells through TGF-β/Smad3 signaling.
This may explain why we observed no obvious effect of PHA-activated
T cells on osteogenic differentiation of MSCs, but it is
complicated. Additionally, there are some limitations to the
current research that merit further study. First, T cells, even
activated T cells, normally have a short lifespan, which may have
partially influenced the experimental outcomes. Thus, in future
experiments, a constant supply of T cells will be necessary to
observe the effects on MSC differentiation. Second, the present
study mainly focused on the downstream regulatory events, and the
precise molecular mechanisms and signaling pathways are still not
clear. Thus, further study is needed to investigate the upstream
regulatory mechanisms, particularly using knockdown techniques to
elucidate the mechanism involved in the differential gene
expression. In spite of these limitations of the present study,
further investigation of the individual effects of activated T
cells and inflammation on MSC differentiation in vivo is
warranted.
Acknowledgements
The authors appreciate the help of the Osteonecrosis
Research Team of Union Hospital, Tongji Medical College, Wuhan,
China. The present study was supported by the National Natural
Science Foundation of China (no. 81201393).
References
|
1
|
Nuttall ME, Patton AJ, Olivera DL, Nadeau
DP and Gowen M: Human trabecular bone cells are able to express
both osteoblastic and adipocytic phenotype: implications for
osteopenic disorders. J Bone Miner Res. 13:371–382. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sottile V, Halleux C, Bassilana F, Keller
H and Seuwen K: Stem cell characteristics of human trabecular
bone-derived cells. Bone. 30:699–704. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lepperdinger G: Inflammation and
mesenchymal stem cell aging. Curr Opin Immunol. 23:518–524. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Garcia-Olmo D, Garcia-Arranz M, Garcia LG,
Cuellar ES, Blanco IF, Prianes LA, Montes JA, Pinto FL, Marcos DH
and Garcia-Sancho L: Autologous stem cell transplantation for
treatment of rectovaginal fistula in perianal Crohn’s disease: a
new cell-based therapy. Int J Colorectal Dis. 18:451–454.
2003.PubMed/NCBI
|
|
5
|
Le Blanc K, Frassoni F, Ball L, Locatelli
F, Roelofs H, Lewis I, Lanino E, Sundberg B, Bernardo ME, Remberger
M, Dini G, Egeler RM, Bacigalupo A, Fibbe W and Ringden O:
Mesenchymal stem cells for treatment of steroid-resistant, severe,
acute graft-versus-host disease: a phase II study. Lancet.
371:1579–1586. 2008.
|
|
6
|
Gonzalez-Rey E, Gonzalez MA, Varela N,
O’Valle F, Hernandez-Cortes P, Rico L, Buscher D and Delgado M:
Human adipose-derived mesenchymal stem cells reduce inflammatory
and T cell responses and induce regulatory T cells in vitro in
rheumatoid arthritis. Ann Rheum Dis. 69:241–248. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gerdoni E, Gallo B, Casazza S, Musio S,
Bonanni I, Pedemonte E, Mantegazza R, Frassoni F, Mancardi G,
Pedotti R and Uccelli A: Mesenchymal stem cells effectively
modulate pathogenic immune response in experimental autoimmune
encephalomyelitis. Ann Neurol. 61:219–227. 2007. View Article : Google Scholar
|
|
8
|
Tabassam FH, Umehara H and Domae N: Beta
2-integrin, LFA-1-mediated p125FAK activation. J Osaka Dent Univ.
33:43–51. 1999.PubMed/NCBI
|
|
9
|
Schmitz ML, Bacher S and Dienz O: NF-κB
activation pathways induced by T cell costimulation. FASEB J.
17:2187–2193. 2003.
|
|
10
|
Russo C, Indiveri F, Quaranta V, Molinaro
GA, Pellegrino MA and Ferrone S: Stimulation of human T lymphocytes
by PHA-activated autologous T lymphocytes: analysis of the role of
Ia-like antigens with monoclonal antibodies. Immunogenetics.
12:267–274. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang L, Li Y, Chen X, Chen J, Gautam SC,
Xu Y and Chopp M: MCP-1, MIP-1, IL-8 and ischemic cerebral tissue
enhance human bone marrow stromal cell migration in interface
culture. Hematology. 7:113–117. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Barry FP and Murphy JM: Mesenchymal stem
cells: clinical applications and biological characterization. Int J
Biochem Cell Biol. 36:568–584. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Duque G: Bone and fat connection in aging
bone. Curr Opin Rheumatol. 20:429–434. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang J, Zhao L, Xing L and Chen D:
MicroRNA-204 regulates Runx2 protein expression and mesenchymal
progenitor cell differentiation. Stem Cells. 28:357–364.
2010.PubMed/NCBI
|
|
15
|
Le Blanc K: Immunomodulatory effects of
fetal and adult mesenchymal stem cells. Cytotherapy. 5:485–489.
2003.PubMed/NCBI
|
|
16
|
Krampera M, Glennie S, Dyson J, Scott D,
Laylor R, Simpson E and Dazzi F: Bone marrow mesenchymal stem cells
inhibit the response of naive and memory antigen-specific T cells
to their cognate peptide. Blood. 101:3722–3729. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bocelli-Tyndall C, Bracci L, Spagnoli G,
Braccini A, Bouchenaki M, Ceredig R, Pistoia V, Martin I and
Tyndall A: Bone marrow mesenchymal stromal cells (BM-MSCs) from
healthy donors and auto-immune disease patients reduce the
proliferation of autologous- and allogeneic-stimulated lymphocytes
in vitro. Rheumatology. 46:403–408. 2007. View Article : Google Scholar
|
|
18
|
Scapini P, Lamagna C, Hu Y, Lee K, Tang Q,
DeFranco AL and Lowell CA: B cell-derived IL-10 suppresses
inflammatory disease in Lyn-deficient mice. Proc Natl Acad Sci USA.
108:E823–E832. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Oh JY, Kim MK, Shin MS, Lee HJ, Ko JH, Wee
WR and Lee JH: The anti-inflammatory and anti-angiogenic role of
mesenchymal stem cells in corneal wound healing following chemical
injury. Stem Cells. 26:1047–1055. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yew TL, Hung YT, Li HY, Chen HW, Chen LL,
Tsai KS, Chiou SH, Chao KC, Huang TF, Chen HL and Hung SC:
Enhancement of wound healing by human multipotent stromal cell
conditioned medium: the paracrine factors and p38 MAPK activation.
Cell Transplant. 20:693–706. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kelchtermans H, Billiau A and Matthys P:
How interferon-gamma keeps autoimmune diseases in check. Trends
Immunol. 29:479–486. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Majumdar MK, Thiede MA, Haynesworth SE,
Bruder SP and Gerson SL: Human marrow-derived mesenchymal stem
cells (MSCs) express hematopoietic cytokines and support long-term
hematopoiesis when differentiated toward stromal and osteogenic
lineages. J Hematother Stem Cell Res. 9:841–848. 2000. View Article : Google Scholar
|
|
23
|
Pricola KL, Kuhn NZ, Haleem-Smith H, Song
Y and Tuan RS: Interleukin-6 maintains bone marrow-derived
mesenchymal stem cell stemness by an ERK1/2-dependent mechanism. J
Cell Biochem. 108:577–588. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kawai M and Rosen CJ: PPARγ: a circadian
transcription factor in adipogenesis and osteogenesis. Nat Rev
Endocrinol. 6:629–636. 2010.
|
|
25
|
Bennett CN, Longo KA, Wright WS, Suva LJ,
Lane TF, Hankenson KD and MacDougald OA: Regulation of
osteoblastogenesis and bone mass by Wnt10b. Proc Natl Acad Sci USA.
102:3324–3329. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rosen ED and MacDougald OA: Adipocyte
differentiation from the inside out. Nat Rev Mol Cell Biol.
7:885–896. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Reese-Wagoner A, Thompson J and Banaszak
L: Structural properties of the adipocyte lipid binding protein.
Biochim Biophys Acta. 1441:106–116. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Distel RJ, Robinson GS and Spiegelman BM:
Fatty acid regulation of gene expression. Transcriptional and
post-transcriptional mechanisms. J Biol Chem. 267:5937–5941.
1992.PubMed/NCBI
|
|
29
|
Platt ID and El-Sohemy A: Regulation of
osteoblast and adipocyte differentiation from human mesenchymal
stem cells by conjugated linoleic acid. J Nutr Biochem. 20:956–964.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kitamura S, Ohgushi H, Hirose M, Funaoka
H, Takakura Y and Ito H: Osteogenic differentiation of human bone
marrow-derived mesenchymal cells cultured on alumina ceramics.
Artif Organs. 28:72–82. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
van Dinther M, Visser N, de Gorter DJ,
Doorn J, Goumans MJ, de Boer J and ten Dijke P: ALK2 R206H mutation
linked to fibrodysplasia ossificans progressiva confers
constitutive activity to the BMP type I receptor and sensitizes
mesenchymal cells to BMP-induced osteoblast differentiation and
bone formation. J Bone Miner Res. 25:1208–1215. 2010.
|
|
32
|
Tan X, Weng T, Zhang J, Wang J, Li W, Wan
H, Lan Y, Cheng X, Hou N, Liu H, Ding J, Lin F, Yang R, Gao X, Chen
D and Yang X: Smad4 is required for maintaining normal murine
postnatal bone homeostasis. J Cell Sci. 120:2162–2170. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Roelen BA and Dijke P: Controlling
mesenchymal stem cell differentiation by TGFβ family members. J
Orthop Sci. 8:740–748. 2003.
|
|
34
|
Tsurutani Y, Fujimoto M, Takemoto M,
Irisuna H, Koshizaka M, Onishi S, Ishikawa T, Mezawa M, He P, Honjo
S, Maezawa Y, Saito Y and Yokote K: The roles of transforming
growth factor-β and Smad3 signaling in adipocyte differentiation
and obesity. Biochem Biophys Res Commun. 407:68–73. 2011.
|
|
35
|
Choy L, Skillington J and Derynck R: Roles
of autocrine TGF-β receptor and Smad signaling in adipocyte
differentiation. J Cell Biol. 149:667–682. 2000.
|