Introduction
Gliomas are the most common primary tumors of the
central nervous system (CNS). They are aggressive, highly invasive,
and neurologically destructive tumors. Among these, glioblastoma
multiforme (GBM) is the most malignant phenotype. Several studies
on a variety of cancers have suggested that the activity of ion
channels is closely related to a tumor cell’s ability to migrate
and proliferate (1–3). It has been reported that glioma cells
express higher levels of potassium, sodium and chloride channels
compared to normal astrocytes (4–6),
suggesting these ion channels may play a role in the progression of
glioma. Several members of the ENaC/Degenerin family are expressed
in a variety of GBM cell lines, including U251-MG (7). The ENaC/Degenerin superfamily includes
multiple members of the ENaC and ASICs (ASIC1-4) subfamilies
(8). The ENaC/Degenerin superfamily
members can be specifically inhibited by amiloride (9). Previous studies report that glioma
cells are unable to regulate their volume subsequent to cell
shrinkage by hyperosmolar solutions when treated with either
amiloride or PcTX-1, a specific ASIC1 blocker (40-amino acid
peptide, purified from the venom of the West Indies tarantula)
(10,11). These studies suggest that ASIC1 may
be involved in the regulation of glioma cell volume, thereby
affecting the migration ability of glioma cells.
Unlike ENaC, ASICs are non-selective cation channels
permeable to both monovalent and divalent cations, including
Ca2+(12,13). Four ASIC genes (ASIC1, ASIC2, ASIC3
and ASIC4) and splice variants for ASIC1 (ASIC1a and ASIC1b) have
been identified in a variety of cell types (14,15).
ASIC channels can be transiently activated by extracellular
acidosis (14). Although a
constitutively activated, amiloride-sensitive Na+
current was reported in GBM cells, this current is not seen in
normal astrocytes or low-grade gliomas (4). Later studies suggest that this
amiloride-sensitive, constitutively-activated cation current is
mediated by a cross-clade channel composed of ASIC1 and two
subunits of ENaC, αENaC and γENaC. Knockdown of any of the three
ENaC/Degenerin subunits (ASIC1, αENaC and γENaC) eliminates this
current and attenuates migration of GBM cells (11). However, no typical extracellular
acidosis-activated, whole-cell current was detected in GBM cells
(16).
CaMKII catalyzes the phosphorylation of ASIC1a to
activate ASIC1a channels and subsequently increase cell death in
rodent ischemic CNS neurons (17).
This Ca2+-sensitive kinase may also play an important
role in glioma biology, particularly because glioma cells require
Ca2+, which acts as a second messenger to support cell
migration (18). Although these
functional data suggest the importance of ASIC1 and CaMKII in
ischemic CNS, the functional interaction of the two within GBM
cells remains unclear. We hypothesized that ASIC1 is constitutively
activated in GBM cells, and that CaMKII regulates this process.
Furthermore, we hypothesized that the activity of ASIC1 is
associated with the ability of glioma cells to migrate.
Materials and methods
Cell culture
The glioma cell line U251-MG (GBM, derived from a
World Health Organization grade IV tumor) (ATCC, Rockville, MD,
USA) was routinely cultured in 1:1 Dulbecco’s modified Eagle’s/F-12
medium (Hyclone, Logan, UT, USA) supplemented with 15% fetal calf
serum (Hyclone) and 1% penicillin/streptomycin (Invitrogen,
Carlsbad, CA, USA). The cells were incubated in an atmosphere
containing 5% CO2 and 95% air at 37°C. The medium was
changed every three days. Cells were split 48 h prior to
electrophysiological recording onto 35-mm dishes containing
flame-sterilized coverslips. Cells were split and starved 24 h and
4–6 h before transfection, respectively.
Immunofluorescence
U251-MG cells were grown on 25-mm glass coverslips
and processed for indirect immunofluorescence after reaching
confluence. Cells were fixed and subsequently incubated for 2 h
with the following antibodies: a goat polyclonal anti-ASIC1 (1:50
dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and a
rabbit monoclonal anti-CaMKII (1:200 dilution; Abcam, Cambridge,
MA, USA). Following incubation with the primary antibodies, the
cells were then visualized using either Alexa Fluor 594 Donkey
Anti-Goat IgG (H+L) or Alexa Fluor 488 Donkey Anti-Rabbit IgG (H+L)
(Invitrogen). Nuclei were stained with DAPI (1:1000 dilution,
Invitrogen) for 15 min. Images were captured at room temperature
using a 63x/1.2 oil immersion objective on a confocal microscope
(Fluoview1000, Olympus).
Patch clamp electrophysiology
The whole-cell patch-clamp technique was used to
record the amiloride-sensitive whole-cell current. The steady-state
current-voltage relationships were determined using a holding
potential of −60 mV. The membrane potential was stepped from −160
mV to +100 mV in 20 mV increments. The peak current at each voltage
was measured. Resistance of patch pipettes ranged from 1.5 to 2 MΩ
when filled with the pipette solution. Whole-cell recordings were
performed with an Axopatch 200B amplifier operated with the pCLAMP
software, filtered at 10K Hz, acquired at 2 kHz with pCLAMP, and
analyzed with the Clampfit 9.0.
Solutions
Amiloride-sensitive currents were recorded in an
external solution of RPMI-1640 medium containing the following (in
mM): 133 Na+, 5.3 K+ and 108.3
Cl−. The pipette solution contained (in mM) 100
potassium gluconate, 30 KCl, 20 HEPES, 0.5 EGTA, 4 ATP and 10 nM
free calcium at a pH of 7.2 (adjusted by HCl).
ACCN2 genes silencing
Using the BLOCK-iT pol II miR RNAi expression vector
kit from Invitrogen, we designed miRNA mimics that were 21 nt in
length. The miRNA target and negative control sequences were
defined as 5′-TCAGGATGTAGCCTACAGCAC-3′ and
5′-AAATGTACTGCGCGTGGAGAC-3′, respectively. The miRNAs were cloned
into the pcDNA 6.2-GW/EmGFP-miR vector by inserting them into the
3′-UTR of the EmGFP gene. The U251-MG cells were seeded at 40–60%
confluency in 6-well plates a day before transfection. The cells
were transfected using lipofectamine 2000 (Invitrogen). After
transfection, cells were continuously cultured in the presence of
10 ng/ml blasticidine S hydrochloride (Sigma, St. Louis, MO, USA).
The cells were used for other experiments three weeks after
transfection.
Western blotting
Cells were lysed and equal concentrations of
proteins were separated on 10% SDS-polyacrylamide gels (Invitrogen)
and transferred onto polyvinylidene difluoride (PVDF) membranes.
The blots were incubated in anti-ASIC1 (1:500 dilution; Abcam),
anti-CaMKII (1:1000 dilution; Abcam), and anti-β-actin (1:5000
dilution; Santa Cruz Biotechnology) antibodies for 2 h followed by
incubation with a peroxidase-conjugated secondary antibody (1:5000
dilution; Santa Cruz Biotechnology) for 1 h at room temperature.
Labeled proteins were visualized with ECL (Invitrogen).
Immunoprecipitation
Cells were lysed and centrifuged at 10,000 × g for
10 min to remove cellular debris. The supernatant was removed and
aliquots of proteins (500 μg) were incubated with 5 μg of
anti-CaMKII antibody or rabbit IgG overnight at 4°C.
Immunoprecipitates were captured with 40 μl of protein A/G beads
(Santa Cruz Biotechnology) at 4°C for 1 h. Samples were centrifuged
and then washed three times with 1 ml of lysis buffer. The proteins
were separated by SDS-PAGE electrophoresis and transferred to PVDF
membrane. Blots were incubated in anti-ASIC1 or anti-CaMKII
antibodies overnight at 4°C.
Migration assay
In vitro migration assays were performed
using Costar transwell inserts (Costar, NY, USA; pore size 8-μm) in
24-well plates. A total of ~1–1.5×105 cells in 200 μl
DMEM were seeded on the upper chamber of a 6.5 mm-transwell with
8.0 μm-pore polycarbonate membrane inserts. DMEM (600 μl) with 10%
fetal bovine serum was added into the lower chamber as a
chemoattractant. Cells were allowed to adhere for 30 min before
treatment with 10 nM PcTX-1 (Abcam) to block ASIC1 or 1 μM
myristoylated AIP (Enzo Life Sciences, Farmingdale, NY, USA) to
block CaMKII. The plates were incubated for 12 h at 37°C in 5%
CO2, followed by fixation in 4% buffered
paraformaldehyde for 15 min and staining with DAPI for 15 min.
Migrated cells were counted in five random areas of the membrane
using a confocal microscope.
Results
ASIC1 is expressed in U251-MG cells
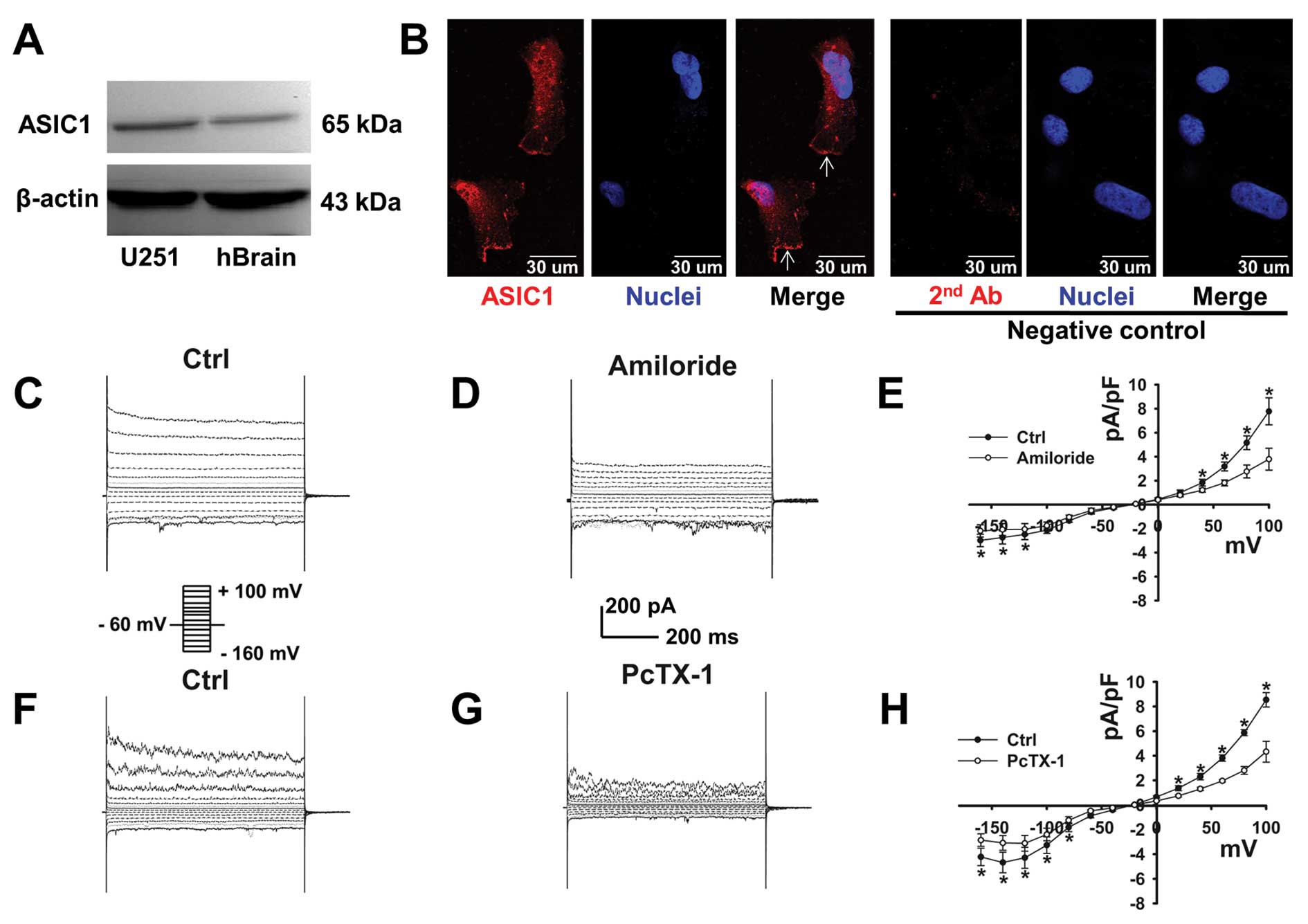
We first attempted to use western blotting to
determine whether U251-MG cells express ASIC1. As shown in Fig. 1A, protein bands of ~65 kDa were
detected in U251-MG cells and human brain tissue. These data
indicate that ASIC1 is present in U251-MG cells. The cellular
distribution of ASIC1 was determined by immunofluorescence
staining. Our results clearly show that ASIC1 is localized at the
plasma membrane (the white arrow heads) of U251-MG cells (Fig. 1B).
A constitutively activated,
amiloride-sensitive current was detected in U251-MG cells
Using a whole-cell patch-clamp configuration, we
detected a constitutively activated current in U251-MG cells
(Fig. 1C). This current was
significantly inhibited by 100 μM amiloride as previously described
(11) (Fig. 1D). The current densities measured at
different voltages in the absence or presence of amiloride were
plotted as a function of voltage; the data show that the current
density was significantly reduced (Fig.
1E; n=4 paired experiments).
To confirm whether this amiloride-sensitive current
is carried by ASIC1 in U251-MG cells, an ASIC1 specific blocker,
PcTX-1, was applied to the bath solution after a control current
was generated (14). This current
was significantly inhibited by treatment with 10 nM PcTX-1
(Fig. 1F and G), which is shown by
the summarized I-V relationships (Fig.
1H; n=6 paired experiments). These data are consistent with the
notion that this constitutively activated current in U251-MG cells
is most likely mediated by ENaC/Degenerin subunits, and that ASIC1
may constitute the central core of the channel (11).
ASIC1 mediates the constitutively
activated current in U251-MG cells
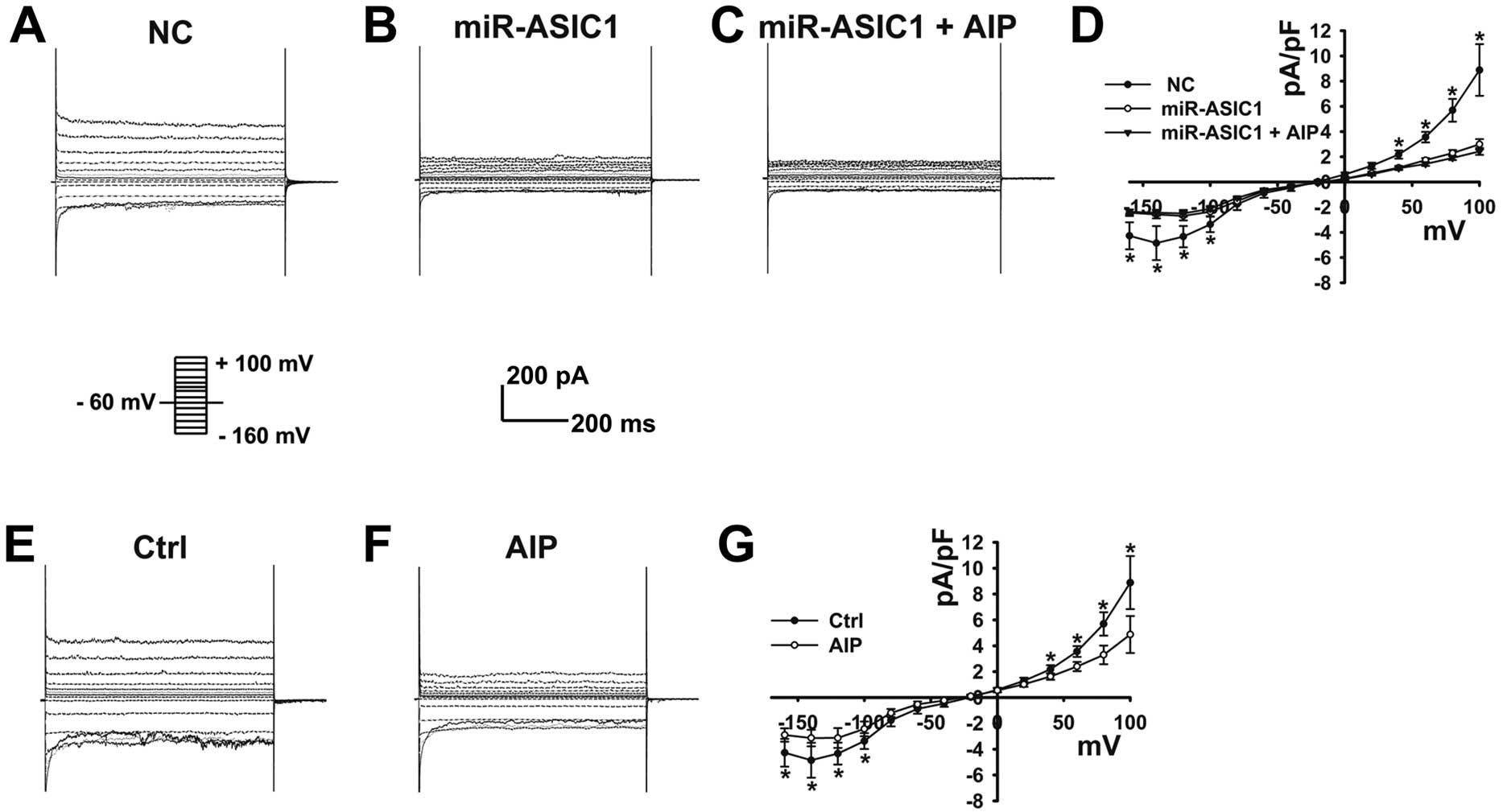
To confirm that this constitutively activated
current in U251-MG cells is mediated by ASIC1, we used a
gene-silencing technique to knock down ASIC1 and performed
patch-clamp experiments in these cells (knockdown efficiency shown
in Fig. 4C). Upon knockdown of
ASIC1, the constitutively activated current in U251-MG cells was
dramatically decreased (Fig. 2A, B and
D; n=6), strongly suggesting that this current is carried by
ASIC1.
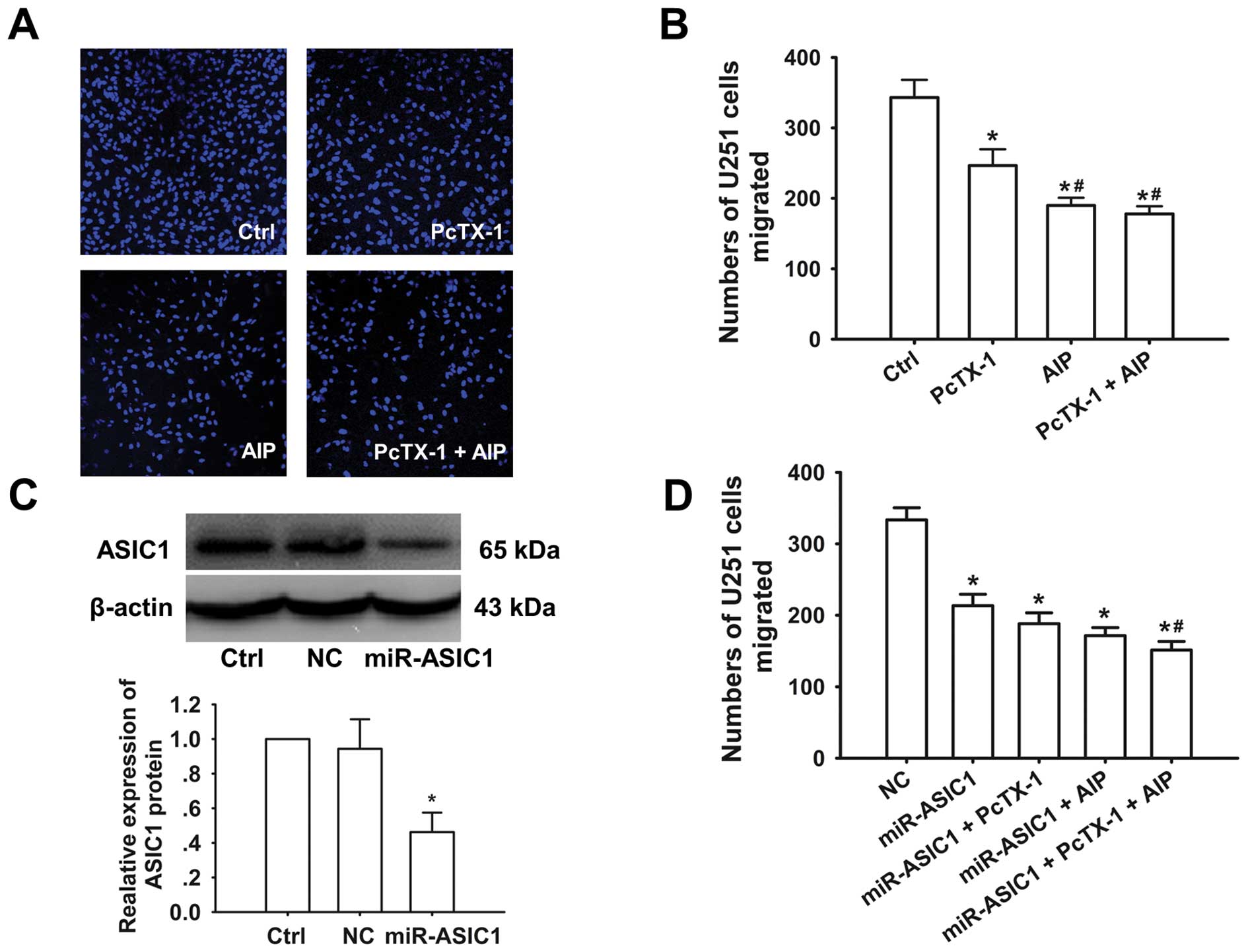 | Figure 4CaMKII mediated activation of ASIC1
plays a role in U251-MG cell migration. (A) Representative images
of migrated U251-MG cells under control conditions, in the presence
of 10 nM PcTX-1, 1 μM AIP, and 10 nM PcTX-1 + 1 μM AIP,
respectively. (B) Summarized numbers of migrated U251-MG cells
before and after application of PcTX-1, AIP and PcTX-1 + AIP (n=3).
Ctrl represents normal U251-MG cells; PcTX-1, AIP, PcTX-1 + AIP
indicate normal U251-MG cells treated with PcTX-1, AIP, and AIP +
PcTX-1, respectively. *p<0.05 compared with control
cells. #p<0.05 compared with PcTX-1 treated group
(n=3 for each condition). (C) Representative western blot
demonstrating the knockdown efficiency of ASIC1 by miRNA (top), and
the expression level of ASIC1 was reduced ~55% in miRNA-transfected
U251-MG cells (n=3). *p<0.05 compared with control
and negative control cells. (D) ASIC1 knockdown resulted in a
decrease in U251-MG cell migration; either PcTX-1 or AIP failed to
further decrease the migration ability in ASIC1 knockdown cells;
however, PcTX-1 and AIP together led to a slight but significant
decrease in migration in ASIC1 knockdown cells. NC represents the
scramble miRNA transfected group; miR-ASIC1, miR-ASIC1 + PcTX-1,
miR-ASIC1 + AIP, and miR-ASIC1 + PcTX-1 + AIP indicate specific
miRNA against ASIC transfected group, specific miRNA against ASIC
transfected group treated with PcTX-1, AIP, and AIP + PcTX-1,
respectively. *p<0.05 compared with negative control
cells. #p<0.05 compared with miR-ASIC1 group (n=3 for
each condition). |
Since it was previously reported that CaMKII
regulates ASIC1a in rats (17), we
reasoned that CaMKII might regulate ASIC1 currents in U251-MG
cells. We tested whether AIP, a highly specific and potent
inhibitor of CaMKII, could inhibit this current in U251-MG cells.
As seen in Fig. 2F, the ASIC1
currents were significantly decreased upon treatment with AIP
(Fig. 2E–2G; n=6 paired
experiments). Moreover, AIP failed to further decrease ASIC1
currents when ASIC1 was knocked down in U251-MG cells, suggesting
the specificity of CaMKII-mediated regulation of ASIC1 currents
(Fig. 2A–D).
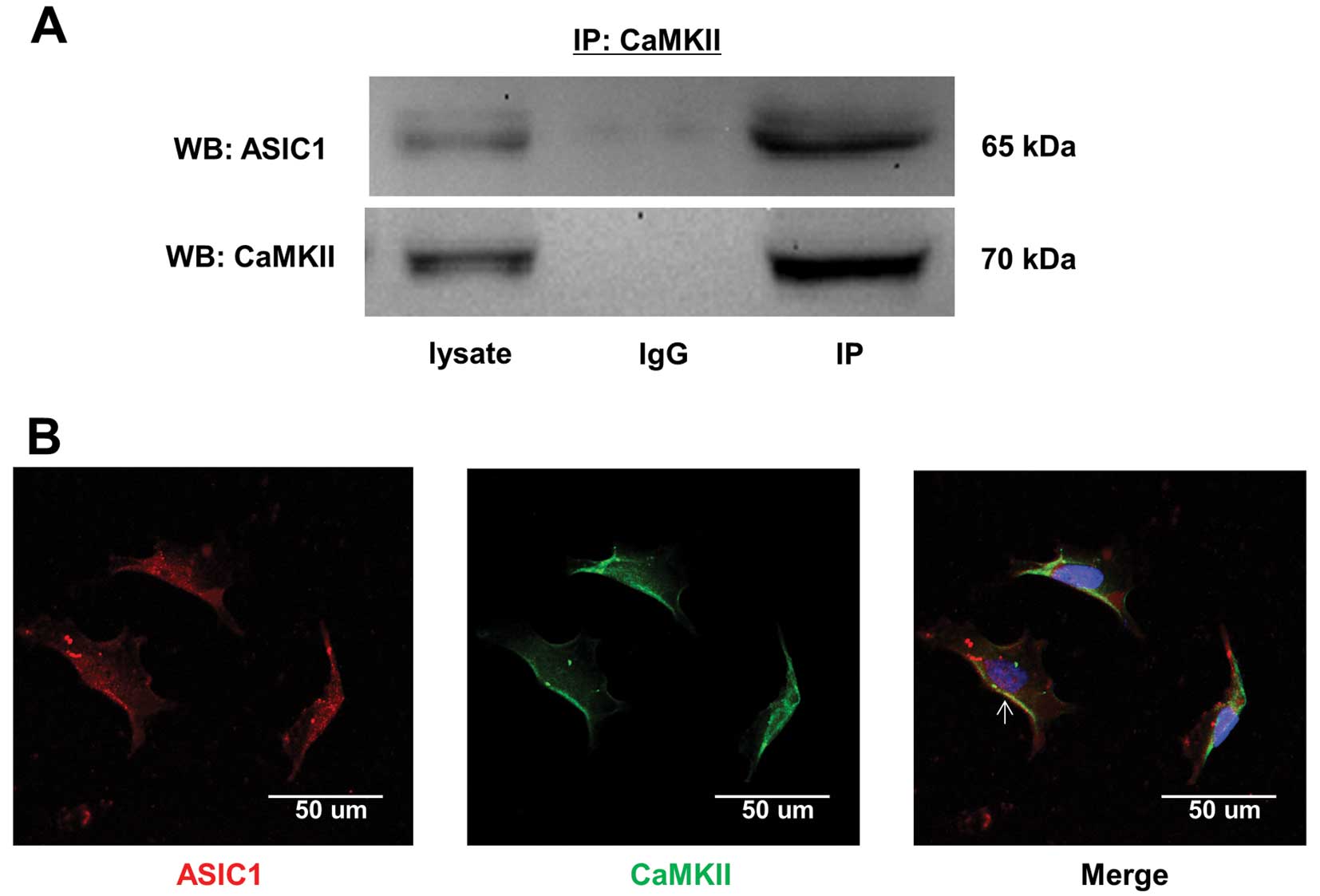
CaMKII physically associates with ASIC1
and regulates ASIC1 in U251-MG cells
Since we found that AIP inhibits ASIC1 (Fig. 2E–G), we performed immunofluorescence
staining and co-IP experiments to determine whether these proteins
functionally interact. Our co-IP results show that ASIC1 and CaMKII
associate in U251-MG cells (Fig.
3A). The immunofluorescence staining shows that CaMKII and
ASIC1 co-localize predominantly at the plasma membrane of U251-MG
cells (Fig. 3B; the white arrow
head). These data, along with the data shown in Fig. 2D and G, suggest that CaMKII and
ASIC1 assemble to form a functional complex in U251-MG cells, and
that CaMKII regulates ASIC1.
CaMKII-mediated activation of ASIC1
contributes to the ability of GBM cells to migrate
Previous studies have demonstrated that ASIC1 plays
a role in the malignant behavior of glioma cells (11,16,19,20),
and that CaMKII regulates many ion channels (21). Therefore, we hypothesized that
inhibiting CaMKII and/or ASIC1 activity would reduce the ability of
GBM cells to migrate. Therefore, we used a transwell migration
assay in combination with a CaMKII inhibitor and ASIC1 blocker to
test this hypothesis. Fig. 4A shows
representative images of migrated U251-MG cells under different
experimental conditions. It appeared that treatment with PcTX-1,
AIP, or PcTX-1 + AIP was able to significantly attenuate cell
migration (Fig. 4A and B). Under
control conditions 343±25 cells migrated through the pores; in the
presence of PcTX-1, AIP, or PcTX-1 + AIP the number of migrated
cells was reduced to 247±23, 190±11, and 178±11, respectively
(Fig. 4A and B; n=3 for each
condition).
We also performed migration assays when ASIC1 was
knocked down in U251-MG cells (knockdown efficiency shown in
Fig. 4C); our results indicate that
the number of migrated cells was significantly reduced in these
cells compared with the negative control (NC) (Fig. 4D). Of note, we found that neither
PcTX-1 nor AIP treatment further reduced cell migration in U251-MG
cells when ASIC1 was knocked down (188±15 and 171±11 cells vs.
213±16 cells); however, PcTX-1 + AIP treatment slightly but
significantly reduced the number of migrated cells (151±12 vs.
213±16 cells) (Fig. 4A and D; n=3
for each condition). Taken together, our data suggest that
CaMKII-mediated activation of ASIC1 affects the ability of GBM
cells to migrate, and that inhibition of ASIC1 or CaMKII can
attenuate this migration.
Discussion
The aim of current study was to determine whether
CaMKII-mediated activation of ASIC1 contributes to the ability of
GBM cells to migrate. The major findings of the current study are
as follows: i) ASIC1 and CaMKII physically interact and
co-localized at the plasma membrane in U251-MG cells; ii) CaMKII
regulates ASIC1 in U251-MG cells; iii) CaMKII-mediated activation
of ASIC1 affects the ability of U251-MG cells to migrate.
It has been reported that ASIC1a functions as a
non-selective transient channel in rat C6 glioma cells; activation
of ASIC1a induces a short depolarization or transient calcium
influx in these cells, even if the acidic stimulus is persistent
(22). Glioma cells appear to not
exhibit the typical acid induced ASIC1 current (11,16),
which may be due to the channel already being maximally activated
by the native acidic condition (13). In this study, we recorded a
constitutively activated, amiloride-sensitive current in U251-MG
cells. This current is inhibited by treatment with PcTX-1, a
specific ASIC1 blocker. Moreover, upon knockdown of ASIC1, the
current was dramatically decreased. These data strongly suggest
that this constitutively activated current recorded in U251-MG
cells is carried by ASIC1.
CaMKII catalyzes the phosphorylation of ASIC1a at
Ser478 and Ser479 residues, which activates the ASIC channels. This
process may contribute to ischemia-induced cell death in rodent
ischemic CNS neurons (17). GBM
cells and ischemic CNS neurons share similar acidic and low oxygen
microenvironments (23,24). Therefore, we hypothesized that
CaMKII may regulate ASIC1 currents in GBM cells. We found that
CaMKII interacted with ASIC1 and co-localized at the plasma
membrane in U251-MG cells. Furthermore, we discovered that the
ASIC1 currents were significantly decreased in U251-MG cells upon
treatment with AIP, a specific CaMKII inhibitor. Moreover, AIP
treatment did not further decrease ASIC1 currents in cells where
ASIC1 was knocked down, suggesting that CaMKII specifically
regulates ASIC1 currents. ASIC1 plays a role in GBM cell migration
ability (11,20), cell cycle progression (20), and volume regulation (19). However, it is not known whether this
CaMKII-mediated activation of ASIC1 affects the ability of GBM
cells to migrate.
To this end, we tested whether inhibition of CaMKII
and/or ASIC1 would lead to reduced GBM cell migration. We reasoned
that if ASIC1 is the most relevant target for CaMKII, then
downregulation of ASIC1 expression or pharmacological inhibition of
its activity should result in significantly reduced migration of
U251-MG. The migration assay results show that both reduced
expression of ASIC1 or pharmacological inhibition of ASIC1 caused a
significant reduction of cell migration. Furthermore, inhibition of
CaMKII led to a greater reduction of cell migration, suggesting
CaMKII-mediated ASIC1 contributes to a reduction of the cell’s
ability to migrate. Since it has been reported that the
volume-gated chloride channel ClC-3 is involved in glioma invasion
(25) and that phosphorylation of
ClC-3 by CaMKII is important in glioma cell migration (26), the synergetic effect of PcTX-1 and
AIP on cell migration might be attributed to inhibition of ClC-3.
Although AIP treatment induced a greater reduction in cell
migration, this treatment did not further reduce migration in cells
where ASIC1 was knocked down.
Moreover, a combined PcTX-1 and AIP treatment
exhibited an additive effect on the reduction of cell migration in
cells where ASIC1 was knocked down. This additive effect might be a
result of residual ASIC1s in these cells. Specifically, AIP
treatment likely inhibits the activity of the residual ASIC1s and
further reduces cell migration. Taken together, these data suggest
that the migration of U251-MG cells is primarily regulated by
CaMKII-mediated, constitutively activated ASIC1 channels. Since it
has been suggested that ASIC1a is permeable to calcium (27), it is possible that these
constitutively activated ASIC1 channels may also allow calcium to
permeate and activate CaMKII in U251-MG cells, thereby regulating
ASIC1 channels. Nevertheless, our data may provide potential
therapeutic targets for preventing the invasiveness of gliomas.
Acknowledgements
This study was supported by the National High
Technology Research and Development Program of China (863 Program,
2012AA02A508), the National Basic Research Program of China (973
Program, 2012CB517803), the National Natural Science Foundation of
China (81070217 and 81270340), and the Research Foundation of
Chinese Ministry of Health (w2011bx059).
References
|
1
|
Soroceanu L, Manning TJ Jr and Sontheimer
H: Modulation of glioma cell migration and invasion using Cl(−) and
K(+) ion channel blockers. J Neurosci. 19:5942–5954.
1999.PubMed/NCBI
|
|
2
|
Kunzelmann K: Ion channels and cancer. J
Membr Biol. 205:159–173. 2005. View Article : Google Scholar
|
|
3
|
Roderick HL and Cook SJ: Ca2+
signalling checkpoints in cancer: remodelling Ca2+ for
cancer cell proliferation and survival. Nat Rev Cancer. 8:361–375.
2008.
|
|
4
|
Bubien JK, Keeton DA, Fuller CM, et al:
Malignant human gliomas express an amiloride-sensitive
Na+ conductance. Am J Physiol. 276:C1405–C1410.
1999.PubMed/NCBI
|
|
5
|
Olsen ML, Schade S, Lyons SA, Amaral MD
and Sontheimer H: Expression of voltage-gated chloride channels in
human glioma cells. J Neurosci. 23:5572–5582. 2003.PubMed/NCBI
|
|
6
|
Weaver AK, Bomben VC and Sontheimer H:
Expression and function of calcium-activated potassium channels in
human glioma cells. Glia. 54:223–233. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Berdiev BK, Xia J, McLean LA, et al:
Acid-sensing ion channels in malignant gliomas. J Biol Chem.
278:15023–15034. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kellenberger S and Schild L: Epithelial
sodium channel/degenerin family of ion channels: a variety of
functions for a shared structure. Physiol Rev. 82:735–767.
2002.PubMed/NCBI
|
|
9
|
Eaton DC and Hamilton KL: The
amiloride-blockable sodium channel of epithelial tissue. Ion
Channels. 1:251–282. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Escoubas P, De Weille JR, Lecoq A, et al:
Isolation of a tarantula toxin specific for a class of proton-gated
Na+ channels. J Biol Chem. 275:25116–25121. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kapoor N, Bartoszewski R, Qadri YJ, et al:
Knockdown of ASIC1 and epithelial sodium channel subunits inhibits
glioblastoma whole cell current and cell migration. J Biol Chem.
284:24526–24541. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Waldmann R and Lazdunski M: H(+)-gated
cation channels: neuronal acid sensors in the NaC/DEG family of ion
channels. Curr Opin Neurobiol. 8:418–424. 1998.
|
|
13
|
Wemmie JA, Price MP and Welsh MJ:
Acid-sensing ion channels: advances, questions and therapeutic
opportunities. Trends Neurosci. 29:578–586. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Krishtal O: The ASICs: signaling
molecules? Modulators Trends Neurosci. 26:477–483. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ettaiche M, Guy N, Hofman P, Lazdunski M
and Waldmann R: Acid-sensing ion channel 2 is important for retinal
function and protects against light-induced retinal degeneration. J
Neurosci. 24:1005–1012. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kapoor N, Lee W, Clark E, et al:
Interaction of ASIC1 and ENaC subunits in human glioma cells and
rat astrocytes. Am J Physiol Cell Physiol. 300:C1246–C1259. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gao J, Duan B, Wang DG, et al: Coupling
between NMDA receptor and acid-sensing ion channel contributes to
ischemic neuronal death. Neuron. 48:635–646. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bordey A, Sontheimer H and Trouslard J:
Muscarinic activation of BK channels induces membrane oscillations
in glioma cells and leads to inhibition of cell migration. J Membr
Biol. 176:31–40. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ross SB, Fuller CM, Bubien JK and Benos
DJ: Amiloride-sensitive Na+ channels contribute to
regulatory volume increases in human glioma cells. Am J Physiol
Cell Physiol. 293:C1181–C1185. 2007.
|
|
20
|
Rooj AK, McNicholas CM, Bartoszewski R,
Bebok Z, Benos DJ and Fuller CM: Glioma-specific cation conductance
regulates migration and cell cycle progression. J Biol Chem.
287:4053–4065. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nerbonne JM: Repolarizing cardiac
potassium channels: multiple sites and mechanisms for
CaMKII-mediated regulation. Heart Rhythm. 8:938–941. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Weng XC, Zheng JQ, Li J and Xiao WB:
Underlying mechanism of ASIC1a involved in acidosis-induced
cytotoxicity in rat C6 glioma cells. Acta Pharmacol Sin.
28:1731–1736. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Allen NJ and Attwell D: Modulation of ASIC
channels in rat cerebellar Purkinje neurons by ischaemia-related
signals. J Physiol. 543:521–529. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xu L, Fukumura D and Jain RK: Acidic
extracellular pH induces vascular endothelial growth factor (VEGF)
in human glioblastoma cells via ERK1/2 MAPK signaling pathway:
mechanism of low pH-induced VEGF. J Biol Chem. 277:11368–11374.
2002. View Article : Google Scholar
|
|
25
|
Ransom CB, O’Neal JT and Sontheimer H:
Volume-activated chloride currents contribute to the resting
conductance and invasive migration of human glioma cells. J
Neurosci. 21:7674–7683. 2001.PubMed/NCBI
|
|
26
|
Cuddapah VA and Sontheimer H: Molecular
interaction and functional regulation of ClC-3 by
Ca2+/calmodulin-dependent protein kinase II (CaMKII) in
human malignant glioma. J Biol Chem. 285:11188–11196. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Waldmann R: Proton-gated cation channels -
neuronal acid sensors in the central and peripheral nervous system.
Adv Exp Med Biol. 502:293–304. 2001. View Article : Google Scholar : PubMed/NCBI
|