Introduction
Signal transducer and activator of transcription 3
(STAT3) is one of the seven members of the Stat protein family that
mediates the actions of many cytokines and growth factors. STAT3
shows constitutive activity in many different types of cancers,
including breast, prostate, head and neck, lung, colon, liver and
pancreatic cancers, and large granular lymphocytic leukemia and
multiple myeloma (1–3). In addition, human tumor xenograft
studies in mice have repeatedly demonstrated that inhibition of
STAT3 signaling results in decreased tumor growth and improved
animal survival by inducing apoptosis in tumor cells, inhibiting
angiogenesis (4), and enhancing
antitumor immune-mediated cytotoxicity (1,5). Thus,
STAT3 has been identified as a potential high-yield target for
pharmaceutical prevention in treating many types of cancers
(6).
Hepatocellular carcinoma (HCC) is the fifth most
common cancer and the third leading cause of cancer-related
mortality worldwide (7). The
diagnosis of HCC is difficult due to the lack of early screening
methods, and treatment is arduous due to its aggressive nature and
the absence of therapeutic targets. Numerous studies regarding
preventative and curative strategies for HCC have been conducted in
recent years, leading to significant discoveries (6,8,9). HCC
patients are found to have high levels of IL-6 that promote the
survival of HCC cells through the upregulation of the STAT3
signaling pathway (10). Thus,
abnormal levels of IL-6 have profound impacts on cancer occurrence,
development and progression. Heightened expression of IL-6 may be
blocked by disruption of the STAT3 pathway that in turn blocks cell
transformation, inhibits angiogenesis and suppresses tumor growth
(11); epigallocatechin-3-gallate
(EGCG) can promote this disruption.
According to epidemiologic studies, the risk of HCC,
along with that of many types of cancers, can be reduced through
tea consumption. Although green tea is a promising dietary source
of chemopreventive and chemoprotective chemicals (12–14),
its mechanism is still not fully understood. However, many reports
have identified EGCG as the ingredient of green tea that
contributes to the tea’s anticancer function.
The purpose of this study was to gain insight into
the molecular mechanism involved in the effects of STAT3. Surface
plasmon resonance (SPR) detection, in silico docking
simulations, MTT assay, FACS-based apoptosis assay, immunoblotting,
and RT-PCR were among the techniques used to validate our findings.
EGCG was found to disrupt Stat3-phosphorylated peptide binding,
inhibit the expression of phosphorylated Stat3 protein as well as
many downstream genes regulated by STAT3, induce HCC cell
apoptosis, and suppress HCC cell growth, by possibly inhibiting the
STAT3 signaling pathway to directly interfere with the Stat3
protein.
Materials and methods
Reagents
EGCG was obtained from Catch Bio-Science &
Technology Ltd. (Jiangsu, China) with a purity of >99.99%. All
cell culture reagents were purchased from Biowest (USA). MTT
[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide]
detection kit, Annexin V-FITC apoptosis detection kit and cell
cycle detection kit were obtained from BestBio (Shanghai, China).
Western blotting antibodies specific to p-Stat3, total-Stat3 and
β-actin were purchased from Cell Signaling Technology, Inc.
(Beverly, MA, USA). Cell-based ELISA kit [human/mouse phospho-Stat3
(Y705) immunoassay] was purchased from R&D Systems
(Minneapolis, MN, USA).
Cell lines
The BEL-7402 and QGY-7703 human HCC cell lines were
provided by Shanghai Institute of Biochemistry and Cell Biology.
They were cultured in RPMI-1640 medium supplemented with 10% (v/v)
fetal bovine serum, 1X penicillin-streptomycin solution in a
humidified 5% CO2 atmosphere at 37°C.
Stat3/pY-peptide binding assay
Stat3 binding assays were performed at 25°C with a
BIAcore 3000 biosensor using 20 mM Tris-buffer pH 8.0 that
contained 2 mM β-mercaptoethanol and 5% DMSO as running buffer.
Phosphorylated and non-phosphorylated control biotinylated
EGFR-derived dodecapeptides based on the sequence surrounding Y1068
were immobilized on a streptavidin-coated sensor chip (BIAcore
Inc., Piscataway, NJ, USA). The binding of Stat3 was conducted in
20 mM Tris-buffer pH 8.0 containing 2 mM β-mercaptoethanol at a
flow rate of 10 μl/min for 1–2 min. Aliquots of Stat3 at 500 nM
were premixed with compound to achieve a final concentration of
1–1,000 μM, and incubated at 4°C prior to being injected onto the
sensor chip. The chip was regenerated by injecting 10 μl of 100 mM
glycine at pH 1.5 after each sample injection. A control (Stat3
with DMSO but without compound) was run at the beginning and the
end of each cycle (40 sample injections in total) to maintain the
integrity of the sensor chip throughout the cycle run. The average
of the 2 controls was normalized to 100% and used to evaluate the
effect of each compound on Stat3 binding. Responses were normalized
by dividing the value at 2 min by the response obtained in the
absence of compounds at 2 min and multiplying by 100.
IC50 values were determined by plotting the percentage
of the maximum response as a function of the log concentration of
the compound, and fitting the experimental points to a competitive
binding model using a four-parameter logistic equation: R =
Rhigh − (Rhigh − Rlow)/(1 +
conc/A1)A2, where R is the percent response
at the inhibitor concentration; Rhigh is the percent
response with no compound; Rlow is the percent response
at the highest compound concentration; A2 is the fitting
parameter (slope); and A1 is the IC50
(BIAevaluation software version 4.1).
Molecular docking between EGCG and
Stat3
Both molecular structures of Stat3 and EGCG were
retrieved from the Protein Data Bank (PDB Code: 1BG1 and ENG5) and
optimized in the implicit solvent for docking preparation. The
docking region mainly constructed by the residues of Arg-609 and
K-591 was localized based on the interface between the STAT3 SH2
domain and the phosphopeptide. The docking procedure was executed
using CHARMm force field on the CDOCKER module platform in
Discovery Studio (Accelrys Inc.). The conformation search space was
limited to a spherical region with a center of 104.8, 74.3, 63.3
and a radius of 13 Å. The other parameters were determined based on
the default setting of the module, with a grid extension of 8.0.
The ligand partial charge method was performed by CHARMm. Ten top
hits were obtained from the docking simulation. Simulated annealing
method was employed for the final conformation treatment (the
system was heated to 700 K with 2,000 steps and then cooled to 300
K with 5,000 steps). Finally, the best conformation was selected as
the analysis object according to the values of the scores.
Cell proliferation assay
Cell proliferation was determined using MTT assay
according to the manufacturer’s instructions. Briefly, BEL-7402 and
QGY-7703 cells were then seeded into 96-well plates at a density of
5×103/well (100 μl). After 24 h, the indicated
concentrations of EGCG were added. After incubation for 24 and 48
h, respectively, cells were washed twice with PBS. Ten microliters
of MTT medium was then added into each well, at which time cells
were incubated for another 3 h. The medium was removed, and 150 μl
of dissolution was added into each well. The plate was gently
rotated on an orbital shaker for 10 min to dissolve the precipitate
completely. The absorbance was detected at 492 nm with a microplate
reader.
Flow cytometry and detection of
apoptosis
QGY-7703 cells were treated with EGCG in complete
medium for 48 h as previously described. Following treatment, the
cells were harvested by trypsin (not containing EDTA) and rinsed
twice with PBS at 4°C. Cells were then resuspended in 1X Annexin V
binding buffer. Five microliters of Annexin V-FITC solution was
added to each tube. All tubes were incubated for 15 min at 4°C in
darkness. Fifteen microliters of PI solution was added to each
tube. All tubes were incubated for another 5 min at 4°C in
darkness. Cells were then analyzed using flow cytometry (Accuri C6;
BD Biosciences, USA).
Western blotting and ELISA
To detect protein expression and modification in
response to treatment with EGCG, HCC cells, which were treated with
various concentrations of EGCG, were plated onto 6-well plates at a
density of 2×105 cells/ml. After incubation for 24 h,
cells were lysed in cold RIPA lysis buffer. Total protein was
extracted with high-salt buffer (0.5% sodium deoxycholate, 1% SDS,
1 mM sodium orthovanadate, 1 mM β-glycerol phosphate, 1 mM sodium
fluoride, 2.5 mM sodium pyrophosphate) containing a protease
inhibitor cocktail (Roche, Nutley, NJ, USA). Protein samples were
separated by SDS-PAGE, transferred onto PVDF membranes, and
immunoblotted with the corresponding antibodies. The signals were
visualized with Enhanced Chemiluminescence Plus (ECL Plus)
detection system (Dingguo, China). The ELISA procedure was used
according to the manufacturer’s instructions (Cell Signaling
Technology, Inc.). Briefly, 100 μl of 15,000 BEL-7402 cells was
seeded into each well of a black 96-well microplate with a clear
bottom, and incubated overnight at 37°C. Cells were then treated
with different concentrations of EGCG in complete medium for 24 h.
Subsequently, cells were stimulated with interleukin 6 (IL-6) (50
ng/ml) to induce Stat3 phosphorylation. Following the treatments,
cells were treated and tested with the cell-based ELISA kit.
RT-PCR
The BEL-7402 and QGY-7703 cells were treated with
EGCG at 40, 80 and 160 μM for 48 h as previously described. Total
RNAs were extracted from the cells using a commercially available
RNA-Bee isolation kit (Tel-Test). Standard reverse transcription
was performed with 500 ng of total RNA using TIANScriptRT kit
(Tiangen Beijing, China). Reverse transcription-PCR was performed
using 1 μl of cDNA template, 10 pmol of primers, and a PCR premix
(1 U Taq DNA polymerase, 250 mM dNTPs, 10 mM Tris-HCl, 40 mM KCl
and 1.5 mM MgCl2; Tiangen). The following primers were
used in the PCR reactions: Bcl-xL forward, 5′-agctggtggt
tgactttctctc-3′ and reverse, 5′-ccggaagagttcattcactacc-3′; c-Myc
forward, 5′-ctaccctctcaacgacagcag-3′ and reverse, 5′-gtgtgtt
cgcctcttgacatt-3′; VEGF forward, 5′-gcagaatcatcacgaagtggt-3′ and
reverse, 5′-catttgttgtgctgtaggaagc-3′; cyclin D1 forward,
5′-atctacaccgacaactccatcc-3′ and reverse, 5′-gcattttggaga
ggaagtgttc-3′; β-actin forward, 5′-agagctacgagctgcctgctg-3′ and
reverse, 5′-agtacttgcgctcaggagga-3′.
The amplified products obtained from the
β-actin-specific primers served as internal controls. PCR was
conducted using Bio-Rad T-100 (Bio-Rad, Hercules, CA, USA) with a
5-min denaturation step at 94°C; 30 cycles of 94°C for 30 sec, 62°C
for 30 sec and 72°C for 30 sec; and a final extension at 72°C for
10 min. PCR amplifications were verified to be in the linear
range.
Statistical analysis
Data are presented as means ± SD for 3 separate
experiments. One-way ANOVA was employed for statistical analysis
using SPSS 17.0. P<0.05 was considered to indicate a
statistically significant result.
Results
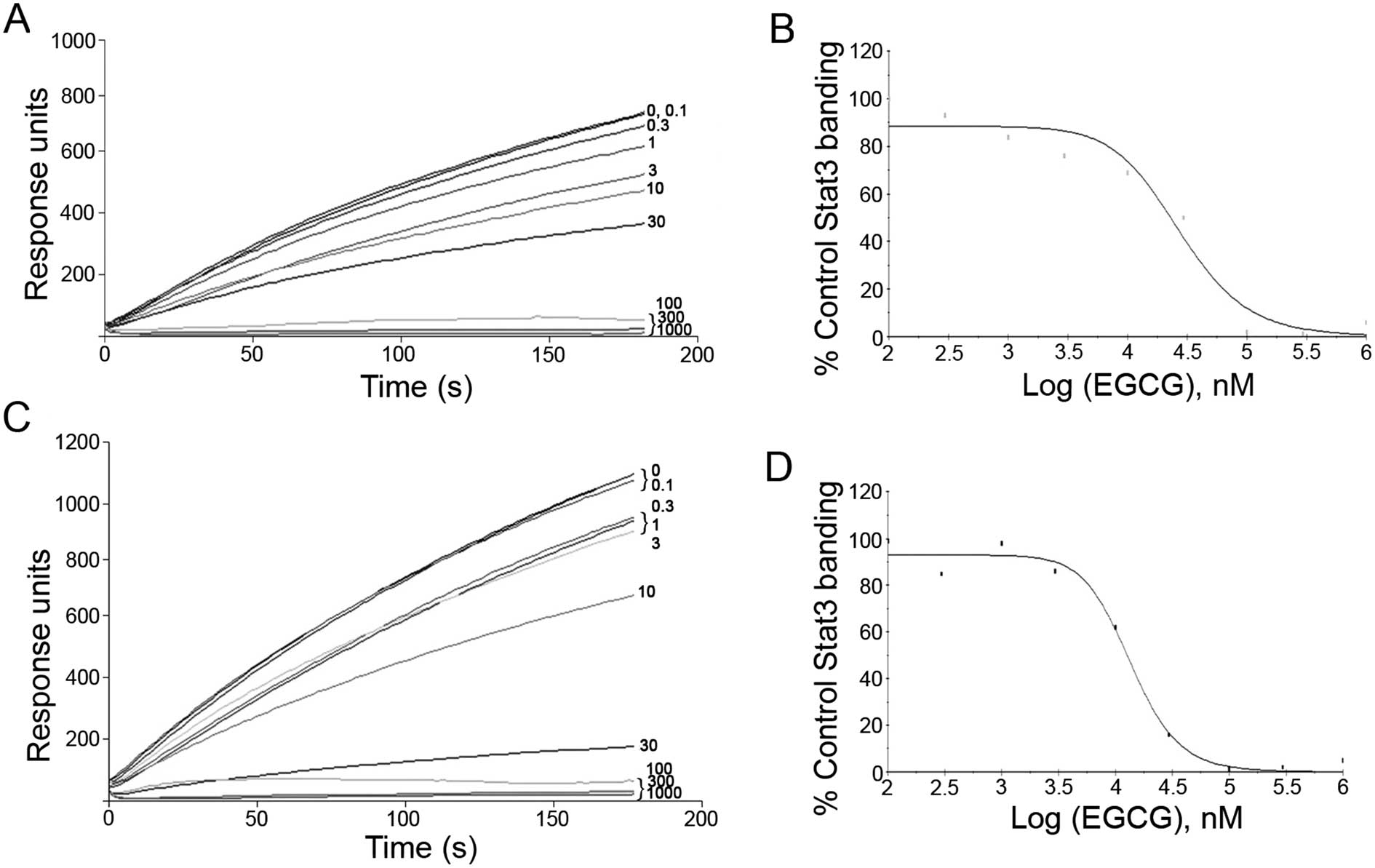
EGCG blocks Stat3 binding to its
phosphopeptide ligand
EGCG was tested for its ability to block Stat3
binding to its phosphopeptide ligand using SPR binding assay
(15). SPR experiments showed that
EGCG was able to directly compete with pY-peptide for binding with
Stat3 at an IC50 value of 10–30 μM (Fig. 1).
Molecular docking between EGCG and
Stat3
Fig. 2A is a
computer model image of EGCG on Stat3. According to the figure,
EGCG is located in a phosphopeptide binding pocket formed by the
STAT3 SH2 fold. Fig. 2B shows the
spatial matching results of EGCG and Stat3; the 3-D structure of
EGCG matches perfectly with the phosphopeptide binding site of
STAT3 SH2. Fig. 2C and D depict the
specific interactions between EGCG and STAT3 SH2. According to
these two figures, the -NH3 group of LYS591 is located between the
2 aromatic rings of EGCG and the 2 hydrogen atoms from the -NH3
group, resulting in the formation of cation-π bonds. The other
hydrogen atom in the -NH3 group forms a hydrogen bond with the -O-
in EGCG. On the other hand, the -C=O group functions as a hydrogen
receptor in EGCG, forming hydrogen bonds with -NH in GLU612 and -OH
in SER613, respectively. While the 2 aromatic rings both function
as hydrogen donors in EGCG, one also forms hydrogen bonds with the
-NH2 group in ARG60, while the other forms hydrogen bonds with the
-NH2 group in Glu638. The other part of EGCG forms a van der Waals
interaction with the phosphopeptide binding pocket of STAT3 SH2
(LYS591, GLU594, ARG609, SER611, GLU612, SER613, THR620, VAL637,
GLU638, PRO639) (Fig. 2C). These
multiple interactions result in a steady locked relationship
(15–18).
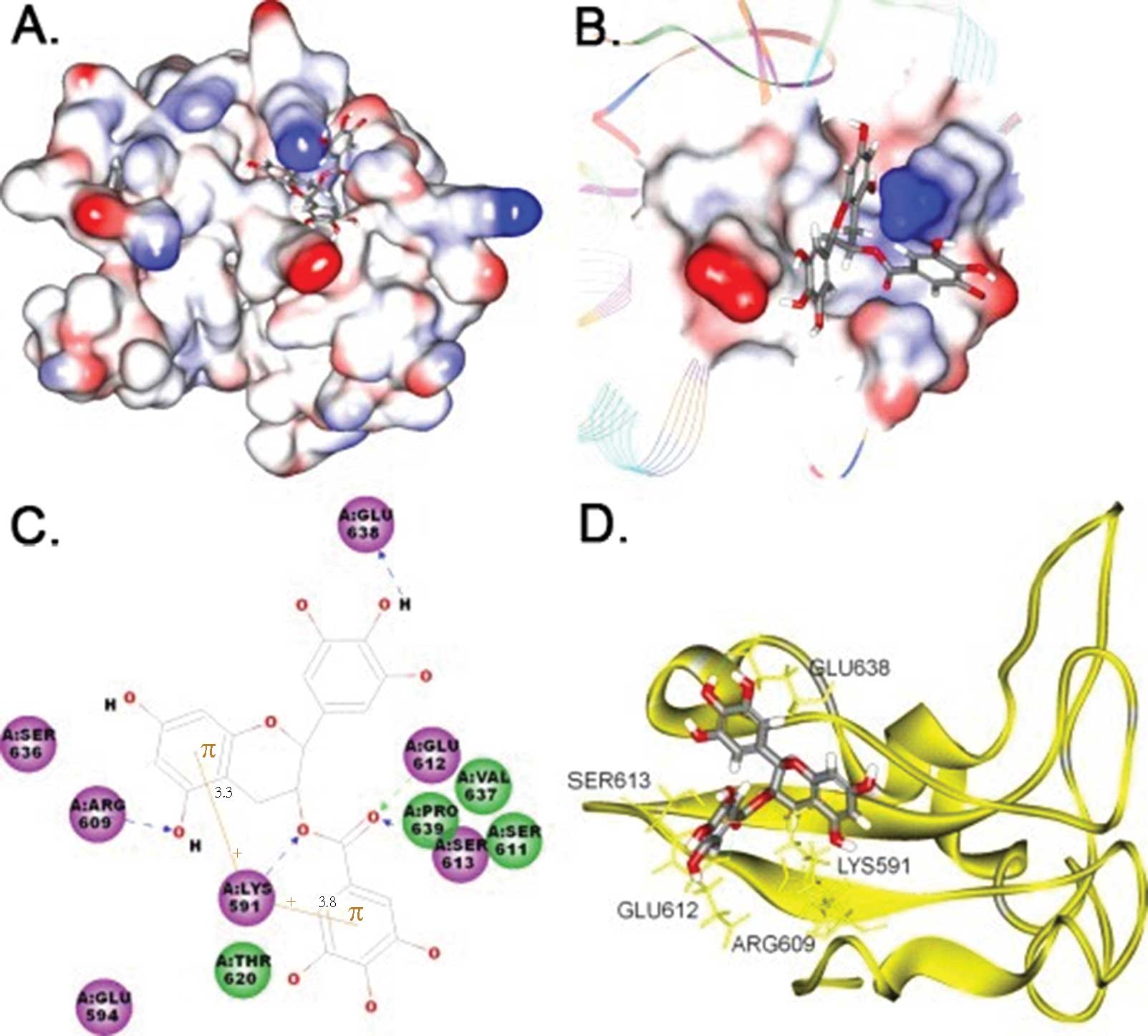 | Figure 2Computer modeling of EGCG bound by the
SH2 domain of STAT3. The 3-D structures and results of computer
docking of EGCG to the STAT3 SH2 domain are shown in A, B and D.
The left side of C shows the 2-D structure. The middle portion of A
and B shows EGCG binding to an electrostatic molecular surface
model of the STAT3 SH2 domain; blue represents areas with
positive-charge; red represents areas with negative-charge and the
stick model depicts EGCG. C is a closer view of this interaction
with hydrogen bonds indicated by dotted lines. Cation-π
interactions are shown in brown color and critical residues that
contribute mainly to the EGCG-Stat3 binding are represented by
circles in green or purple colors, and EGCG molecules are presented
by a line model. In D, STAT3 SH2 skeletal configuration is
presented in a ribbon model, EGCG is represented by stick models
and all atoms of critical residues in STAT3 SH2 domains are
represented in gold color. The carbon, oxygen and hydrogen atoms of
EGCG are represented by silver, red and white color, respectively,
in A, B, C and D. |
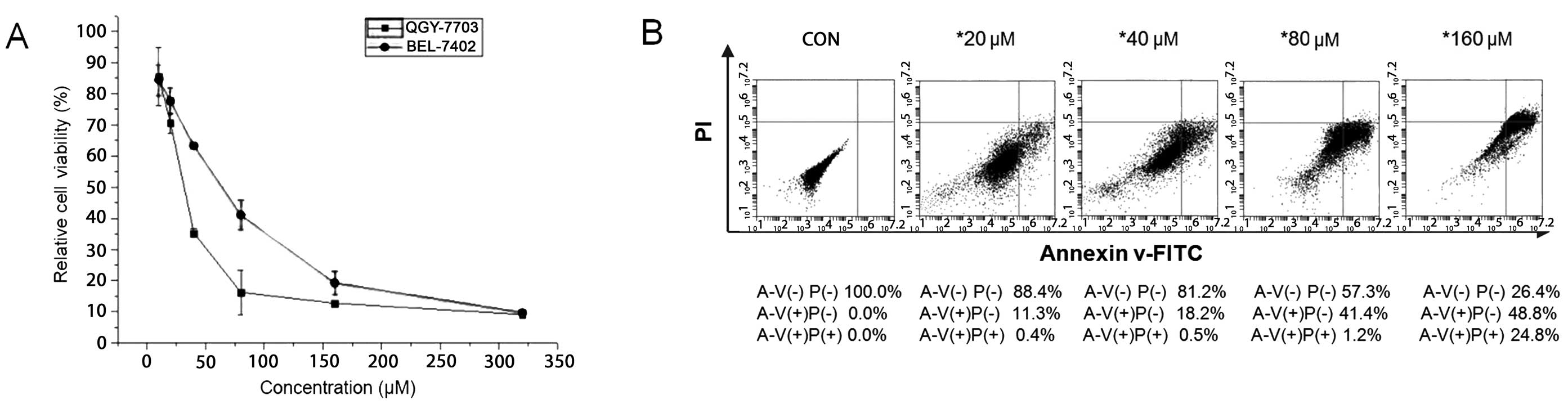
EGCG suppresses BEL-7402 and QGY-7703
cell growth, and induces apoptosis in QGY-7703 cells
To determine the potential cytotoxic and
anti-proliferative effects of EGCG, the human HCC cell lines
BEL-7402 and QGY-7703 were cultured with EGCG at various
concentrations (0–320 μM). Cell viability was then determined by
MTT assay. Results showed that treatment with EGCG led to a
significant dose-dependent inhibition of HCC cell growth in
vitro (Fig. 3A). The half
maximal (50%) inhibitory concentrations (IC50) for
BEL-7402 and QGY-7703 cells were ~55 and 35 μM, respectively.
Induction of cell apoptosis was confirmed by Annexin V-FITC
staining in QGY-7703 cells. Results showed that treatment with EGCG
led to significant dose-dependent apoptosis-inducing effects on HCC
cell growth in vitro. According to Fig. 3B, the upper right quadrant
represents late apoptosis, while the lower right quadrant
represents early apoptosis. Increasing concentrations of EGCG at
20, 40, 80 and 160 μM, respectively, were added to the QGY-7703
cell line for 48 h. As a result, the rates of cell apoptosis were
11.7, 18.7, 42.6 and 73.6%, respectively. Thus, as the
concentration of EGCG increased, the rate of apoptosis of the
QGY-7703 cells also increased. The standard deviations were
calculated based on 3 independent experiments.
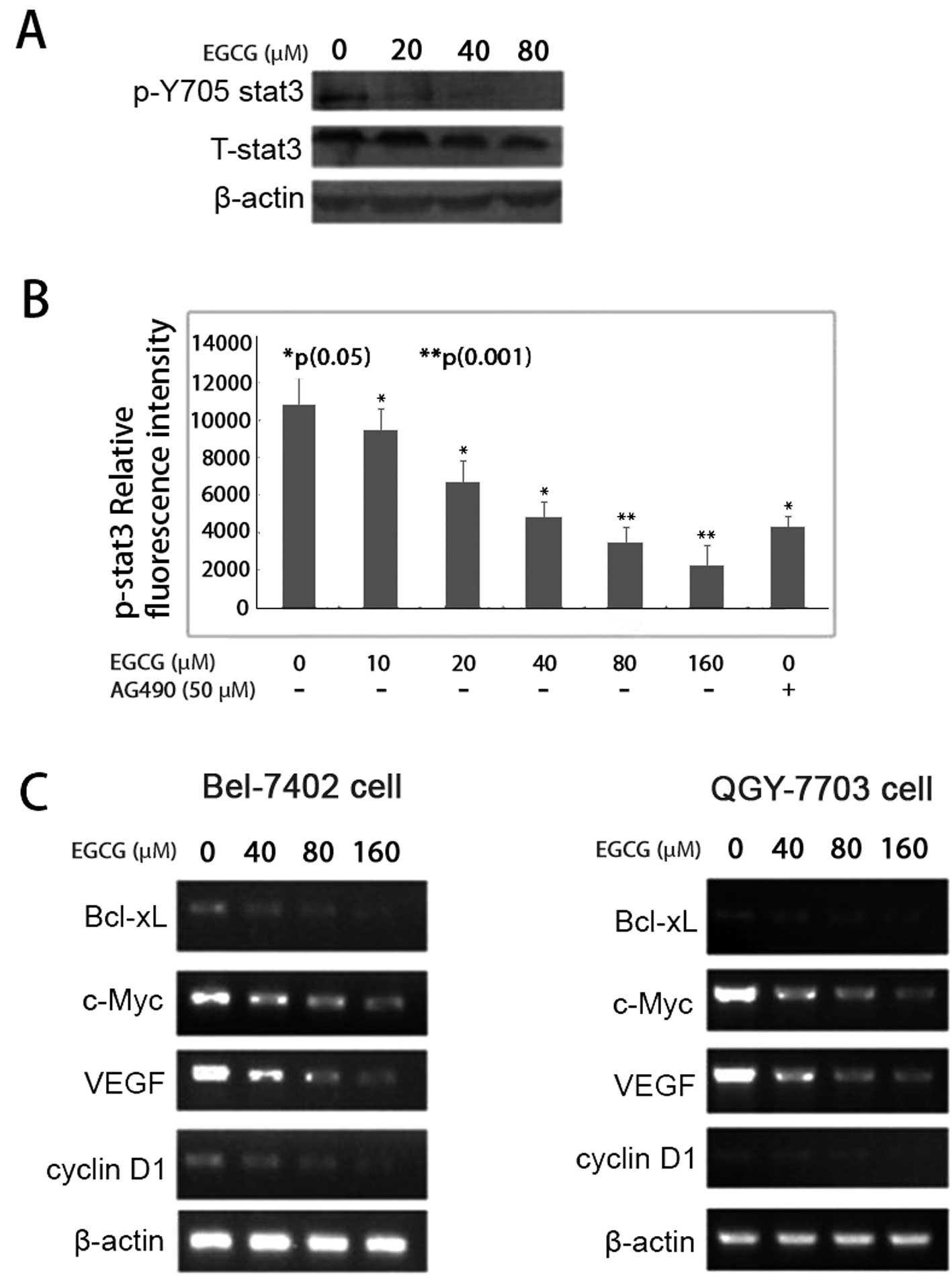
EGCG inhibits IL-6-induced Stat-3
phosphorylation
To examine whether EGCG has inhibitory effects on
IL-6-induced Stat3 phosphorylation, QGY-7703 cells were cultured
and pretreated with different concentrations of EGCG for 48 h, and
were then treated with 50 ng/ml of IL-6 stimulation for 30 min.
After treatment, the phosphorylated Stat3 and total Stat3 were
analyzed by western blotting and cell-based ELISA. EGCG inhibited
Stat3 phosphorylation on tyrosine 705 in a dose-dependent manner
(Fig. 4A). The p-Stat3 relative
florescence intensity was significantly reduced following EGCG
treatment. When QGY-7703 cells were treated with EGCG at
concentrations of 10, 20, 40, 80 and 160 μM, respectively, the
p-Stat3 relative average fluorescence intensities were 7,400,
6,600, 4,500, 3,400 and 2,200, respectively. Statistical analysis
showed a P-value of <0.05 for EGCG at 10, 20 and 40 μM in
relation to their corresponding fluorescence intensity; EGCG at 80
and 160 μM had a P-value of <0.001 in relation to their
corresponding fluorescence intensity (Fig. 4B).
EGCG downregulates the expression of
cancer-related genes
Exposure to EGCG resulted in a dose-dependent
decrease in cyclin D1 mRNA expression in both BEL-7402 and QGY-7703
cells, as demonstrated by RT-PCR analysis. Furthermore, the
expression levels of Bcl-xL, c-Myc and VEGF were also significantly
reduced at the transcriptional levels (Fig. 4C).
Discussion
Tea is one of the most popular beverages in the
world and has been well known to promote good health in numerous
ways for over two thousand years. Daily consumption of tea may
reduce cholesterol and the incidence of heart disease, boost
immunity and benefit human skin. Particularly, tea may lower the
risk of various types of cancers, including gastric, pancreatic and
colorectal, in the human population (19–21).
EGCG, which contributes to more than 40% of the total polyphenol
mixture in tea, plays an essential role in its chemotherapeutic and
chemopreventive effects. In fact, the anti-oxidative activity and
metal chelating functions of EGCG may contribute to the inhibitory
activity of tea against carcinogenesis (22). Additionally, there is considerable
evidence that EGCG has an anticancer nature by modulating the
intracellular signaling network.
To study the mechanism of the inhibitory effects of
EGCG on carcinoma cells, we conducted molecular binding computation
and related experiments. Based on our study from the BIAcore
binding assay in micromoles, EGCG blocked Stat3 binding to its
phosphopeptide ligand on SPR testing. Furthermore, the EGCG
molecule had major interactions with two key residues, R609 and
K591, localized in the STAT3 SH2 domain, which we found through
docking simulation analysis. We then confirmed that EGCG
significantly inhibited carcinoma cell growth in vitro in
two human HCC cell lines, BEL-7402 and QGY-7703, in a
dose-dependent trend by MTT assay. Additionally, EGCG interrupted
Stat3 phosphorylation on tyrosine 705 in a dose-dependent manner as
detected by western blotting and cell-based ELISA immunoblot
testing in micromoles. As shown in Fig.
4B, EGCG at 40 μM had the same effects on p-Stat3
phosphorylation inhibition as the well-known EGFR inhibitor AG490
at 50 μM. HCC cells treated with EGCG exhibited a significant
transcriptional decrease in the expression of many genes related to
cell growth, survival and apoptosis, including Bcl-xL, c-Myc, VEGF
and cyclin D1, as determined by RT-PCR analysis. This in turn led
to HCC cell apoptosis, as demonstrated by flow cytometry.
Our research data support that the anticancer
function of green tea is the result of the inhibition of the STAT3
signaling pathway by EGCG. However, additional studies suggest that
EGCG is not only a multiple effector that regulates cell signaling
such as STAT1 and ERK1/2 (23), but
is also a general binder that binds to STAT1 and other
bio-molecules, including RNA. Based on our conclusion, EGCG is a
STAT3 signaling inhibitor that competitively binds to the STAT3 SH2
domain, contributing to the regulation of the cellular signaling
network and the anticancer effects of green tea. However, further
research is needed before a full understanding of the mechanism of
EGCG in tea is achieved in order to benefit the health of the
general population.
Acknowledgements
We thank Shanghai Institute of Biochemistry and Cell
Biology for contributing the BEL-7402 and QGY-7703 human HCC cell
lines.
References
|
1
|
Darnell JE Jr, Kerr IM and Stark GR:
Jak-STAT pathways and transcriptional activation in response to
IFNs and other extracellular signaling proteins. Science.
264:1415–1421. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Buettner R, Mora LB and Jove R: Activated
STAT signaling in human tumors provides novel molecular targets for
therapeutic intervention. Clin Cancer Res. 8:945–954.
2002.PubMed/NCBI
|
|
3
|
Aggarwal BB, Sethi G, Ahn KS, Sandur SK,
Pandey MK, Kunnumakkara AB, Sung B and Ichikawa H: Targeting
signal-transducer-and-activator-of-transcription-3 for prevention
and therapy of cancer: modern target but ancient solution. Ann NY
Acad Sci. 1091:151–169. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Redell MS and Tweardy DJ: Targeting
transcription factors in cancer: challenges and evolving
strategies. Drug Discov Today Technol. 3:261–267. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Whittaker S, Marais R and Zhu AX: The role
of signaling pathways in the development and treatment of
hepatocellular carcinoma. Oncogene. 29:4989–5005. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Berishaj M, Gao SP, Ahmed S, Leslie K,
Al-Ahmadie H, Gerald WL, Bornmann W and Bromberg JF: Stat3 is
tyrosine-phosphorylated through the interleukin-6/glycoprotein
130/Janus kinase pathway in breast cancer. Breast Cancer Res.
9:R322007. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lin L, Amin R, Gallicano GI, Glasgow E,
Jogunoori W, Jessup JM, Zasloff M, Marshall JL, Shetty K, Johnson
L, Mishra L and He AR: The STAT3 inhibitor NSC 74859 is effective
in hepatocellular cancers with disrupted TGF-β signaling. Oncogene.
28:961–972. 2009.PubMed/NCBI
|
|
8
|
Soresi M, Giannitrapani L, D’Antona F,
Florena AM, La Spada E, Terranova A, Cervello M, D’Alessandro N and
Montalto G: Interleukin-6 and its soluble receptor in patients with
liver cirrhosis and hepatocellular carcinoma. World J
Gastroenterol. 12:2563–2568. 2006.PubMed/NCBI
|
|
9
|
Berasin C, Castillo J, Perugorria MJ,
Latasa MU, Prieto J and Avila MA: Inflammation and liver cancer:
new molecular links. Ann NY Acad Sci. 1155:206–221. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Calvisi DF, Ladu S, Gorden A, Farina M,
Conner EA, Lee JS, Factor VM and Thorgeirsson SS: Ubiquitous
activation of Ras and Jak/Stat pathways in human HCC.
Gastroenterology. 130:1117–1128. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
To KF, Chan MW, Leung WK, Ng EK, Yu J, Bai
AH, Lo AW, Chu SH, Tong JH, Lo KW, Sung JJ and Chan FK:
Constitutional activation of IL-6-mediated JAK/STAT pathway through
hypermethylation of SOCS-1 in human gastric cancer cell line. Br J
Cancer. 91:1335–1341. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li HC, Yashiki S, Sonoda J, Lou H, Ghosh
SK, Byrnes JJ, Lema C, Fujiyoshi T, Karasuyama M and Sonoda S:
Green tea polyphenoles induce apoptosis in vitro in peripheral
blood T lymphocytes of adult T-cell leukemia patients. Jpn J Cancer
Res. 91:34–40. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Park G, Yoon BS, Moon JH, Kim B, Jun EK,
Oh S, Kim H, Song HJ, Noh JY, Oh C and You S: Green tea polyphenol
epigallocatechin-3-gallate suppresses collagen production and
proliferation in keloid fibroblasts via inhibition of the
STAT3-signaling pathway. J Invest Dermatol. 128:2429–2441. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ahn HY, Hadizadeh KR, Seul C, Yun YP,
Vetter H and Sachinidis A: Epigallocathechin-3 gallate selectively
inhibits the PDGF-BB-induced intracellular signaling transduction
pathway in vascular smooth muscle cells and inhibits transformation
of sis-transfected NIH 3T3 fibroblasts and human glioblastoma
cells. Mol Biol Cell. 10:1093–1104. 1999. View Article : Google Scholar
|
|
15
|
Xu X, Kasembeli MM, Jiang X, Tweardy BJ
and Tweardy DJ: Chemical probes that competitively and selectively
inhibit Stat3 activation. PLoS One. 4:e47832009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kasembeli MM, Xu X and Tweardy DJ: SH2
domain binding to phosphopeptide ligands: potential for drug
targeting. Front Biosci. 14:1010–1022. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shao H, Xu X, Jing N and Tweardy DJ:
Unique structural determinants for Stat3 recruitment and activation
by the granulocyte colony-stimulating factor receptor at
phosphotyrosine ligands 704 and 744. J Immunol. 176:2933–2941.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shao H, Xu X, Mastrangelo MA, Jing N, Cook
RG, Legge GB and Tweardy DJ: Structural requirements for signal
transducer and activator of transcription 3 binding to
phosphotyrosine ligands containing the YXXQ motif. J Biol Chem.
279:18967–18973. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Takada M, Nakamura Y, Koizumi T, Toyama H,
Kamigaki T, Suzuki Y, Takeyama Y and Kuroda Y: Suppression of human
pancreatic carcinoma cell growth and invasion by
epigallocatechin-3-gallate. Pancreas. 25:45–48. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu B, Chen H, Zhan W, Wang CY, Cai SR,
Wang Z, Zhang CH and He YL: (−)-Epigallocatechin-3-gallate inhibits
VEGF expression induced by IL-6 via Stat3 in gastric cancer. World
J Gastroenterol. 7:2315–2325. 2011.
|
|
21
|
Yang GY, Liao J, Kim K, Yurkow EJ and Yang
CS: Inhibition of growth and induction of apoptosis in human cancer
cell lines by tea polyphenols. Carcinogenesis. 19:611–616. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang CS, Lambert JD, Hou Z, Ju J, Lu G and
Hao X: Molecular targets for the cancer preventive activity of tea
polyphenols. Mol Carcinog. 45:431–435. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shankar S, Suthakar G and Srivastava RK:
Epigallocatechin-3-gallate inhibits cell cycle and induces
apoptosis in pancreatic cancer. Front Biosci. 12:5039–5051. 2007.
View Article : Google Scholar : PubMed/NCBI
|