Introduction
Currently, resistance to chemotherapy is a major
issue in the clinical treatment of tumors. The mechanism of tumor
resistance to chemotherapy is extremely complex, and high
expression of anti-apoptotic protein Bcl-2 is considered to be a
major reason why tumor cells escape from apoptosis (1–3).
Bcl-2-specific inhibitors can increase the sensitivity of tumor
cells to chemotherapeutics (4–7).
Therefore, exploring the activity and mechanism of Bcl-2 inhibitors
can provide new insights into the treatment of tumors, including
drug-resistant tumors.
Bcl-2 is involved in not only the mitochondrial
apoptotic pathway, but also in autophagy and endoplasmic reticulum
(ER) stress. During tunicamycin- and thapsigargin-induced ER
stress, Bcl-2 was found to mediate the stability of the ER membrane
and to be involved in ER stress-mediated apoptosis (8,9).
HA14-1, a Bcl-2 inhibitor, significantly increased proteasome
inhibitor bortezomib-induced cell death by increasing JNK- and
caspase-4-mediated ER stress-induced apoptosis (10). In addition, Bcl-2, which is located
at the ER, binds to Beclin-1 containing a BH3 region, thereby
inhibiting Beclin-1-dependent autophagy (11–13). A
natural BH3 mimetic was found to release Beclin-1 and then induce
autophagy through inhibition of Bcl-2 or Bcl-XL expression
(14). Although ER stress and
autophagy are two independent response mechanisms in cells, there
is an important link between them. Inhibition of autophagy can
enhance chemotherapeutic effects by upregulating ER stress-mediated
apoptosis (15,16).
S1, a Bcl-2-specific inhibitor, is a BH3-only
protein mimetic (17). S1 has been
proven to induce apoptosis of liver cancer cells and breast cancer
cells via the mitochondrial pathway by interfering with the
interactions between Bcl-2/Bax and Mcl-1/Bak, and thus exhibits
antitumor activity (18,19). Moreover, S1 was shown to induce
autophagy and ER stress in human glioma cells (20).
In the present study, S1 was found to inhibit the
survival of SKOV3 ovarian cancer cells and their related
cisplatin-resistant SKOV-3/DDP cells, and a significantly higher
level of autophagy was detected in S1-treated SKOV3/DDP cells
compared with SKOV3 cells. We also observed that activation of
autophagy delayed S1-mediated apoptosis in SKOV3/DDP cells at early
time points. In addition, we found that although S1 activated
autophagy, it induced apoptosis in drug-resistant tumor cells via
the ER stress-mediated caspase-4 pathway. Therefore, Bcl-2 family
protein targeted therapy is a promising therapeutic strategy for
the treatment of human ovarian cancer.
Materials and methods
Cell culture
SKOV3 human ovarian cancer cells and their related
cisplatin-resistant SKOV3/DDP cells were obtained from the Chinese
Academy of Medical Sciences and Peking Union Medical College. Both
cell lines were cultured at 37°C under 5% CO2 in Roswell
Park Memorial Institute (RPMI)-1640 culture medium (Gibco,
Carlsbad, CA, USA) supplemented with 10% fetal bovine serum
(Invitrogen, Carlsbad, CA, USA). In addition, SKOV3/DDP cells were
maintained in RPMI-1640 medium supplemented with 10% fetal bovine
serum plus 1 μg/ml cisplatin (Sigma-Aldrich, St. Louis, MO, USA) to
maintain their resistance.
Cell viability assays
SKOV3 and SKOV3/DDP cells were plated in
96-multi-well plates at 1×104 cells/well 24 h before
treatment. The cells were then treated with increasing
concentrations of S1 for 12 or 24 h, or with 10 μM S1 for different
time periods. Each treatment was repeated in three wells. The cell
viability was assessed using the MTT colorimetric assay. Briefly,
MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide)
(10 μl; 5 mg/ml in PBS; Sigma-Aldrich) was added and incubated for
4 h. Subsequently, 150 μl of dimethyl sulfoxide was added to
dissolve the formazan crystals. After shaking for 10 min, the
absorbance values were measured at a wavelength of 570 nm using a
microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Western blot analysis
Whole-cell proteins were extracted from the human
ovarian cancer cells using RIPA buffer. After two sonications for
10 sec each on ice, the cells were lysed at 4°C for 45 min. The
cell lysates were centrifuged at 3,000 × g for 15 min, and the
protein concentrations were determined using a protein assay kit
(Bio-Rad Laboratories Hercules, CA, USA). For western blot
analysis, equivalent amounts of proteins (30–90 μg) were separated
by 12% SDS-polyacrylamide gel electrophoresis and transferred onto
nitrocellulose membranes (Whatman, Maidstone, UK). The membranes
were blocked with 5% non-fat dry milk in buffer (10 mM Tris-HCl pH
7.6, 100 mM NaCl and 0.1% Tween-20) for 1 h at room temperature and
then incubated with the relevant primary antibody overnight at 4°C.
The anti-PDI, anti-Beclin-1, anti-GRP78, anti-caspase-4, anti-JNK
and anti-p-JNK antibodies (all used at a 1:200 dilution) were
obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The
anti-β-actin antibody (1:1,000 dilution) was obtained from
Epitomics Inc. (Burlingame, CA, USA). The anti-LC3 antibody (1:500
dilution) was obtained from Abcam Hong Kong Ltd. (Hong Kong,
China). On the following day, the membranes were incubated with a
horseradish peroxidase-conjugated secondary antibody (Thermo Fisher
Scientific, Waltham, MA, USA) at a 1:2,000 dilution for 1 h at room
temperature. The immunoreactive bands were visualized by a
diaminobenzidine (Sigma) coloration method. The reactive bands were
measured with a Tanon GIS gel imager system, and the protein levels
were quantified by densitometry using Quantity One software
(Bio-Rad Laboratories).
Immunofluorescence staining and confocal
laser microscopy
Cells were seeded onto coverslips in 24-well plates
at a density of 5×104 cells/well 24 h before treatment.
After exposure to 10 μM S1 for 0 or 24 h, the cells were fixed with
4% paraformaldehyde for 30 min, stained with the nuclear stain
Hoechst 33342 (2 μg/ml; Sigma-Aldrich) for 2 min, washed with PBS,
and examined using an FV1000 confocal laser microscope (Olympus,
Tokyo, Japan) to reveal the chromatin condensation.
The expression levels of microtubule-associated
protein light chain 3 (LC3) and protein disulfide isomerase (PDI)
were examined by an indirect immunofluorescence method. Cells were
cultured on coverslips overnight, treated with 10 μM S1 for
different time periods, and fixed with 4% paraformaldehyde for 30
min. After permeabilization with 0.1% Triton X-100 for 5 min, the
cells were blocked with bovine serum albumin for 30 min, and
incubated with a primary antibody against LC3 or PDI (1:100
dilution) overnight at 4°C. On the following day, the cells were
incubated with FITC/Texas Red-conjugated secondary antibodies
(1:400 dilution; Santa Cruz Biotechnology) for 1 h, stained with
Hoechst 33342 (2 μg/ml) for 2 min, washed with PBS three times, and
examined using the Olympus FV1000 confocal laser microscope.
Statistical analysis
The data are representative of three independent
experiments performed as triplicate determinations. Statistical
analyses of the data were performed by one-way ANOVA. The Tukey
post-hoc test was used to determine the significance of all
pairwise comparisons of interest. Values of P<0.05 were
considered to indicate statistically significant differences.
Results
S1 inhibits the viability of SKOV3 and
SKOV3/DDP cells
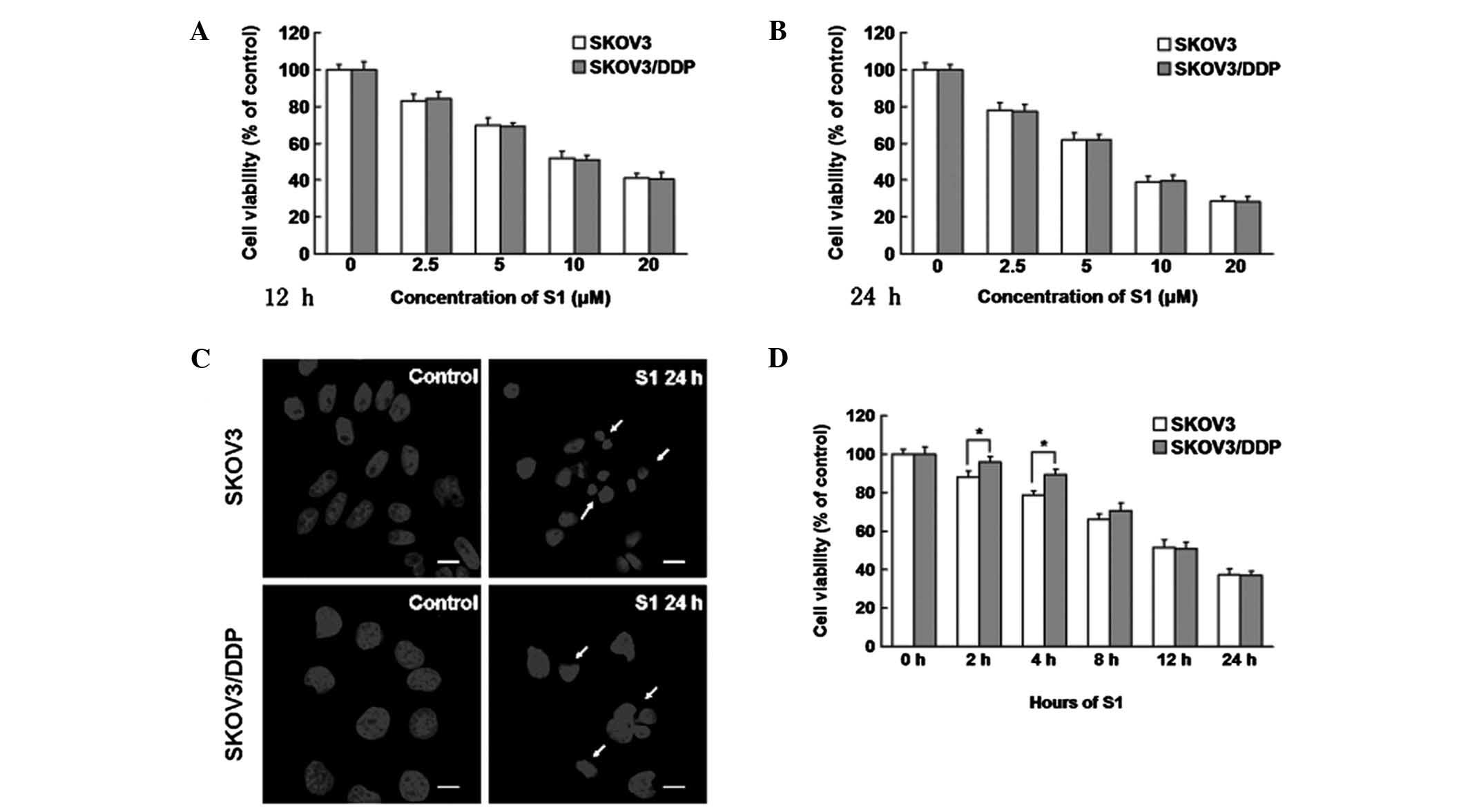
We treated cisplatin-sensitive SKOV3 and
cisplatin-resistant SKOV3/DDP cells with increasing doses of S1 for
12 or 24 h, and examined the growth inhibition using MTT assays. We
found that S1 inhibited the viability of both cell lines (Fig. 1A and B). Based on the MTT assay
results, we examined the apoptotic chromatin condensation by
Hoechst 33342 staining and confocal microscopy. Compared with the
control cells, S1 induced apoptotic chromatin in SKOV3 and
SKOV3/DDP cells (Fig. 1C). After
treatment with 10 μM S1 for different time periods, it was
interesting to note that SKOV3 cells were more sensitive to S1 than
SKOV3/DDP cells at early time points (2 or 4 h) (Fig. 1D), but in the end there was no
difference in cell viability. These findings demonstrate that S1
has time- and dose-dependent effects on SKOV3 and SKOV3/DDP cells,
and that SKOV3 cells are more sensitive at early time points.
S1 induces ER stress in SKOV3 and
SKOV3/DDP cells
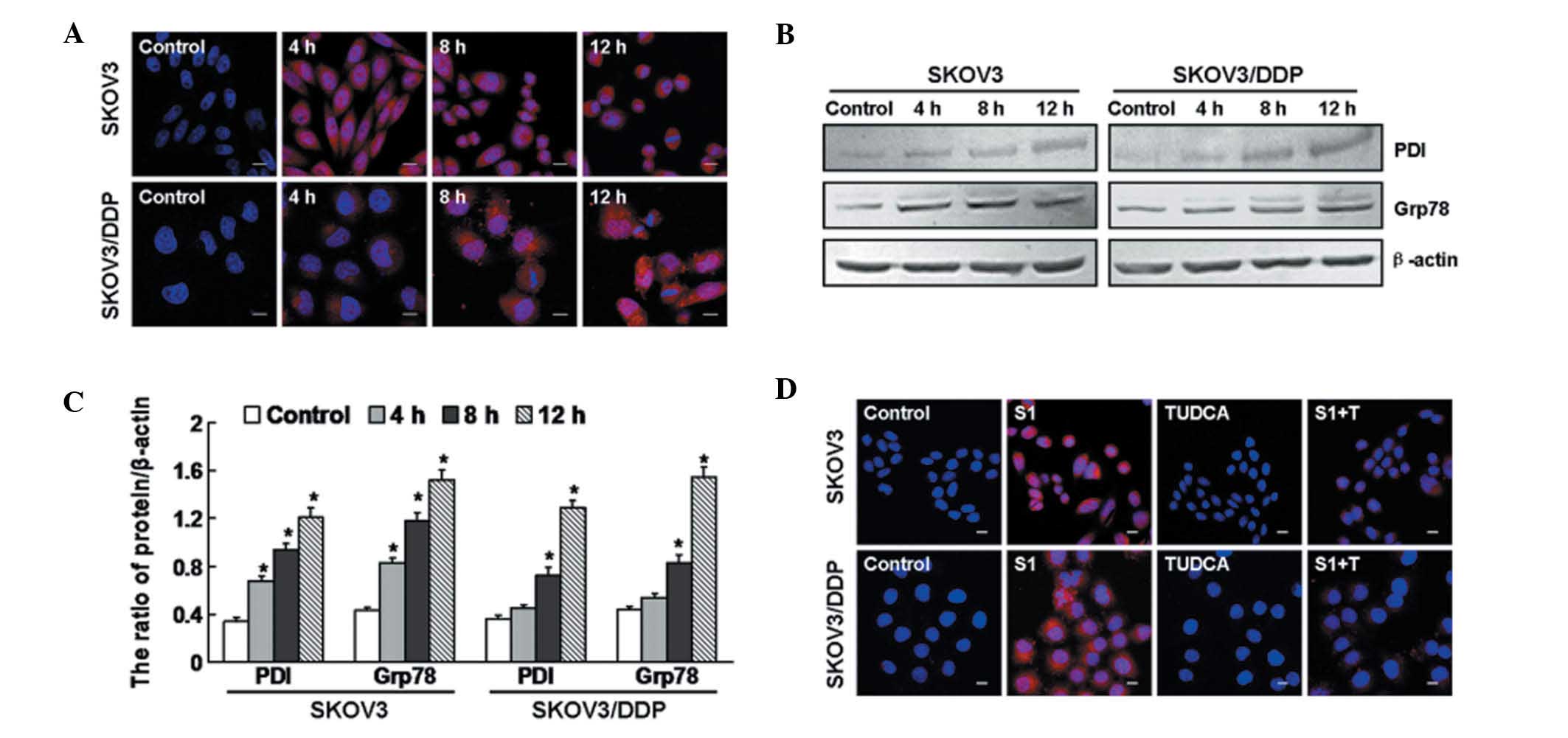
To evaluate whether S1 induces ER stress, we
examined the expression of PDI, an ER-specific protein that
accumulates under ER stress (21).
Using confocal microscopy, we observed that PDI began to accumulate
after 4 h in S1-treated SKOV3 cells, but only at 8 h in S1-treated
SKOV3/DDP cells (Fig. 2A). By
conducting western blot analysis, we found that the expression
levels of PDI and Grp78, an ER chaperone protein (22), were upregulated at 4, 8 and 12 h in
SKOV3 cells, but at 8 and 12 h in SKOV3/DDP cells (Fig. 2B and C). To demonstrate that PDI
accumulation was ER stress-dependent, we treated cells with
tauroursodeoxycholic acid (TUDCA), a known inhibitor of ER stress
(23,24). As shown in Fig. 2D, PDI accumulation was downregulated
at 12 h in both cell lines treated with S1 plus TUDCA, compared
with cells treated with S1 alone. These findings indicate that the
S1 induces ER stress in both SKOV3 and SKOV3/DDP cells.
S1-induced ER stress-associated apoptosis
in SKOV3 and SKOV3/DDP cells
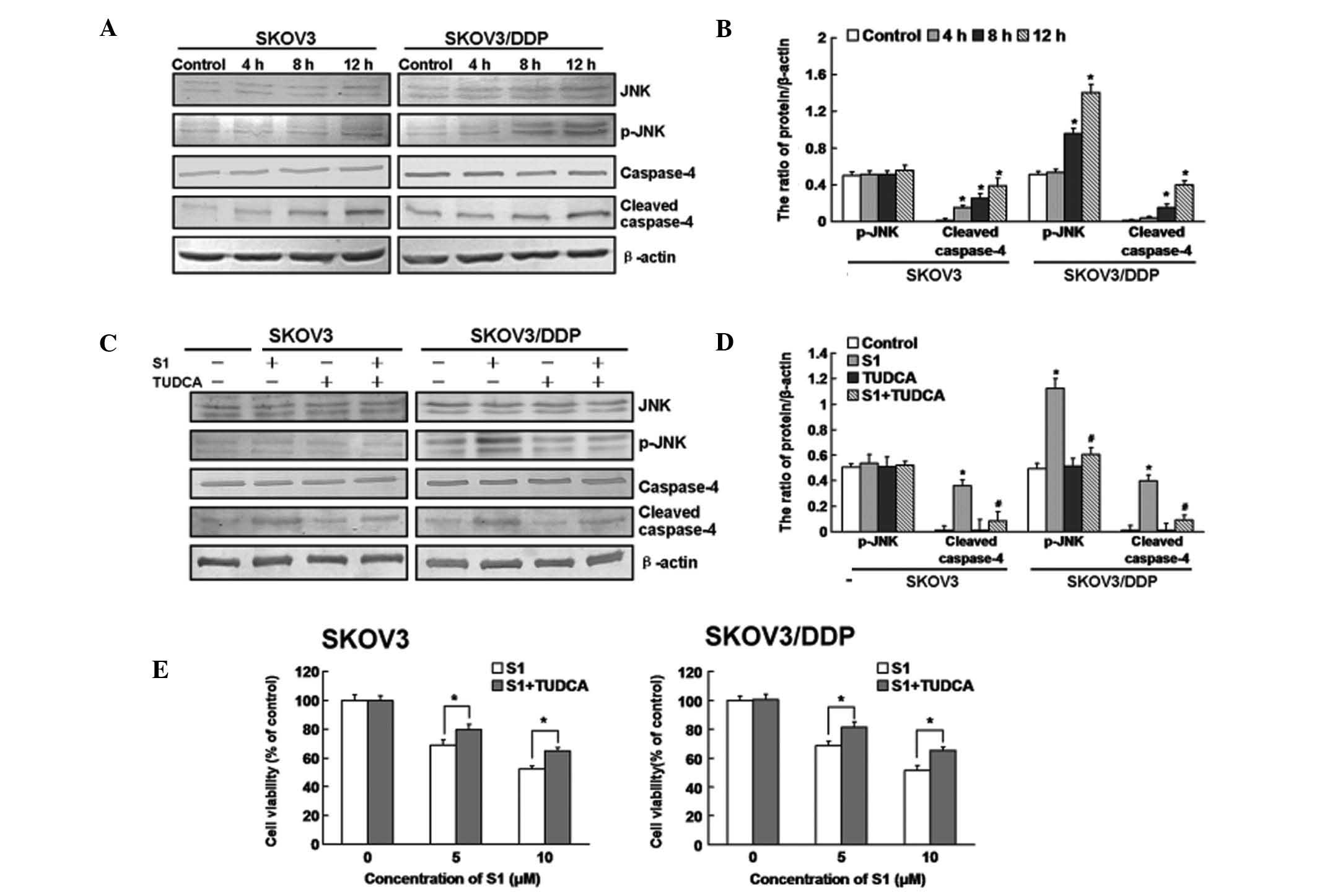
To determine the relevance of ER stress to
S1-induced apoptosis, we investigated whether ER-resident caspases
are activated by S1. Caspase-4 is an ER-resident caspase, activated
in response to ER stress and is required for ER stress-induced
apoptosis (similar to caspase-12 in murine cells) (25–27).
As show in Fig. 3A and B, cleaved
caspase-4 was significantly increased in SKOV3 cells at 4 h
following S1 treatment, compared with 8 h in SKOV3/DDP cells, and
both types of cells showed significant increases at 12 h. Many
signaling pathways are involved in ER stress. Among them, the
IRE1/JNK pathway is extremely important in regulating apoptosis and
is closely related to the Bcl-2 family (28). Therefore, we examined the expression
of JNK protein. The expression of p-JNK was increased in SKOV3/DDP
cells, but not in SKOV3 cells (Fig. 3A
and B). As shown in Fig. 3C and
D, cleaved caspase-4 was downregulated in both cell lines
treated with S1 plus TUDCA, compared with cells treated with S1
alone for 12 h, while p-JNK was downregulated only in the SKOV3/DDP
cells. MTT assays indicated that TUDCA treatment attenuated the
cytotoxic effects of S1 in both SKOV3 and SKOV3/DDP cells (Fig. 3E).
These findings indicate that ER stress-associated
apoptosis is involved in S1-induced apoptosis in both SKOV3 and
SKOV3/DDP cells, and S1-induced ER stress is delayed in SKOV3/DDP
cells.
S1 treatment activates autophagy in
SKOV3/DDP cells
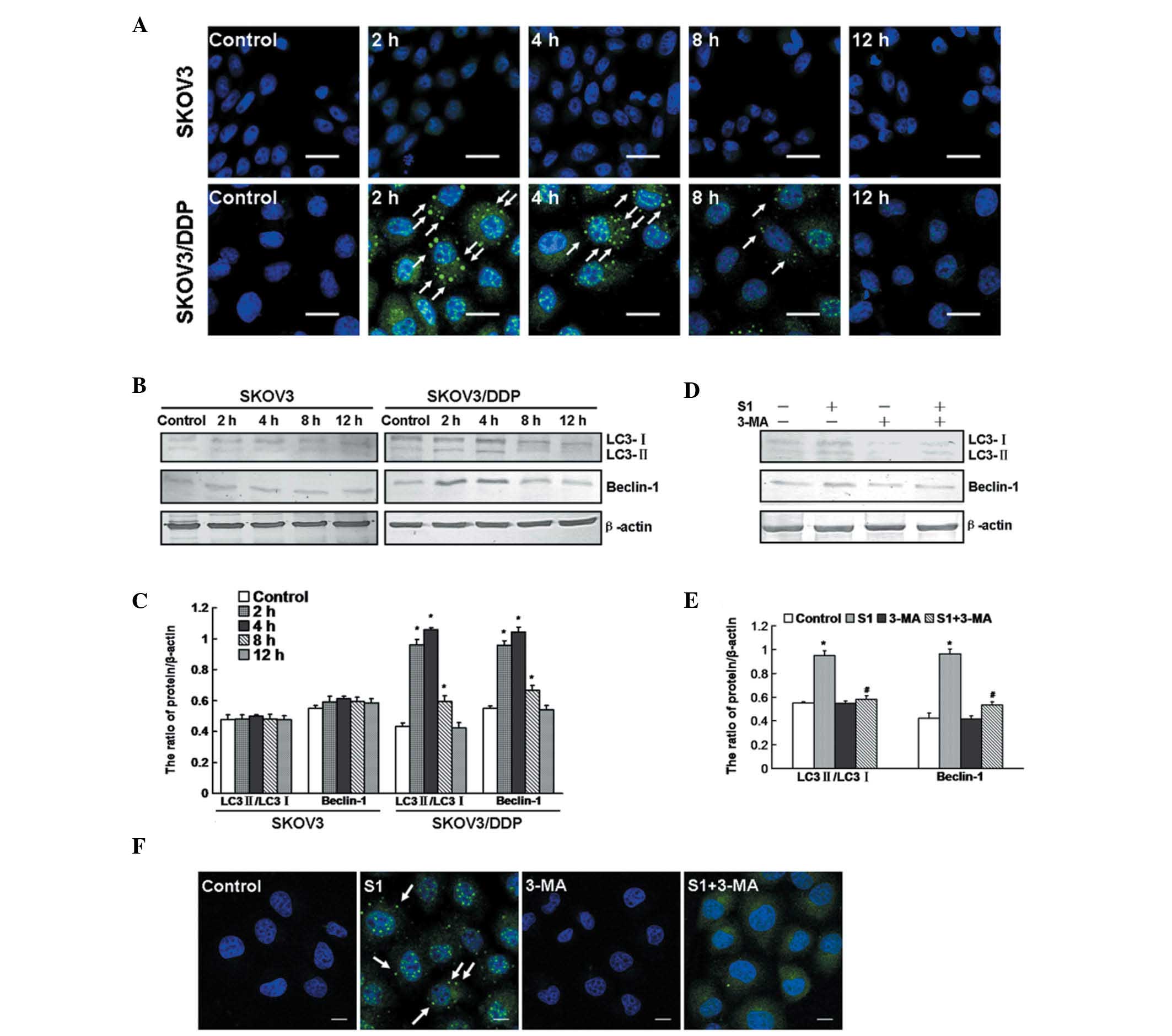
Previous reports have suggested that autophagy can
be induced by S1 (20). Therefore,
we used indirect fluorescence technology to detect the activation
of autophagy. We observed significant puncta of LC3, a molecular
marker of autophagy, in SKOV3/DDP cells following S1 treatment at
2, 4 and 8 h, but did not observe this effect in SKOV3 cells
(Fig. 4A). We detected the
transformation of LC3-I to LC3-II and expression of Beclin-1 by
western blot analysis. Similarly, the ratio of LC3-II/LC3-I and
expression of Beclin-1 were increased in SKOV3/DDP cells at 2, 4
and 8 h, but were not increased in SKOV3 cells (Fig. 4B and C).
We used the autophagy-specific inhibitor
3-methyladenine (3-MA) to inhibit the autophagy induced by S1 in
SKOV3/DDP cells. Western blot analysis demonstrated that SKOV3/DDP
cells treated with S1 plus 3-MA showed a low ratio of LC3-II/LC3-I
and low expression of Beclin-1, compared with cells treated with S1
alone (Fig. 4D and E). Using
confocal microscopy, fewer LC3 puncta were observed in the cells
after 4 h of treatment with S1 plus 3-MA (Fig. 4F). These findings demonstrate that
S1 activates autophagy in SKOV3/DDP cells at early time points, but
there is no activation in SKOV3 cells.
Inhibition of autophagy increases
S1-induced ER stress-associated apoptosis in SKOV3/DDP cells
The above-described findings showed that S1
treatment induced both an ER stress response and autophagy.
Moreover, these events occurred in a defined time sequence in
SKOV3/DDP cells, since we detected autophagy at early time points
(2 and 4 h) and ER stress-mediated apoptosis at later time points
(8 and 12 h). We subsequently further investigated the relationship
of autophagy and ER stress-associated apoptosis following S1
treatment in SKOV3/DDP cells.
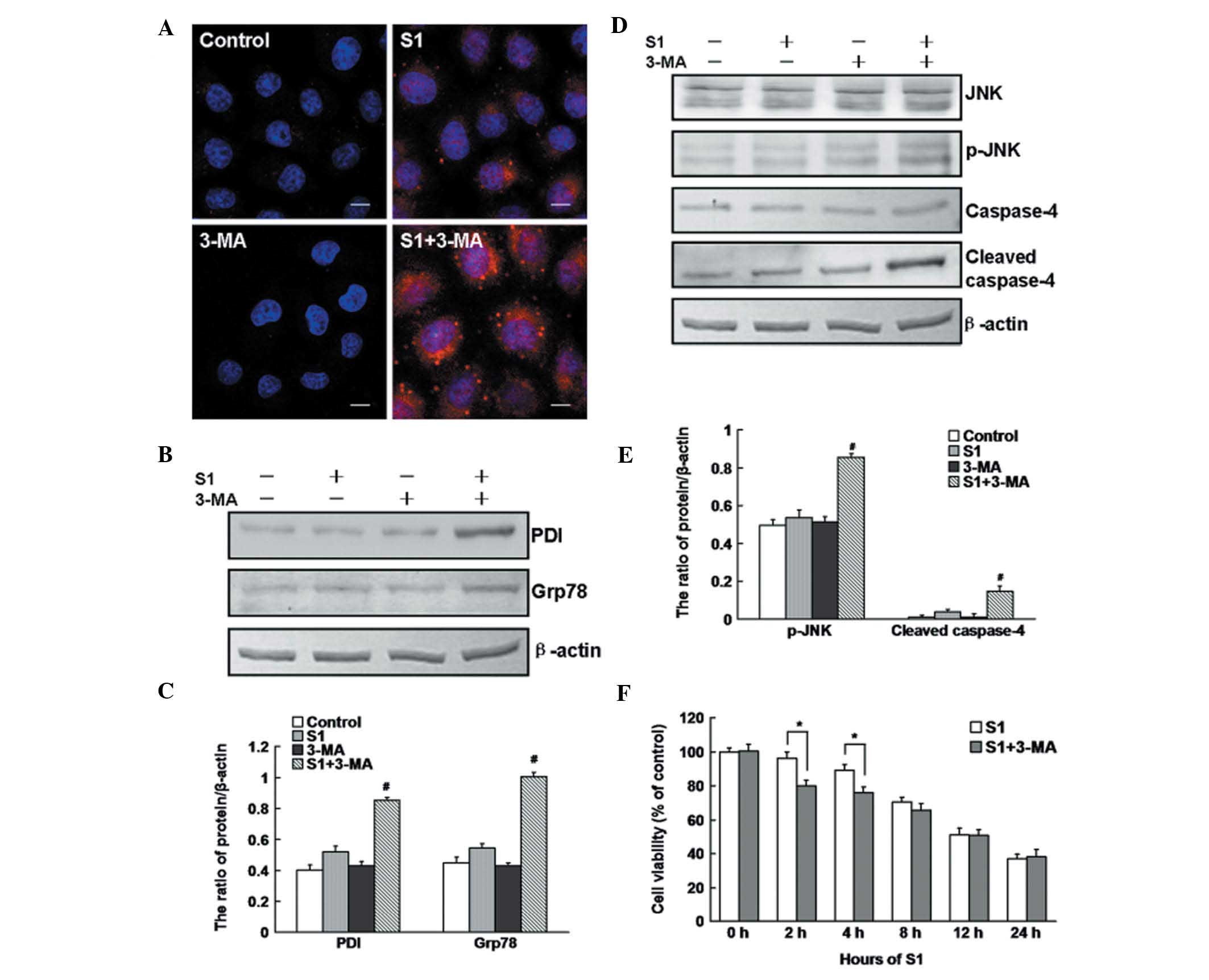
Using confocal microscopy and western blot analysis,
we found that PDI was accumulated (Fig.
5A) and the expression levels of PDI and Grp78 were enhanced
(Fig. 5B and C) after treatment
with S1 plus 3-MA in SKOV3/DDP cells. To further confirm the role
of autophagy in ER stress-mediated apoptosis, we detected the
expression levels of p-JNK and cleaved caspase-4 by western blot
analysis. As shown in Fig. 5D and
E, p-JNK and cleaved caspase-4 were upregulated in SKOV3/DDP
cells treated with S1 plus 3-MA, compared with cells treated with
S1 alone for 4 h. MTT assays indicated that 3-MA treatment enhanced
the cytotoxic effect of S1 at early time points (2 and 4 h), while
a 12-h treatment with 3-MA combined with S1 had no significant
toxic effect compared with S1 treatment alone (Fig. 5F).
These findings demonstrate that S1 leads to
autophagy activation that attenuates ER stress-mediated apoptosis
in SKOV3/DDP cells, and that inhibition of autophagy increases
S1-induced ER stress-associated apoptosis at early time points. In
the end, the activation of autophagy did not protect SKOV3/DDP
cells from S1-induced cell death.
Discussion
Currently, the resistance of ovarian cancer cells to
chemotherapeutic-induced apoptosis is a difficult issue that
remains to be solved in the treatment of ovarian cancer (29,30).
High Bcl-2 expression may be involved in the process of cancer cell
resistance to chemotherapeutics (31–33).
Therefore, treatments targeting Bcl-2 in tumors have been given a
high priority. In the present study, treatment with the
small-molecule BH3-only protein mimetic S1 for 12 or 24 h killed
both cisplatin-sensitive SKOV3 cells and cisplatin-resistant
SKOV3/DDP cells, with no significant difference in the mortality of
the cells following treatment for 12 or 24 h, indicating that S1
effectively inhibited the survival of cisplatin-resistant human
ovarian cancer cells through inhibition of Bcl-2 expression.
Bcl-2 is a crossover point of multiple signaling
pathways, as the Bcl-2-specific inhibitor, S1, induces not only
apoptosis via the mitochondrial pathway (17,19),
but also ER stress (20). When
cells receive low-level stimulation, the ER resists the stimulation
by maintaining cell homeostasis (34). When cells are severely damaged, ER
stress initiates a cell death program via the JNK, caspase-4, and
GADD153 signaling pathways (26,35).
Our findings showed that S1 treatment induced elevated expression
of PDI, Grp78 and caspase-4 in SKOV3 and SKOV3/DDP cells,
suggesting that S1 may induce apoptosis of human ovarian cancer
cells via the ER stress pathway. However, the apoptosis of
SKOV3/DDP cells induced by ER stress occurred at a significantly
later time point than that of SKOV3 cells. The ER stress-mediated
apoptosis of SKOV3/DDP cells induced by S1 triggered JNK
activation. After TUDCA-mediated inhibition of ER stress, JNK
phosphorylation decreased, suggesting that S1 induced ER
stress-associated apoptosis in SKOV3/DDP cells through activation
of JNK and caspase-4. It was previously reported that JNK triggers
apoptosis by mediating Bcl-2 phosphorylation (36). However, our findings showed that the
activation of the JNK signaling pathway was mediated by the
S1-induced ER stress pathway.
Importantly, the present study revealed a
significant difference in the survival rates of SKOV3 and SKOV3/DDP
cells following S1 treatment for short periods (2 and 4 h). Further
analyses showed different levels of autophagy activation in the two
types of cells. Following S1 treatment for 2, 4 or 8 h, punctate
aggregation of LC3 was observed, the ratio of LC3II/LC3I was
increased, and obvious autophagy was present in SKOV3/DDP cells,
while no obvious punctate aggregation of LC3 was observed in SKOV3
cells. It was reported that autophagy activation mediates apoptosis
in apoptosis-deficient cells, which is termed autophagic cell death
(37,38). However, when cells with normal
apoptosis receive a specific stimulation, activation of autophagy
protects cells by inhibiting apoptosis (39–43),
and this protective autophagy is activated in a time-dependent
manner (44), suggesting that
autophagy activation may be involved in the survival and death of
SKOV3/DDP cells. Combined treatment with 3-MA and S1 reduced cell
survival compared with S1 treatment alone, demonstrating that
autophagy activation facilitated drug-resistant cell resistance to
drug stimulation.
Recently, the roles of ER stress and autophagy in
cells and their interaction have become a hot topic of research.
Our findings showed that S1 treatment induced autophagy activation
and ER stress-associated apoptosis in SKOV3/DDP cells. To assess
the effect of the autophagy caused by S1 treatment on ER
stress-associated apoptosis, 3-MA was used to inhibit autophagy,
and significantly enhanced caspase-4 activation and increased ER
stress were observed. These observations indicate that S1
treatment-induced autophagy of SKOV3/DDP cells may be a resistant
stress to the external environment. However, such protection by
autophagy only functions within a short period. With prolonged ER
stress, autophagy activation is attenuated, and its protection is
diminished.
In summary, our findings showed that S1, an
inhibitor of Bcl-2, effectively induced ER stress-associated
apoptosis in both SKOV3 and SKOV3/DDP cells. Activation of
autophagy within a short period provided protection for SKOV3/DDP
cells and facilitated their resistance to stress. However, with
prolongation of the S1 treatment, autophagy activation was
attenuated and ER stress-mediated apoptosis played a leading role,
effectively killing SKOV3/DDP cells. These results suggest that
transient activation of autophagy is inadequate to resist
S1-induced apoptosis, demonstrating that S1 inhibits the growth of
human ovarian cancer SKVO3 and SKOV3/DDP cells, thereby effectively
killing drug-resistant tumor cells.
Acknowledgements
This research was supported by the National Natural
Science Foundation of China (nos. 81272876, 81141099, 81100808, and
81202552).
References
|
1
|
Karnak D and Xu L: Chemosensitization of
prostate cancer by modulating Bcl-2 family proteins. Curr Drug
Targets. 11:699–707. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gul O, Basaga H and Kutuk O: Apoptotic
blocks and chemotherapy resistance: strategies to identify Bcl-2
protein signatures. Brief Funct Genomic Proteomic. 7:27–34. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu JR, Opipari AW, Tan L, Jiang Y, Zhang
Y, Tang H and Nuñez G: Dysfunctional apoptosome activation in
ovarian cancer: implications for chemoresistance. Cancer Res.
62:924–931. 2002.PubMed/NCBI
|
|
4
|
Klymenko T, Brandenburg M, Morrow C, Dive
C and Makin G: The novel Bcl-2 inhibitor ABT-737 is more effective
in hypoxia and is able to reverse hypoxia-induced drug resistance
in neuroblastoma cells. Mol Cancer Ther. 10:2373–2383. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang L, Ming L and Yu J: BH3 mimetics to
improve cancer therapy: mechanisms and examples. Drug Resist Updat.
10:207–217. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kang MH and Reynolds CP: Bcl-2 inhibitors:
targeting mitochondrial apoptotic pathways in cancer therapy. Clin
Cancer Res. 15:1126–1132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Premkumar DR, Jane EP, DiDomenico JD,
Vukmer NA, Agostino NR and Pollack IF: ABT-737 synergizes with
bortezomib to induce apoptosis, mediated by Bid cleavage, Bax
activation, and mitochondrial dysfunction in an Akt-dependent
context in malignant human glioma cell lines. J Pharmacol Exp Ther.
341:859–872. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang X, Olberding KE, White C and Li C:
Bcl-2 proteins regulate ER membrane permeability to luminal
proteins during ER stress-induced apoptosis. Cell Death Differ.
18:38–47. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bhavya BC, Indira D, Seervi M, Joseph J,
Sobhan PK, Mathew KA, Varghese S and Santhoshkumar TR: Endoplasmic
reticulum-targeted Bcl-2 inhibitable mitochondrial fragmentation
initiates ER stress-induced cell death. Adv Exp Med Biol.
749:83–95. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dasmahapatra G, Lembersky D, Rahmani M,
Kramer L, Friedberg J, Fisher RI, Dent P and Grant S: Bcl-2
antagonists interact synergistically with bortezomib in DLBCL cells
in association with JNK activation and induction of ER stress.
Cancer Biol Ther. 8:808–819. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Szegezdi E, Macdonald DC, Ní Chonghaile T,
Gupta S and Samali A: Bcl-2 family on guard at the ER. Am J Physiol
Cell Physiol. 296:C941–C953. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhou F, Yang Y and Xing D: Bcl-2 and
Bcl-xL play important roles in the crosstalk between autophagy and
apoptosis. FEBS J. 278:403–413. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Marquez RT and Xu L: Bcl-2: Beclin 1
complex: multiple, mechanisms regulating autophagy/apoptosis toggle
switch. Am J Cancer Res. 2:214–221. 2012.PubMed/NCBI
|
|
14
|
Lian J, Wu X, He F, Karnak D, Tang W, Meng
Y, Xiang D, Ji M, Lawrence TS and Xu L: A natural BH3 mimetic
induces autophagy in apoptosis-resistant prostate cancer via
modulating Bcl-2-Beclin1 interaction at endoplasmic reticulum. Cell
Death Differ. 18:60–71. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu Y, Yu H, Qin H, Kang J, Yu C, Zhong J,
Su J, Li H and Sun L: Inhibition of autophagy enhances cisplatin
cytotoxicity through endoplasmic reticulum stress in human cervical
cancer cells. Cancer Lett. 314:232–243. 2011. View Article : Google Scholar
|
|
16
|
Shi YH, Ding ZB, Zhou J, Hui B, Shi GM, Ke
AW, Wang XY, Dai Z, Peng YF, Gu CY, Qiu SJ and Fan J: Targeting
autophagy enhances sorafenib lethality for hepatocellular carcinoma
via ER stress-related apoptosis. Autophagy. 7:1159–1172. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang Z, Song T, Zhang T, Gao J, Wu G, An
L and Du G: A novel BH3 mimetic S1 potently induces
Bax/Bak-dependent apoptosis by targeting both Bcl-2 and Mcl-1. Int
J Cancer. 128:1724–1735. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang Z, Wu G, Gao J and Song T: Inclusion
complex of a Bcl-2 inhibitor with cyclodextrin: characterization,
cellular accumulation, and in vivo antitumor activity. Mol Pharm.
7:1348–1354. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Z, Wu G, Xie F, Song T and Chang X:
3-Thiomorpholin-8-oxo-8H-acenaphtho[1,2-b]pyrrole-9-carbonitrile
(S1) based molecules as potent, dual inhibitors of B-cell lymphoma
2 (Bcl-2) and myeloid cell leukemia sequence 1 (Mcl-1):
structure-based design and structure-activity relationship studies.
J Med Chem. 54:1101–1105. 2011.
|
|
20
|
Zhong JT, Xu Y, Yi HW, Su J, Yu HM, Xiang
XY, Li XN, Zhang ZC and Sun LK: The BH3 mimetic S1 induces
autophagy through ER stress and disruption of Bcl-2/Beclin 1
interaction in human glioma U251 cells. Cancer Lett. 323:180–187.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Muller C, Bandemer J, Vindis C, Camaré C,
Mucher E, Guéraud F, Larroque-Cardoso P, Bernis C, Auge N, Salvayre
R and Negre-Salvayre A: Protein Disulfide Isomerase Modification
and Inhibition potentiates ER stress and apoptosis induced by
oxidized low density lipoproteins. Antioxid Redox Signal.
18:731–742. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee AS: The glucose-regulated proteins:
stress induction and clinical applications. Trends Biochem Sci.
26:504–510. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Miller SD, Greene CM, McLean C, Lawless
MW, Taggart CC, O’Neill SJ and McElvaney NG: Tauroursodeoxycholic
acid inhibits apoptosis induced by Z alpha-1 antitrypsin via
inhibition of bad. Hepatology. 46:496–503. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang JY, Diao YF, Kim HR and Jin DI:
Inhibition of endoplasmic reticulum stress improves mouse embryo
development. PLoS One. 7:e404332012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nakagawa T, Zhu H, Morishima N, Li E, Xu
J, Yankner BA and Yuan J: Caspase-12 mediates
endoplasmic-reticulum-specific apoptosis and cytotoxicity by
amyloid-β. Nature. 403:98–103. 2000.PubMed/NCBI
|
|
26
|
Binet F, Chiasson S and Girard D: Evidence
that endoplasmic reticulum (ER) stress and caspase-4 activation
occur in human neutrophils. Biochem Biophys Res Commun. 391:18–23.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mao ZG, Jiang CC, Yang F, Thorne RF,
Hersey P and Zhang XD: TRAIL-induced apoptosis of human melanoma
cells involves activation of caspase-4. Apoptosis. 15:1211–1222.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Verfaillie T, Salazar M, Velasco G and
Agostinis P: Linking ER stress to autophagy: potential implications
for cancer therapy. Int J Cell Biol. 2010:9305092010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Eliopoulos AG, Kerr DJ, Herod J, Hodgkins
L, Krajewski S, Reed JC and Young LS: The control of apoptosis and
drug resistance in ovarian cancer: influence of p53 and Bcl-2.
Oncogene. 11:1217–1228. 1995.PubMed/NCBI
|
|
30
|
Mano Y, Kikuchi Y, Yamamoto K, Kita T,
Hirata J, Tode T, Ishii K and Nagata I: Bcl-2 as a predictor of
chemosensitivity and prognosis in primary epithelial ovarian
cancer. Eur J Cancer. 35:1214–1219. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
van Delft MF and Huang DC: How the Bcl-2
family of proteins interact to regulate apoptosis. Cell Res.
16:203–213. 2006.PubMed/NCBI
|
|
32
|
Simonian PL, Grillot DA and Nuñez G: Bcl-2
and Bcl-XL can differentially block chemotherapy-induced cell
death. Blood. 90:1208–1216. 1997.PubMed/NCBI
|
|
33
|
Reed JC, Miyashita T, Takayama S, Wang HG,
Sato T, Krajewski S, Aimé-Sempé C, Bodrug S, Kitada S and Hanada M:
BCL-2 family proteins: regulators of cell death involved in the
pathogenesis of cancer and resistance to therapy. J Cell Biochem.
60:23–32. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schönthal AH: Endoplasmic reticulum stress
and autophagy as targets for cancer therapy. Cancer Lett.
275:163–169. 2009.PubMed/NCBI
|
|
35
|
Schönthal AH: Pharmacological targeting of
endoplasmic reticulum stress signaling in cancer. Biochem
Pharmacol. 85:653–666. 2013.PubMed/NCBI
|
|
36
|
Zhang YX, Kong CZ, Wang LH, Li JY, Liu XK,
Xu B, Xu CL and Sun YH: Ursolic acid overcomes Bcl-2-mediated
resistance to apoptosis in prostate cancer cells involving
activation of JNK-induced Bcl-2 phosphorylation and degradation. J
Cell Biochem. 109:764–777. 2010.PubMed/NCBI
|
|
37
|
Shimizu S, Konishi A, Nishida Y, Mizuta T,
Nishina H, Yamamoto A and Tsujimoto Y: Involvement of JNK in the
regulation of autophagic cell death. Oncogene. 29:2070–2082. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT,
Liu B and Bao JK: Programmed cell death pathways in cancer: a
review of apoptosis, autophagy and programmed necrosis. Cell
Prolif. 45:487–498. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yu H, Su J, Xu Y, Kang J, Li H, Zhang L,
Yi H, Xiang X, Liu F and Sun L: p62/SQSTM1 involved in cisplatin
resistance in human ovarian cancer cells by clearing ubiquitinated
proteins. Eur J Cancer. 47:1585–1594. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sun WL, Chen J, Wang YP and Zheng H:
Autophagy protects breast cancer cells from epirubicin-induced
apoptosis and facilitates epirubicin-resistance development.
Autophagy. 7:1035–1044. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sirichanchuen B, Pengsuparp T and
Chanvorachote P: Long-term cisplatin exposure impairs autophagy and
causes cisplatin resistance in human lung cancer cells. Mol Cell
Biochem. 364:11–18. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen N and Karantza V: Autophagy as a
therapeutic target in cancer. Cancer Biol Ther. 11:157–168. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Suh DH, Kim MK, Kim HS, Chung HH and Song
YS: Unfolded protein response to autophagy as a promising druggable
target for anticancer therapy. Ann NY Acad Sci. 1271:20–32. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zeng R, He J, Peng J, Chen Y, Yi S, Zhao F
and Cui G: The time-dependent autophagy protects against apoptosis
with possible involvement of Sirt1 protein in multiple myeloma
under nutrient depletion. Ann Hematol. 91:407–417. 2011. View Article : Google Scholar : PubMed/NCBI
|