Introduction
Recent studies have identified a role for
cyclooxygenase-2 (COX-2) in the development and progression of
various tumor types. In vitro and in vivo
investigations have shown that selective COX-2 inhibitors produce
antiproliferative effects in various malignancies, such as gastric,
esophageal, oral, brain, lung and pancreatic cancers (1). Several studies have demonstrated the
antitumor effects of selective COX-2 inhibitors on endometrial
cancer in vitro(2–4). Moreover, small pilot studies have
described the effects of oral administration of selective COX-2
inhibitors in prostate cancer and endometrial cancer patients
(5,6). In a previous study, we found that the
selective COX-2 inhibitor etodolac showed antiproliferative effects
by suppressing COX-2 and cell-cycle regulator protein expression in
patients with endometrial cancer positive for COX-2 expression
(6).
In addition to the use of COX-2 inhibitors alone,
co-administration of chemotherapeutic agents with selective COX-2
inhibitors has been shown to enhance the effects of the
chemotherapeutic agent in pancreas and lung cancer cell lines and
animal models (6–8), and several clinical trials using
selective COX-2 inhibitors have been conducted for human cancers.
In phase II clinical trials for non-small cell lung cancer, the
COX-2 inhibitor celecoxib was shown to significantly enhance the
response to conventional chemotherapeutic agents, i.e., carboplatin
and paclitaxel (9). Similar phase
II studies of selective COX-2 inhibitors in combination with
chemotherapeutic drugs with good efficacy and safety have been
reported in patients with pancreatic (10) and small cell lung cancer (11).
Moreover, the association between COX-2 expression
and multidrug resistance 1 (MDR1) expression has been reported in
various types of cancers (12–15).
Drug resistance may involve the MDR1 gene, which encodes the
transmembrane glycoprotein p-170 (P-gp). P-gp is a transmembrane
phosphoglycoprotein from the ATP-binding cassette superfamily (ABC)
and functions as an efflux pump, transporting a wide range of
compounds and, therefore, facilitating decreased intracellular drug
concentrations and reduced cancer chemotherapy efficacies of such
drugs as doxorubicin (16,17) and paclitaxel (18). COX-2 inhibitors have been reported
to potentially modulate resistance to chemotherapeutic drugs by
affecting MDR1 expression and enhancing the effects of conventional
chemotherapeutic agents (12–15,19,20).
Ratnasinghe et al(12)
proposed that COX-2 inhibitors may act as chemosensitizers,
improving the efficacy of chemotherapeutic agents in part by
inhibiting MDR1.
In ovarian cancer, COX-2 and MDR1 expression levels
are associated with chemotherapy resistance and poor prognosis
(21,22). Therefore, the combination of a
selective COX-2 inhibitor and a chemotherapeutic drug may enhance
the effects of the chemotherapeutic drug alone.
However, no studies have investigated the
relationship between COX-2 and MDR1 expression in endometrial
cancer. Therefore, in the present study, we investigated the
association between COX-2 and MDR1 mRNA expression in
endometrial cancers and evaluated the effects of the COX-2
inhibitor etodolac in combination with paclitaxel on
paclitaxel-resistant endometrial cancer cells. We also investigated
the possibility of overcoming paclitaxel resistance by modulation
of MDR1 expression and activity.
Materials and methods
Relationship between COX-2 and MDR1 mRNA
expression
Thirty-six patients with pathologically confirmed
endometrial carcinoma who underwent surgery at our institution from
2004 to 2008 were enrolled in the present study after providing
informed consent. Thirty-six surgical specimens were subjected to
real-time quantitative RT-PCR to confirm the expression levels of
COX-2 and MDR1 mRNA, and the relationship between
these transcripts was examined. Pathologically confirmed
histological subtypes included endometrioid adenocarcinoma (n=25),
serous adenocarcinoma (n=5) and clear cell adenocarcinoma (n=6).
Total RNA was extracted from tumor samples using an RNeasy kit
(Qiagen, Tübingen, Germany), following the manufacturer’s
instructions and was then subjected to complementary DNA (cDNA)
synthesis using a High-Capacity cDNA reverse transcription kit
(Applied Biosystems, Foster City, CA, USA). cDNA was subsequently
used for fluorescence-based real-time quantitative RT-PCR (TaqMan
PCR) with an ABI Prism 7900 Sequence Detector System (Applied
Biosystems) according to methods described elsewhere (23,24).
The housekeeping gene glyceraldehyde 3-phosphate dehydrogenase
(GAPDH) served as an internal control due to its stable expression
in different tissues. Primers and TaqMan probes were purchased from
Applied Biosystems, and the following primers were used: COX-2
forward, CCTTCCTCCTGTGCCTGATG and reverse,
ACAATCTCATTTGAATCAGGAAGCT; MDR1 forward, GTGGTGTTTCAGAATGGCAGAGT
and reverse, AGCCTGGACACTGACCATTGA; GAPDH forward,
GAAGGTGAAGGTCGGAGTC and reverse, GAAGATGGTGATGGGATTTC.
TaqMan probes were labeled with the reporter dye
6-carboxyfluorescein (FAM) at the 5′-end of the oligonucleotide and
with the quencher dye 6-carboxytetramethylrhodamine (TAMRA) at the
3′-end. The PCR conditions were as follows: 50°C for 2 min and 95°C
for 10 min, followed by 40 cycles at 95°C for 15 sec and 60°C for 1
min. All assays were run in triplicate. The data were analyzed by
the ΔΔCt method for comparing relative expression results (ratio, 2
− [Ctsample −
CtGAPDH]), where Ct means the threshold
cycle. The relative quantity of mRNA was represented as the mean ±
standard deviation (SD).
Establishment of a paclitaxel-resistant
endometrial cancer cell line
To establish a paclitaxel-resistant cell line, we
used the uterine endometrial cancer cell line, OMC-2 (25), which was derived from a moderately
differentiated tumor and has been shown to express COX-2 mRNA and
protein. OMC-2 cells were grown in Ham’s F-12 medium supplemented
with 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100
μg/ml streptomycin at 37°C in a humidified 5% CO2
atmosphere. A clone of paclitaxel-resistant OMC-2 cells was
selected by incubating ‘native’ OMC-2 cells in the presence of
paclitaxel from 0 to 1 μg/ml, gradually escalating the dose of
paclitaxel over a period of 4 months. The IC50 values
for paclitaxel and doubling times in ‘native’ and ‘resistant’ OMC-2
cells were calculated by MTT assay and growth curves,
respectively.
Analysis of COX-2 and MDR1 mRNA
expression in endometrial cancer cells
OMC-2 and OMC-2P cells were seeded in 5-cm dishes at
1×105 cells/well and were grown in medium containing 1
μg/ml paclitaxel alone, 10 μg/ml etodolac alone or 1 μg/ml
paclitaxel plus 10 μg/ml etodolac for 24 h. Total RNA from these
cells was extracted using an RNeasy kit as previously described.
Real-time RT-PCR was then used to analyze the expression of
COX-2 and MDR1 mRNA as previously described.
Measurement of PGE2
concentrations in the supernatants of endometrial cancer cells
OMC-2 and OMC-2P cells were seeded in 6-well dishes
at 1×105 cells/well and were grown in medium containing
1 μg/ml paclitaxel alone, 10 μg/ml etodolac alone or 1 μg/ml
paclitaxel plus 10 μg/ml etodolac for 24 h. The concentration of
prostaglandin E2 (PGE2) in the conditioned
medium collected from each well was determined by enzyme-linked
immunosorbent assay (ELISA) and was normalized to the total protein
concentration. This experiment was run in triplicate.
Rhodamine 123 efflux assay
The function of MDR1 was determined by intracellular
accumulation of Rhodamine 123 (R123) using flow cytometry (13,17).
Subconfluent OMC-2 and OMC-2P cells were incubated in medium
containing 1 μg/ml paclitaxel with or without 10 μg/ml etodolac at
37°C in a humidified 5% CO2 atmosphere for 24 h. The
cells were then incubated in HEPES-buffered solution consisting of
125 mM NaCl, 5 mM KCl, 1 mM MgSO4, 1.36 mM
Na2HPO4, 10 mM sodium acetate, 5 mM HEPES,
1.8 mM CaCl2, and 8 mM glucose titrated to pH 7.4 and
treated with 1 μM R123 for 1 h at 37°C. The cells were collected by
incubation with trypsin containing phosphate-buffered solution with
1 μM R123, washed 3 times with HEPES-buffered solution and
centrifuged at 1,500 rpm for 5 min at 4°C. After the supernatant
containing extracellular R123 was removed, loading was measured by
flow cytometry using a FACSCalibur (Becton-Dickinson, Franklin
Lakes, NJ, USA) to examine the initial level of intracellular
accumulation of R123. Next, the cells were resuspended in
HEPES-buffered solution and incubated for 2 h at 37°C. After
centrifugation at 1,500 rpm for 5 min at 4°C to remove any
extracellular R123, the level of R123 retained in the cells was
measured by flow cytometry to determine the efflux of R123. The
histogram of immunofluorescence of the cell population was plotted
using CellQuest software (Becton-Dickinson). The lowest
fluorescence intensity of the cells taking up R123 at initial
loading was defined as M1, and the percentage of R123 effluxed
cells to all cells including the M1 value was calculated. All
experiments were carried out in triplicate.
Measurement of intracellular paclitaxel
concentrations
Subconfluent OMC-2 and OMC-2P cells were incubated
in medium containing 1 μg/ml paclitaxel with or without 10 μg/ml
etodolac at 37°C in a humidified 5% CO2 atmosphere for 6
h. The cells were collected by incubation with trypsin in
phosphate-buffered solution, washed 3 times with phosphate-buffered
solution and centrifuged at 3,000 rpm for 5 min at 4°C. The weight
of the cell pellet was measured, and the pellet was homogenized by
ultrasonication in ice-cold water after addition of 1 ml of 0.07 M
phosphate buffer. After centrifugation at 3,000 rpm for 5 min at
4°C, the supernatant was applied to HPLC (L-7100; Hitachi, Tokyo,
Japan), following the manufacturer’s instructions, in order to
determine the concentration of paclitaxel.
Statistical analysis
The statistical analysis was performed using the
Chi-square test or the Student’s t-test. Differences with a P-value
<0.05 were considered to indicate a statistically significant
result.
Results
Relationship between COX-2 and MDR1 mRNA
expression
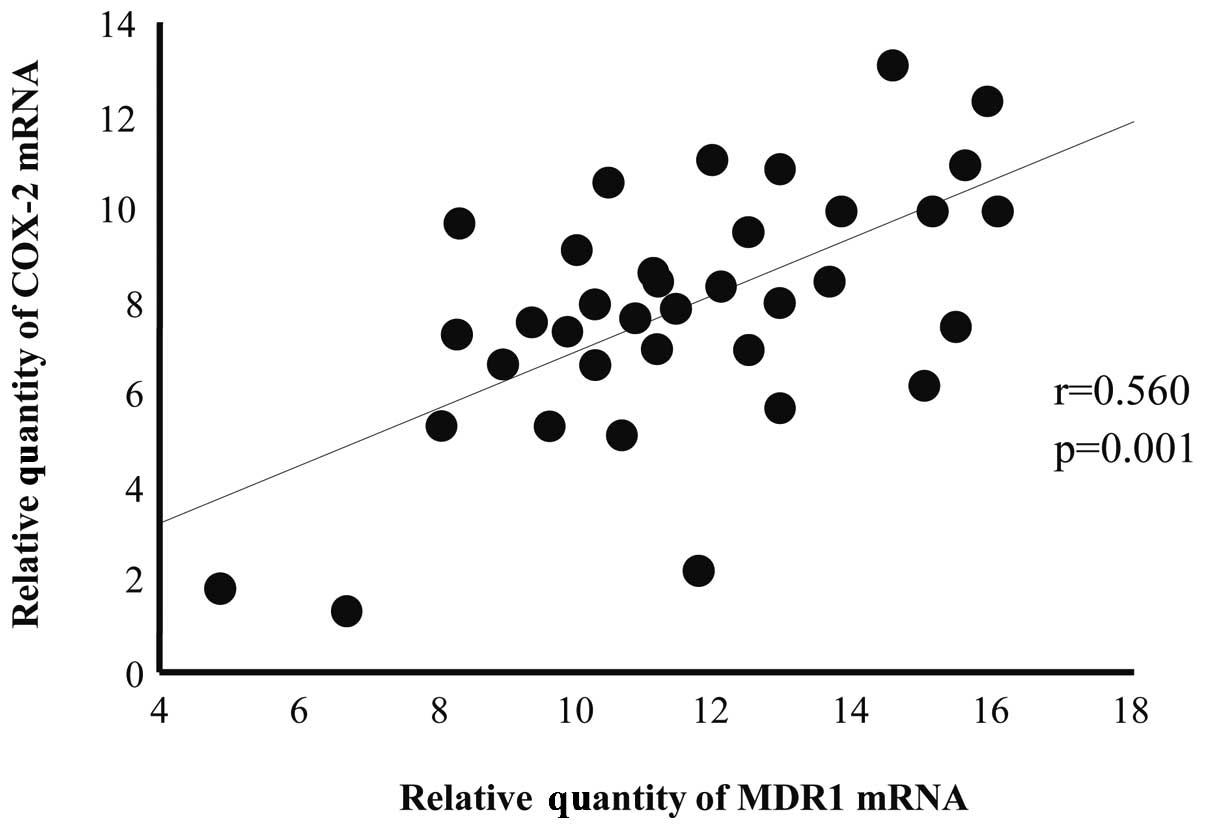
There was a positive correlation between
COX-2 and MDR1 mRNA expression in the 36 endometrial
carcinoma specimens (r=0.560, P=0.001; Fig. 1). Moreover, COX-2 and
MDR1 mRNAs were associated with type 2 non-endometrioid
adenocarcinomas, such as serous or clear cell adenocarcinomas, as
well as type 1 endometrioid adenocarcinomas (data not shown).
Characterization of paclitaxel-resistant
endometrial cancer cells
The obtained paclitaxel-resistant cell line was
designated as OMC-2P. The morphological appearances of the ‘native’
OMC-2 and ‘resistant’ OMC-2P cells were almost identical as
determined by phase-contrast microscopy. The IC50 values
for paclitaxel (after 3 days of exposure) in OMC-2 and OMC-2P
cells, as calculated by MTT assay, were 0.11 and 1.95 μg/ml,
respectively, demonstrating that OMC-2P cells were 17.7 times more
resistant to paclitaxel than the parental cells (data not shown).
The doubling times of OMC-2 and OMC-2P cells, calculated by growth
curves, were 36 and 48 h, respectively (data not shown).
Analysis of COX-2 and MDR1 mRNA
expression in endometrial cancer cell lines
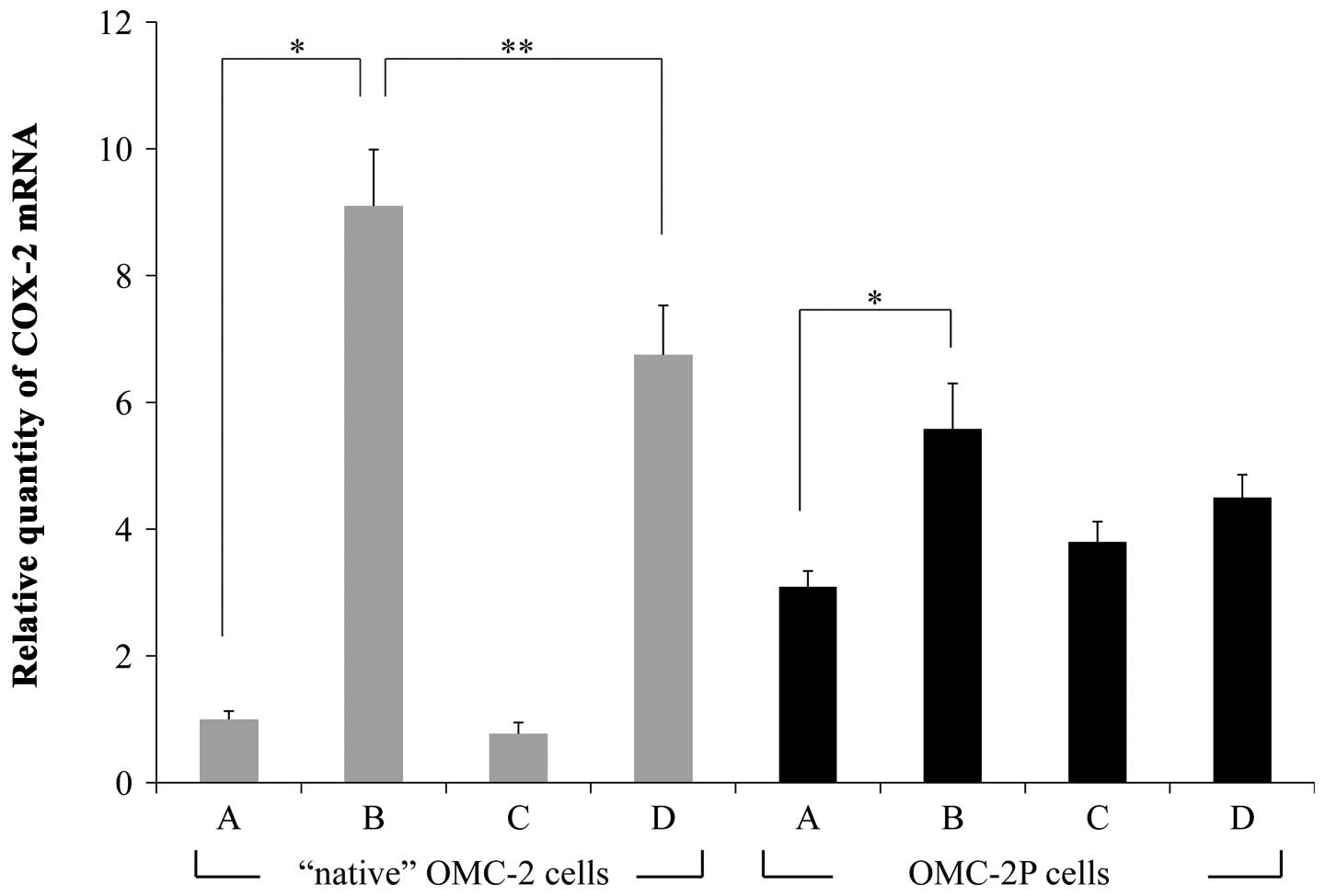
When the relative quantity of COX-2 mRNA in
the control OMC-2 cells was defined as 1.0, the expression levels
were 9.10±0.89, 0.77±0.18 and 6.75±0.78 in the OMC-2 cells treated
with paclitaxel alone, etodolac alone, or co-administration of
paclitaxel and etodolac, respectively. COX-2 expression was
significantly upregulated by paclitaxel treatment (P<0.01)
compared to that of the untreated control and was downregulated by
co-administration with etodolac when compared to that of cells
treated with paclitaxel alone (P<0.05; Fig. 2). In the OMC-2P cells, COX-2
expression levels were 3.09±0.25, 5.58±0.72, 3.80±0.32 and
4.50±0.36 in the control, paclitaxel-treated, etodolac-treated, and
paclitaxel plus etodolac-treated cells, respectively. COX-2
mRNA expression was also significantly upregulated by paclitaxel
treatment (P<0.01) when compared to that of the control and
tended to be downregulated by co-administration with etodolac
(P=0.067; Fig. 2). The expression
of COX-2 in the control OMC-2P cells was about 3-times
higher than that in the control OMC-2 cells.
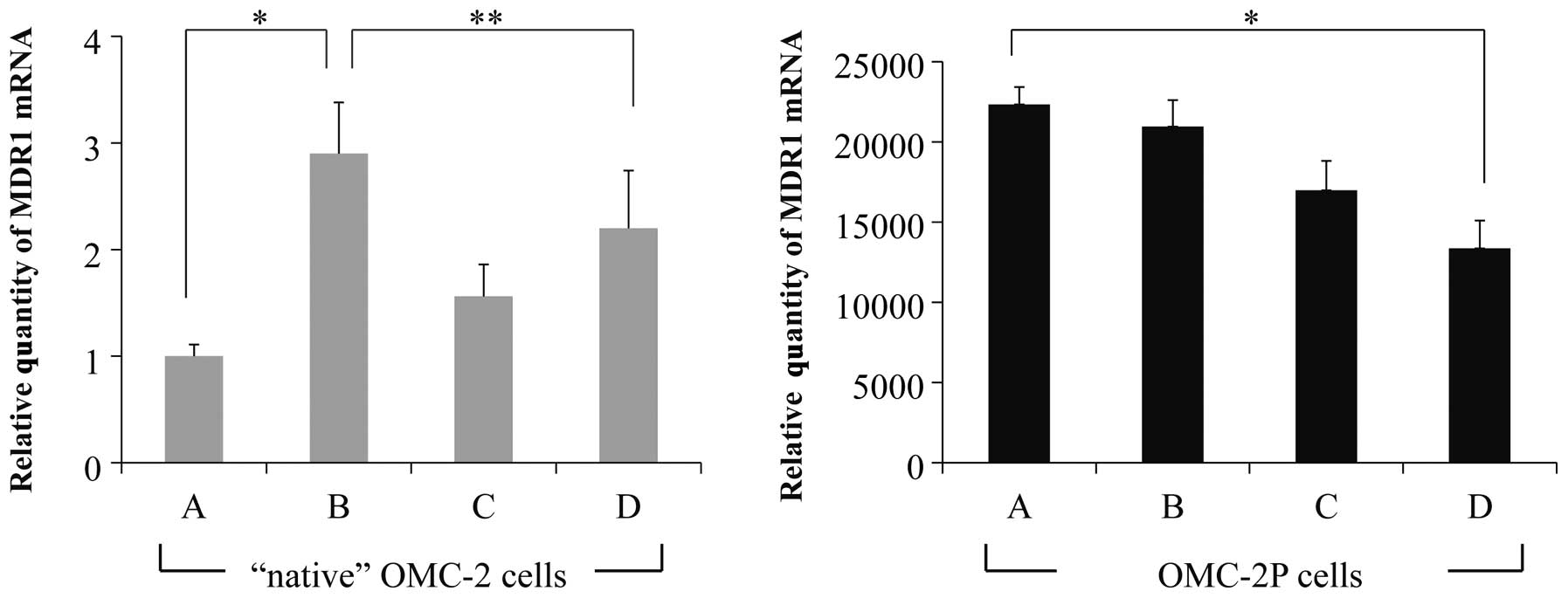
When the relative quantity of MDR1 mRNA in
the control ‘native’ OMC-2 cells was defined as 1.0, the expression
levels of MDR1 mRNAs were 2.90±0.48, 1.56±0.30 and 2.20±0.54
in the OMC-2 cells treated with paclitaxel alone, etodolac alone,
or both paclitaxel and etodolac, respectively. Additionally,
MDR1 expression was significantly upregulated by paclitaxel
treatment (P<0.01) when compared to that of the control cells
and was downregulated by co-administration of paclitaxel and
etodolac when compared to that of cells treated with paclitaxel
alone (P<0.05; Fig. 3). In the
OMC-2P cells, MDR1 expression levels were 22347.0±1078.1,
20965.8±1645.4, 16987.8±1833.2 and 13369.8±1731.5 in the untreated
control, paclitaxel-treated, etodolac-treated, and paclitaxel plus
etodolac-treated cells, respectively. MDR1 mRNA level in the
control OMC-2P cells was markedly higher than that in the control
‘native’ OMC-2 cells and was not affected by paclitaxel or etodolac
treatment, but was downregulated by co-administration of paclitaxel
and etodolac (P<0.01; Fig.
3).
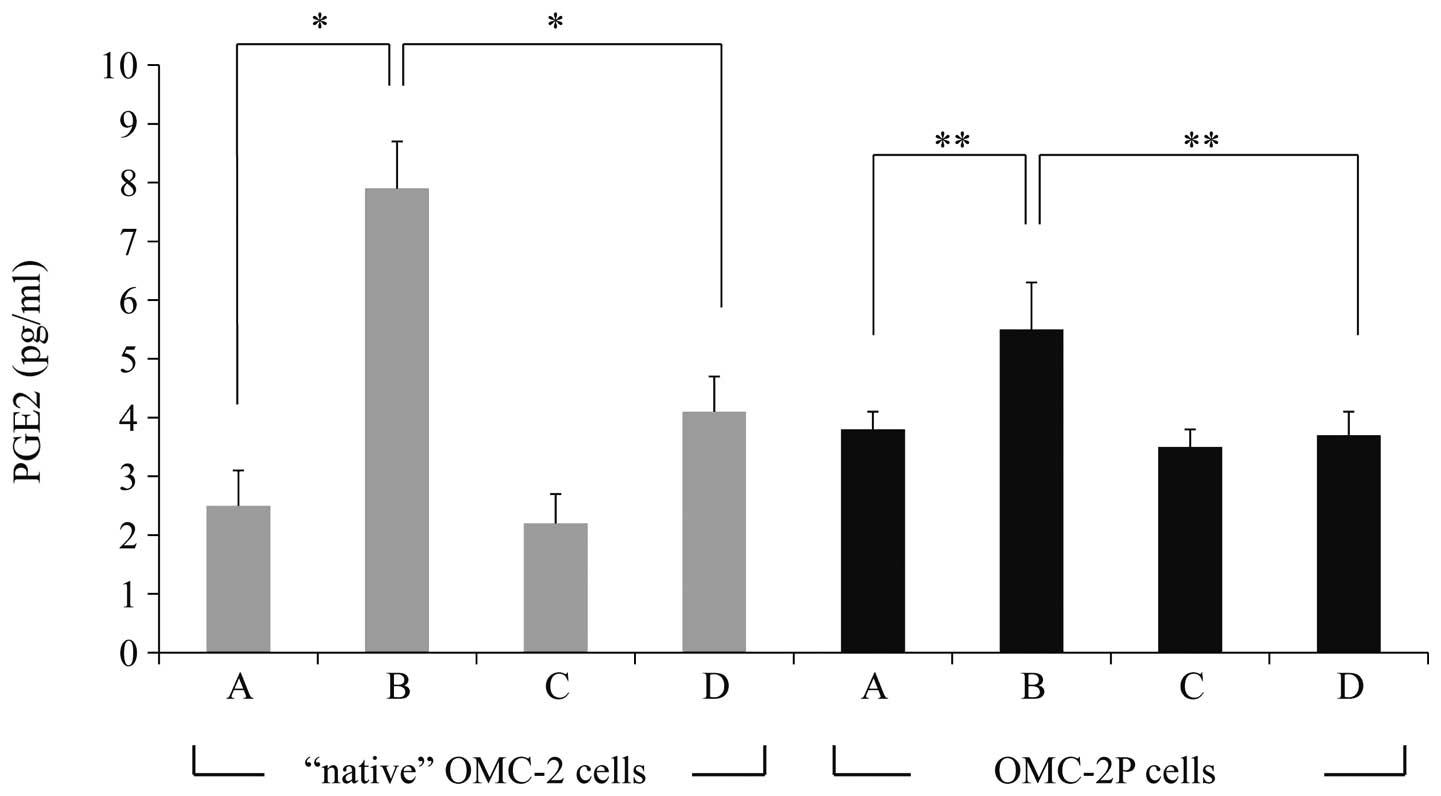
PGE2 concentrations in the
supernatants of OMC-2 and OMC-2P cells
The concentrations of PGE2 in the
supernatants of the untreated control, paclitaxel-treated,
etodolac-treated and paclitaxel plus etodolac-treated OMC-2 cells
were 2.5±0.6, 7.9±0.8, 2.2±0.5 and 4.1±0.6 pg/ml, respectively.
PGE2 was significantly upregulated by paclitaxel
treatment (P<0.01) when compared to that in the untreated
control cells and was downregulated by co-administration of
paclitaxel and etodolac compared to that in cells treated with
paclitaxel alone (P<0.01; Fig.
4). The concentrations of PGE2 in the supernatants
of the untreated control, paclitaxel-treated, etodolac-treated and
paclitaxel plus etodolac-treated OMC-2P cells were 3.8±0.3,
5.5±0.8, 3.5±0.3 and 3.7±0.4 pg/ml, respectively. PGE2
was also upregulated by paclitaxel treatment (P<0.05) when
compared to that in the untreated control OMC-2P cells and was
downregulated by co-administration of paclitaxel and etodolac
(P<0.05; Fig. 4).
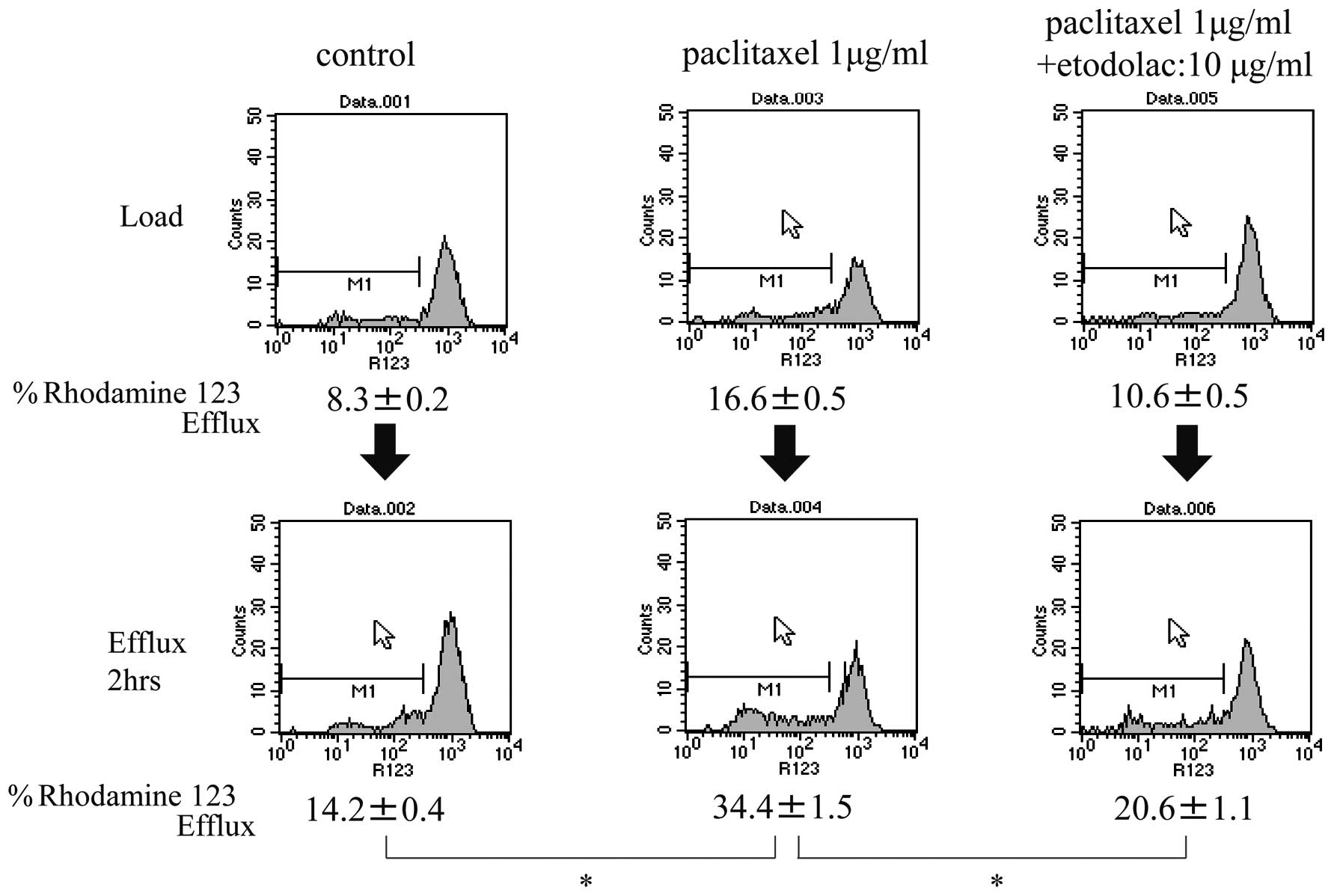
R123 efflux assay
In the ‘native’ OMC-2 cells, the percentages of R123
efflux at time 0 (load) were 8.3±0.2, 16.6±0.5 and 10.5±0.5% in the
untreated control, paclitaxel-treated, and paclitaxel plus
etodolac-treated cells, respectively. These percentages were
increased after 2 h of efflux to 14.2±0.4, 34.4±1.5 and 20.6±1.1%,
respectively (Fig. 5). In the
OMC-2P cells, the percentages of R123 efflux at load were 0.3±0.2,
5.6±0.6 and 4.9±0.9% in the untreated control, paclitaxel-treated,
and paclitaxel plus etodolac-treated cells, respectively. These
percentages were markedly increased after 2 h of efflux to
57.1±2.0, 93.5±1.9 and 73.6±3.3%, respectively (Fig. 6). Intracellular accumulation of R123
after 2 h of efflux was dramatically decreased in the OMC-2P cells
when compared to that in OMC-2 cells, and R123 accumulation was
decreased by paclitaxel treatment and increased by
co-administration of paclitaxel and etodolac in both the OMC-2P and
OMC-2 cells.
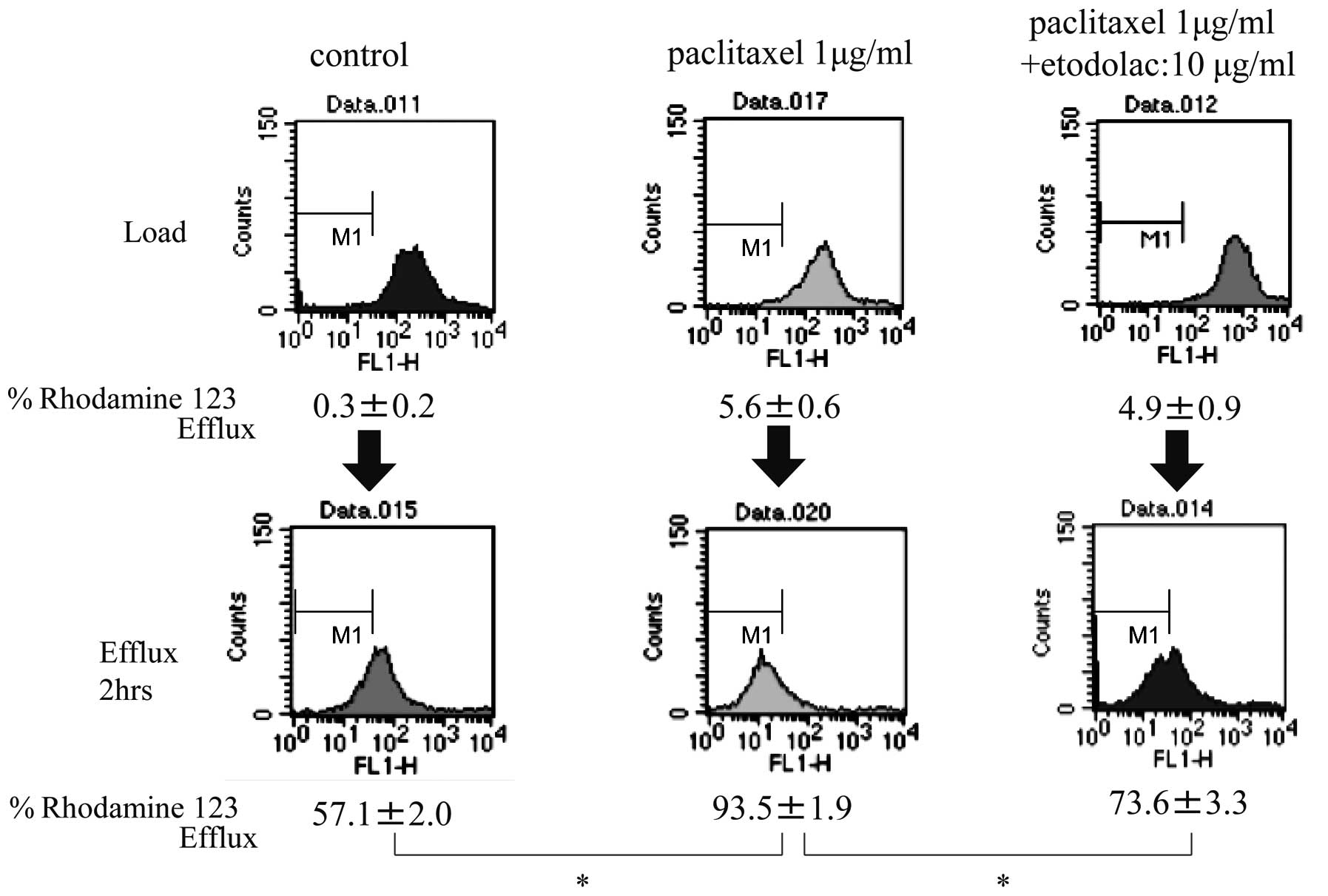 | Figure 6Rhodamine 123 efflux assay in OMC-2P
cells. In the OMC-2P cells, the percentages of R123 efflux at load
were 0.3±0.2, 5.6±0.6 and 4.9±0.9% in the untreated control,
paclitaxel-treated, and paclitaxel plus etodolac-treated cells,
respectively. These percentages were markedly increased after 2 h
of efflux to 57.1±2.0, 93.5±1.9 and 73.6±3.3%, respectively.
Intracellular accumulation of R123 after 2 h of efflux was
dramatically decreased in the OMC-2P cells when compared to that in
the OMC-2 cells, and R123 accumulation was decreased by paclitaxel
treatment and increased by co-administration of paclitaxel and
etodolac in the OMC-2P cells. x-axis, fluorescence intensity;
y-axis, cell number. *P<0.01. |
Concentrations of intracellular
paclitaxel
The concentrations of intracellular paclitaxel were
20.5±2.2 and 47.2±3.8 (ng/g wet weight/104 cells) in the
‘native’ OMC-2 cells treated with paclitaxel or co-administration
of paclitaxel and etodolac, respectively. The concentration of
intracellular paclitaxel in the OMC-2 cells treated with both
paclitaxel and etodolac was significantly higher than that in cells
treated with paclitaxel alone (P<0.01). In the OMC-2P cells, the
concentrations of intracellular paclitaxel were 2.3±0.8 and 9.2±1.3
(ng/g wet weight/104 cells) in cells treated with
paclitaxel or co-administration of paclitaxel and etodolac,
respectively. The concentration of intracellular paclitaxel in the
OMC-2P cells treated with paclitaxel was significant lower than
that in the OMC-2 cells treated with paclitaxel alone (P<0.01)
and was significantly increased after co-administration of
paclitaxel and etodolac (P<0.01; Table I).
 | Table IConcentration of intracellular
paclitaxel in the OMC-2 and OMC-2P cells. |
Table I
Concentration of intracellular
paclitaxel in the OMC-2 and OMC-2P cells.
| Cell lines | Treatment | Intracellular
paclitaxel concentration (ng/g wet weight/104
cells) |
|---|
| OMC-2 | Paclitaxel 1
μg/ml | 20.5±2.2 |
| Paclitaxel 1 μg/ml
+ etodolac 10 μg/ml | 47.2±3.8a |
| OMC-2P | Paclitaxel 1
μg/ml | 2.3±0.8b |
| Paclitaxel 1 μg/ml
+ etodolac 10 μg/ml | 9.2±1.3c |
Discussion
COX-2 has been shown to modulate MDR1 expression,
and inhibition of COX-2 activity results in downregulation of MDR1
expression and function in various types of cancer cells (12–15,19,20).
Moreover, several studies have demonstrated that cytotoxic
drug-induced MDR1 overexpression is effectively downregulated by
COX-2 inhibitors in vitro(16,26,27).
Chen et al(27)reported that
the expression and function of MDR1 in the breast cancer cell line
MCF-7 is upregulated by treatment with doxorubicin, indicating the
chemoresistant phenotype, and its expression and function are also
significantly downregulated by treatment with both doxorubicin and
the COX-2 inhibitor celecoxib. They concluded that celecoxib is
capable of preventing the development of the chemoresistant
phenotype induced by doxorubicin. Zatelli et al(16) also demonstrated that treatment with
a selective COX-2 inhibitor (NS-398) significantly reduced MDR1
expression in doxorubicin-resistant breast cancer cells (rMCF7
cells), and they hypothesized that COX-2 inhibitors can prevent or
reduce the development of the chemoresistant phenotype in breast
cancer cells by inhibiting MDR1 expression and function.
In the present study we demonstrated, for the first
time, the positive relationship between COX-2 and
MDR1 mRNA expression in endometrial cancers. We established
a paclitaxel-resistant cell line, designated OMC-2P, from ‘native’
OMC-2 cells. COX-2 mRNA expression and PGE2
production were elevated in the ‘resistant’ OMC-2P cells when
compared to these values in the OMC-2 cells. Moreover, MDR1
mRNA expression was markedly upregulated in the OMC-2P cells. In
the OMC-2 cells, COX-2 and MDR1 mRNAs were
significantly upregulated by paclitaxel treatment and downregulated
by co-administration with etodolac. In the OMC-2P cells,
COX-2 mRNA expression was also significantly upregulated by
paclitaxel treatment and tended to be downregulated by
co-administration with etodolac. Moreover, the markedly elevated
MDR1 mRNA expression levels in OMC-2P cells were not further
influenced by treatment with paclitaxel or etodolac alone, but were
downregulated by co-administration of paclitaxel and etodolac.
Intracellular accumulation of R123 as determined by flow cytometry
was markedly decreased in the OMC-2P cells when compared to that in
the OMC-2 cells, but was decreased by paclitaxel treatment and
increased by co-administration of paclitaxel plus etodolac in both
cell lines. The actual concentration of intracellular paclitaxel in
the OMC-2P cells as measured by HPLC was significantly lower than
that in the OMC-2 cells when treated with paclitaxel alone, but was
significantly increased after co-administration of paclitaxel and
etodolac. In summary, our data suggest the possible downregulation
of MDR1 by the COX-2 inhibitor etodolac, which may enhance
the accumulation of MDR1 substrates, such as paclitaxel.
Several mechanisms have been proposed to explain the
close association between COX-2 and MDR1 expression
and the downregulation of MDR1 by COX-2 inhibitors. The MDR1
gene promoter contains putative binding sites for the transcription
factors activator protein 1 (AP-1) and nuclear factor-κB (NF-κB),
which appear to be relevant for MDR1 gene induction
(28). Drug resistance in breast
cancer MCF-7 cells has been reported to be accompanied by increases
in AP-1 activity (29). Moreover,
NF-κB, a ubiquitous transcription factor involved in immunity,
inflammation, regulation of cell growth, differentiation, and
apoptosis, was found to induce drug resistance through MDR1
expression (28,30). The inhibition of these factors by
COX-2 inhibitors would induce negative regulation of the
MDR1 gene. Ratnasinghe et al(12) postulated that prostaglandins
modulate the MDR1 gene via the induction of phosphokinase C
(PKC) and subsequent expression of c-Jun (a subunit of AP-1). COX-2
inhibitors could block this cascade, resulting in negative
modulation of the MDR1 gene. Chen et al(27) showed that the COX-2 inhibitor
celecoxib decreased c-Jun and NF-κB expression at the mRNA and
protein level and significantly impaired the DNA-binding activity
of AP-1 and NF-κB, which was partly associated with downregulated
MDR1 expression induced by doxorubicin. Moreover, the activity of
c-Jun NH2-terminal kinase (JNK), a member of the
mitogen-activated protein kinase family that functions downstream
of COX-2, has been implicated in the regulation of MDR1 expression,
and COX-2 and JNK signaling pathways are associated with
MDR1-mediated drug resistance (31,32).
From the above findings, we assumed that cellular
stress caused by paclitaxel treatment induced COX-2 expression and
PGE2 production in the present study, which in turn may
have enhanced the expression of transcription factors, such as AP-1
or NF-κB, and thus ultimately induced the expression of MDR1 in
endometrial cancer cells as well as in other types of cancer cells.
Therefore, our data suggest that the COX-2 inhibitor suppressed
these mediators and paclitaxel efflux, thereby increasing
paclitaxel concentrations in the cells.
Although AP-1 and NF-κB have been shown to enhance
the effects of COX-2 inhibitors on cytotoxic drugs, several studies
have shown conflicting results. Moreover, our data did not exclude
the possibility that other transcription factors, such as Sp1,
nuclear transcription factor Y (NF-Y), Y box binding protein 1
(YB-1), p53, and CCAAT/enhancer binding protein β (C/EBPβ), may be
involved in mediating the downregulation of MDR1 expression by
COX-2 inhibitors. It is also possible that transcription factors
may interact with each other. Therefore, the precise mechanisms
through which COX-2 inhibitors enhance the efficacy of cytostatic
drugs are not fully understood.
In endometrial cancers, chemotherapy is indicated
for patients with advanced or recurrent disease, and platinum-based
regimens in combination with doxorubicin or taxanes have been
recommended in treatment guidelines. However, outcomes for patients
with advanced-stage or recurrent disease are poor and such cancers
are rarely curable. In particular, the prognosis for patients with
recurrent disease showing multiple-drug resistance is markedly
poor. Therefore, it is critical to overcome resistance to cytotoxic
drugs in order to improve the prognoses of these patients. In the
present study, we demonstrated, for the first time, the possibility
of modulating or overcoming paclitaxel resistance by COX-2
inhibitors in endometrial cancers.
In conclusion, the findings of the present study
suggest that paclitaxel resistance in endometrial cancers may be
associated with elevated COX-2 and MDR1 expression in cancer cells.
It is possible that co-administration of paclitaxel and COX-2
inhibitors may play a key role in modulating or overcoming
paclitaxel resistance in endometrial cancers.
References
|
1
|
Dannenberg AJ and Subbaramaiah K:
Targeting cyclooxygenase-2 in human neoplasia: rationale and
promise. Cancer Cell. 4:431–436. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hasegawa K, Ohashi Y, Ishikawa K, et al:
Expression of cyclooxygenase-2 in uterine endometrial cancer and
anti-tumor effects of a selective COX-2 inhibitor. Int J Oncol.
26:1419–1428. 2005.PubMed/NCBI
|
|
3
|
Genç S, Attar E, Gürdöl F, Kendigelen S,
Bilir A and Serdaroğlu H: The effect of COX-2 inhibitor,
nimesulide, on angiogenetic factors in primary endometrial
carcinoma cell culture. Clin Exp Med. 7:6–10. 2007.PubMed/NCBI
|
|
4
|
Wood NJ, Quinton NA, Burdall S, Sheridan E
and Duffy SR: Exploring the potential chemopreventative effect of
aspirin and rofecoxib on hereditary nonpolyposis colorectal
cancer-like endometrial cancer cells in vitro through mechanisms
involving apoptosis, the cell cycle, and mismatch repair gene
expression. Int J Gynecol Cancer. 17:447–454. 2007. View Article : Google Scholar
|
|
5
|
Sooriakumaran P, Coley HM, Fox SB,
Macanas-Pirard P, Lovell DP, Henderson A, et al: A randomized
controlled trial investigating the effects of celecoxib in patients
with localized prostate cancer. Anticancer Res. 29:1483–1488.
2009.PubMed/NCBI
|
|
6
|
Hasegawa K, Torii Y, Ishii R, Oe S, Kato R
and Udagawa Y: Effects of a selective COX-2 inhibitor in patients
with uterine endometrial cancers. Arch Gynecol Obstet.
284:1515–1521. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Soriano AF, Helfrich B, Chan DC, et al:
Synergistic effects of new chemopreventive agents and conventional
cytotoxic agents against human lung cancer cell lines. Cancer Res.
59:6178–6184. 1999.PubMed/NCBI
|
|
8
|
Hida T, Kozaki K, Ito H, et al:
Significant growth inhibition of human lung cancer cells both in
vitro and in vivo by the combined use of a selective cyclooxygenase
2 inhibitor, JTE-522, and conventional anticancer agents. Clin
Cancer Res. 8:2443–2447. 2002.PubMed/NCBI
|
|
9
|
Altorki NK, Keresztes JL, Port JL, Libby
DM, Korst RJ, Flieder DB, et al: Celecoxib, a selective
cyclo-oxygenase-2 inhibitor, enhances the response to preoperative
paclitaxel and carboplatin in early-stage non-small-cell lung
cancer. J Clin Oncol. 21:2645–2650. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lipton A, Campbell-Baird C, Witters L,
Harvey H and Ali S: Phase II trial of gemcitabine, irinotecan, and
celecoxib in patients with advanced pancreatic cancer. J Clin
Gastroenterol. 44:286–288. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Aruajo AM, Mendez JC, Coelho AL, Sousa B,
Barata F, Figueiredo A, et al: Phase II study of celecoxib with
cisplatin plus etoposide in extensive-stage small cell lung cancer.
Cancer Invest. 27:391–396. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ratnasinghe D, Daschner PJ, Anver MR, et
al: Cyclooxygenase-2, P-glycoprotein-170 and drug resistance: Is
chemoprevention against multidrug resistance possible? Anticancer
Res. 21:2141–2147. 2001.PubMed/NCBI
|
|
13
|
Patel VA, Dunn MJ and Sorokin A:
Regulation of MDR-1 (p-glycoprotein) by cyclooxygenase-2. J Biol
Chem. 277:38915–38920. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sorokin A: Cyclooxygenase-2: potential
role in regulation of drug efflux and multidrug resistance
phenotype. Curr Pharm Des. 10:647–657. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zatelli MC, Luchin A, Piccin D, et al:
Cyclooxgenase-2 inhibitors reverse chemoresistance phenotype in
medullary thyroid carcinoma by a permeability glycoprotein-mediated
mechanism. J Clin Endocrinol Metab. 90:5754–5760. 2005. View Article : Google Scholar
|
|
16
|
Zatelli MC, Luchin A, Tagliati F, et al:
Cyclooxygenase-2 inhibitors prevent the development of
chemoresistance phenotype in a breast cancer cell line by
inhibiting glycoprotein p-170 expression. Endocr Relat Cancer.
14:1029–1038. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fontaine M, Elmquist WF and Miller DW: Use
of rhodamine 123 to examine the functional activity of
P-glycoprotein in primary cultured brain microvessel endothelial
cell monolayers. Life Sci. 59:1521–1531. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hille S, Rein DT, Riffelmann M, et al:
Anticancer drugs induce mdr1 gene expression in recurrent ovarian
cancer. Anticancer Drugs. 17:1041–1044. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zrieki A, Farinotti R and Buyse M:
Cyclooxygenase inhibitors down regulate P-glycoprotein in human
colorectal Caco-2 cell line. Pharm Res. 25:1991–2001. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fantappiè O, Masini E, Sardi I, et al: The
MDR phenotype is associated with the expression of COX-2 and iNOS
in a human hepatocellular carcinoma cell line. Hepatology.
35:843–852. 2002.
|
|
21
|
Raspollini MR, Amunni G, Villanucci A,
Boddi V and Taddei GL: Increased cyclooxygenase-2 (COX-2) and
P-glycoprotein-170 (MDR1) expression is associated with
chemotherapy resistance and poor prognosis. Analysis in ovarian
carcinoma patients with low and high survival. Int J Gynecol
Cancer. 15:255–260. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Surowiak P, Materna V, Denkert C, et al:
Significance of cyclooxygenase 2 and MDR1/P-glycoprotein
coexpression in ovarian cancers. Cancer Lett. 235:272–280. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xi H, Baldus SE, Warnecke-Eberz U, et al:
High cyclooxygenase-2 expression following neoadjuvant
radiochemotherapy is associated with minor histopathologic response
and poor prognosis in esophageal cancer. Clin Cancer Res.
11:8341–8347. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Saikawa Y, Sugiura T, Toriumi F, et al:
Cyclooxygenase-2 gene induction causes CDDP resistance in colon
cancer cell line, HCT-15. Anticancer Res. 24:2723–2728.
2004.PubMed/NCBI
|
|
25
|
Yamada T, Ueda M, Maeda T, et al:
Establishment and characterization of CA125 producing cell line
(OMC-2) originating from a human endometrial adenocarcinoma. Asia
Oceania J Obstet Gynecol. 15:403–416. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Puhlmann U, Ziemann C, Ruedell G, et al:
Impact of the cyclooxygenase system on doxorubicin-induced
functional multidrug resistance 1 overexpression and doxorubicin
sensitivity in acute myeloid leukemic HL-60 cells. J Pharmacol Exp
Ther. 312:346–354. 2005. View Article : Google Scholar
|
|
27
|
Chen C, Shen HL, Yang J, Chen QY and Xu
WL: Preventing chemoresistance of human breast cancer cell line,
MCF-7 with celecoxib. J Cancer Res Clin Oncol. 137:9–17. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bentires-Alj M, Barbu V, Fillet M, et al:
NF-κB transcription factor induces drug resistance through MDR1
expression in cancer cells. Oncogene. 22:90–97. 2003.
|
|
29
|
Daschner PJ, Ciolino HP, Plouzek CA and
Yeh GC: Increased AP-1 activity in drug resistant human breast
cancer MCF-7 cells. Breast Cancer Res Treat. 53:229–240. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
van Wijngaarden J, van Beek E, van Rossum
G, et al: Celecoxib enhances doxorubicin-induced cytotoxicity in
MDA-MB231 cells by NF-κB-mediated increase of intracellular
doxorubicin accumulation. Eur J Cancer. 43:433–442. 2007.PubMed/NCBI
|
|
31
|
Bassiouny AR, Zaky A and Neenaa HM:
Synergistic effect of celecoxib on 5-fluorouracil-induced apoptosis
in hepatocellular carcinoma patients. Ann Hepatol. 9:410–418.
2010.PubMed/NCBI
|
|
32
|
Sui H, Zhou S, Wang Y, et al: COX-2
contributes to P-glycoprotein-mediated multidrug resistance via
phosphorylation of c-Jun at Ser63/73 in colorectal cancer.
Carcinogenesis. 32:667–675. 2011. View Article : Google Scholar : PubMed/NCBI
|