Introduction
Our previous study and other authors demonstrated
that As2O3 inhibits the expression of
P-glycoprotein (P-gp) in leukemia cells (unpublished data)
(1); however, the mechanism behind
this inhibition is unclear. Induction of apoptosis is considered to
be the mechanism by which As2O3 acts as a
chemotherapeutic agent. Based on this finding, Mathas et al
(2) found that
As2O3 inhibits the activity of nuclear
factor-κB (NF-κB) and induces apoptosis in Hodgkin/Reed-Sternberg
cell lines. NF-κB is a widely distributed and functional eukaryotic
cell transcription factor that can be activated by many cell
factors and chemotherapeutic agents (2). In addition, a large body of evidence
has demonstrated that NF-κB is related to tumor drug resistance
(3–5).
Previous research suggests that, in addition to
inducing the expression of antiapoptotic genes, NF-κB also induces
the expression of P-gp. Bentires-Alj et al (7) and Kuo et al (8) demonstrated that the promoter of
multi-drug resistance gene 1 (MDR1, expressing P-gp) contained an
NF-κB binding sequence that activates transcription of an MDR1
promoter-driven reporter gene. These findings support the notion
that As2O3 has an effect on the reversal of
drug resistance by inhibition of NF-κB, consequently inducing
apoptosis and expression of MDR1.
Currently, As2O3 is mainly
used to treat promyelocytic leukemia patients and cases for which
chemotherapeutic agents are ineffective; however,
As2O3 is not currently used as a
drug-resistance modulator. According to analyses based on the
clinical effects and proposed mechanism of
As2O3, we hypothesized that it not only
possesses apoptosis-inducing capabilities, but also the ability to
reverse drug resistance. Exploitation of these capabilities could
lead to increasing effectiveness of chemotherapeutics for
treatment-resistant patients.
Therefore, we aimed to study the impact of
As2O3 on the expression and regulation of
P-gp in leukemia cells in order to explore the mechanism of the
reversal of drug resistance. The findings of the present study
provide an important theoretical foundation for the improvement of
chemotherapy. Moreover, study of the function of the reversal of
drug resistance by As2O3 may improve its
therapeutic value and expedite further studies concerning the
mechanism of tumor drug resistance.
Materials and methods
Cell culture
K562/D, K562/S and HEK293T cells were maintained in
RPMI-1640 with 10% fetal bovine serum (FBS), 100 units/ml
penicillin and 100 μg/ml streptomycin in a humidified atmosphere
with 5% CO2 at 37°C. The study was performed in
accordance with the ethical standards of the 1975 Declaration of
Helsinki, as revised in 2000, and was approved by the appropriate
institutional review boards.
In the present study, we used the K562/D and 293T
cell lines in the process of analysis of the inhibition of the MDR1
gene promoter. K562/D cells have a high level of P-gp expression,
and the function of its external input pump leads to low efficiency
of cell transfection; therefore, we choose human embryonic kidney
293T cells instead of K562/D cells for the transfection
experiments.
Identification of the NF-κB response
element of the MDR1 promoter by EMSA
Nuclear extracts were prepared from K562/D cells.
The wild-type probe of the MDR1 promoter was generated by annealing
two complementary oligonucleotides (5′-GCACTGCAGGGGCTTTCCTGTGCGC-3′ and
5′-GCGCACAGGAAAGCCCCTGCAGTGC-3′). The
core sequence of the NF-κB binding site is underlined and the 3′
recessive ends were filled by repair synthesis with dATP, dTTP,
dGTP [α-32P]-dCTP and the Klenow fragment of DNA
polymerase I. The mutated probe of the MDR1 promoter was generated
using the same procedure, except that several oligonucleotides were
mutated (5′-GCACTGCACTCGCTTTCCTGTGCGC-3′ and
5′-GCGCACAGGAAAGCGAGTGCAGTGC-3′). Nuclear extracts of the protein
were preincubated in 20 μl of binding buffer and 4% glycerol with
or without unlabeled excess competitor. For supershift assays, 1 μg
of rabbit IgG (Sigma) was added to the preincubation mixture. After
preincubation on ice for 30 min, the DNA probe labeled with
(α-32P) dCTP was added, and the samples were incubated
at room temperature for 30 min. The reaction mixtures were resolved
on 4% polyacrylamide gels. Antibodies used in the supershift assays
were rabbit polyclonal antibodies against NF-κB p65 from Santa Cruz
Biotechnology.
Western blot analysis
Cells were lysed in RIPA buffer containing a
complete protease inhibitor cocktail, followed by centrifugation at
12,000 × g at 4°C. The protein concentration was determined from
the supernatant by a bicinchoninic acid assay (Beyotime Institute
of Biotechnology, Haimen, China), and the results were assessed
using an ELISA plate reader. Protein extracts were diluted using 5X
SDS loading buffer, boiled and resolved on SDS-PAGE. After
electrophoresis, proteins were transferred to a PVDF membrane by
semidry blotting. Detection was performed using the anti-IκB
antibody (1:1,000); and the anti-actin antibody (1:100) was used as
a loading control. The anti-rabbit and anti-mouse IgG coupled with
horseradish peroxidase were used at a dilution of 1:3,000. Blots
were developed with the Western Lightning Chemiluminescence Reagent
Plus (Pierce, Thermo Fisher Scientific) and chemiluminescence
detection film.
Generation of the MDR1 promoter reporter,
and the IκB and IκB mutant vector constructs
The MDR1 genomic sequence, 986 nucleotides of the
MDR1 TSS, was PCR-amplified using the MDR1 forward primer
5′-GCACTGCAGGGGCTTTCCTGTG-3′ and reverse primer
5′-CTGCAGAAAAATTTCTCCTAGCC-3′. The transcriptional site was defined
as +1, and human genomic DNA was used as the template. Next, 1094
bp nucleotides of IκB were amplified using primers
(5′-GTCCGCGCCATGTTCCAG-3′ and 5′-TGGGCTAGGCAGTGTGCAGT-3′) and the
cDNA from K562/D cells as the template. The PCR product was cloned
into the pMD18-T vector. The MDR1 reporter vector was constructed
into the PGL3-Basic vector with HindIII and SacI
restriction sites. Using the IκB sequence as the template, the IκB
expression vector was constructed in the pcDNA3.1 vector containing
HindIII and EcoRI restriction sites. We constructed
the IκB mutant vector according to the operating instructions of
the Takara MutanBEST kit using the primers
CCTCGTCTTTCATGTAGTCCAGGCCGATGTCGTGGCGGTC and
GACCGCCACGACATCGGCCTGGACTACATGAAAGACGAGG
(underlined are mutated sites that changed the ser 32/36 to ile
32/tyr 36). All of the constructed vectors were confirmed by
sequencing.
Luciferase assay
K562D cells treated with 1 μM
As2O3 and 293T cells treated with or without
40 ng/ml TNF-α stimulation were seeded at 2×105
cells/well in a 24-well plate. After 24 h, the cells were
transfected with the plasmid including IκB, the IκB mutant and p65
siRNA using Lipofectamine 2000 (Invitrogen) according to the
protocol recommended by the manufacturer. Two days after
transfection, the cells were lysed, and the luciferase activities
were assayed using the Dual-Luciferase reporter assay system
(Promega, Madison, WI, USA) and measured with a Lumat LB 9507
luminometer (Bethold Technologies, Bad Wildbad, Germany). The
luciferase levels were calculated by the ratio of firefly
luciferase to Renilla luciferase; the results were averaged
from at least three separate transfection assays in all of the
experiments.
Results
Identification of the NF-κB protein
binding site on the MDR1 promoter
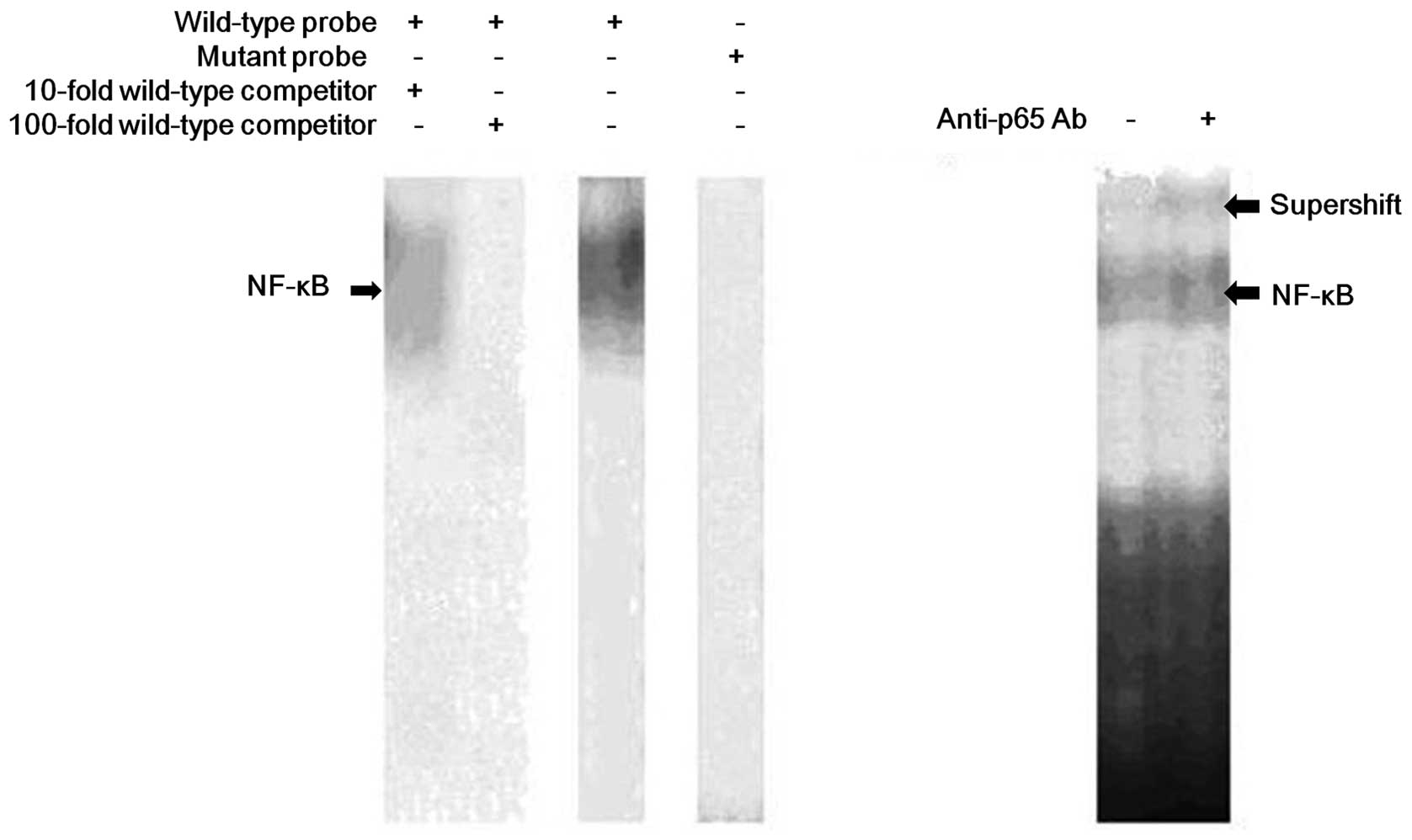
We demonstrated that response elements exist in the
MDR1 promoter by EMSA (Fig. 1). The
wild-type NF-κB-labeled probe formed a complex with the nuclear
extract, and the addition of a cold self-competitor resulted in
weakened binding. However, a mutant competitor could not abolish
the binding. Supershift assays revealed that the binding was
blocked by the p65 antibody. These results indicated that p65 could
bind with the NF-κB binding site of MDR1 specifically in K562/D
cells.
Influence of As2O3
on p65, IκB and phosphorylated IκB in K562/D cells
The results of the western blot analysis
demonstrated that As2O3 reduced the
expression of p65 and phosphorylated IκB but increased the
expression of IκB. We then extracted the nuclear protein and total
protein from untreated K562/D cells and those treated with
As2O3 to identify whether the MDR1 gene was
inhibited by As2O3 via the NF-κB pathway.
Western blot analysis was performed for p65 using
nuclear proteins and IκB and phosphorylated IκB using total
protein. The results demonstrated that the expression of p65 and
phosphorylated IκB was reduced, while the expression of IκB was
increased in K562/D cells treated with As2O3
(Fig. 2). In essence,
phosphorylated IκB was reduced after treatment with
As2O3, resulting in an increased IκB protein
level and decreased NF-κB translocation to the cell nucleus.
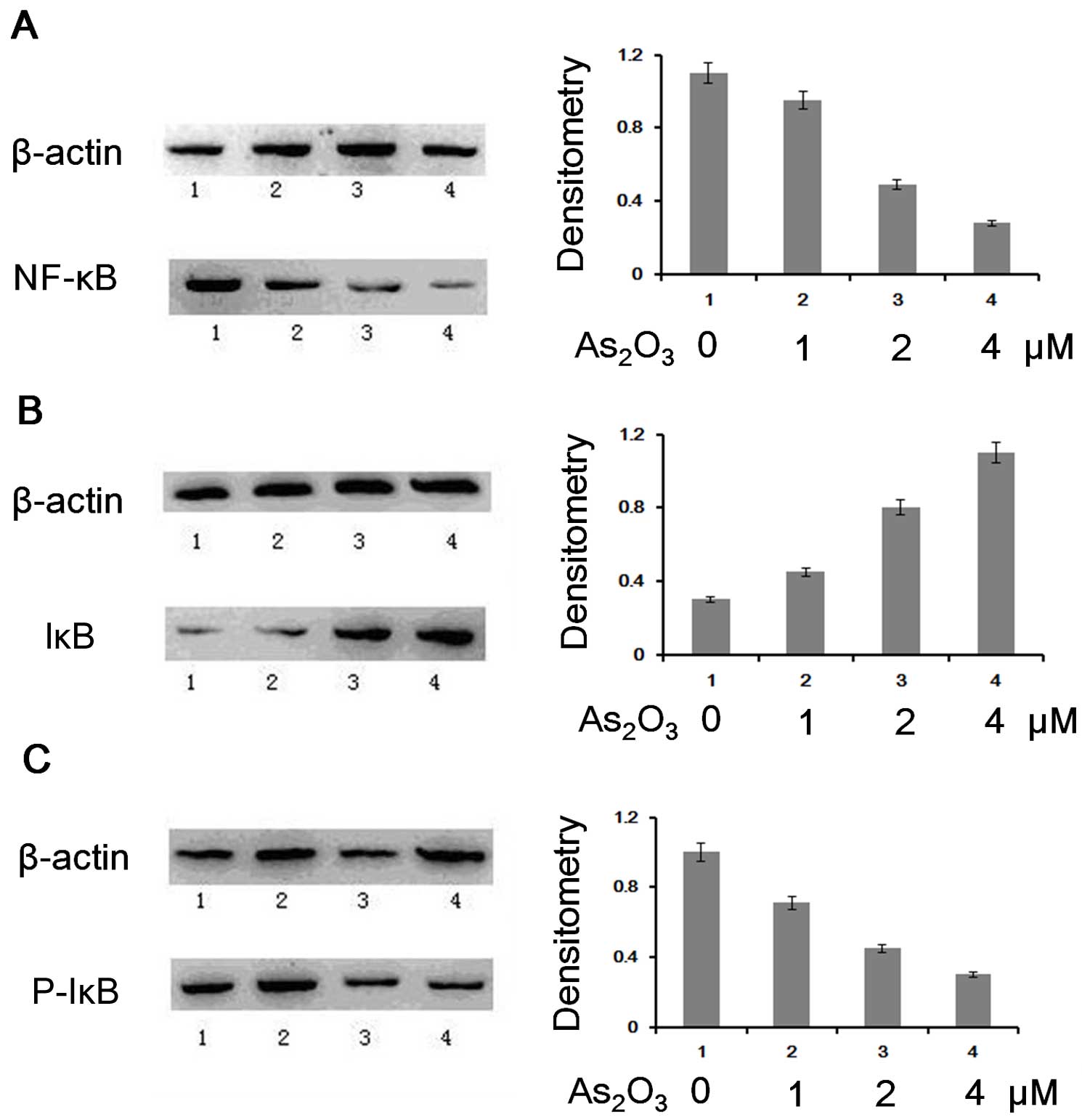 | Figure 2Expression of NF-κB, p65, IκB and
phosphorylated IκB in As2O3-treated-K562/D
cells. Left, western blot analysis of the K562D cells treated with
different concentrations of As2O3 (1,
control; 2, 1 μM As2O3; 3, 2 μM
As2O3; 4, 4 μM As2O3).
Right, analysis of the density by the ratio of NF-κB p65, IκB,
phosphorylated IκB and β-actin. (A) Reduced NF-κB p65 expression,
(B) increased IκB expression, (C) reduced phosphorylated IκB
expression following treatment with
As2O3. |
Influence of TNF-α and
As2O3 treatment on MDR1 promoter
activity
Using the luciferase assay system, we found that
negative regulation of the MDR1 promoter occurred after stimulation
with TNF-α, while As2O3 inhibited this
activity. Luciferase reporter vectors harboring the MDR1 promoter
(Fig. 3A and B) were transfected
into 293T cells after treatment with different concentrations of
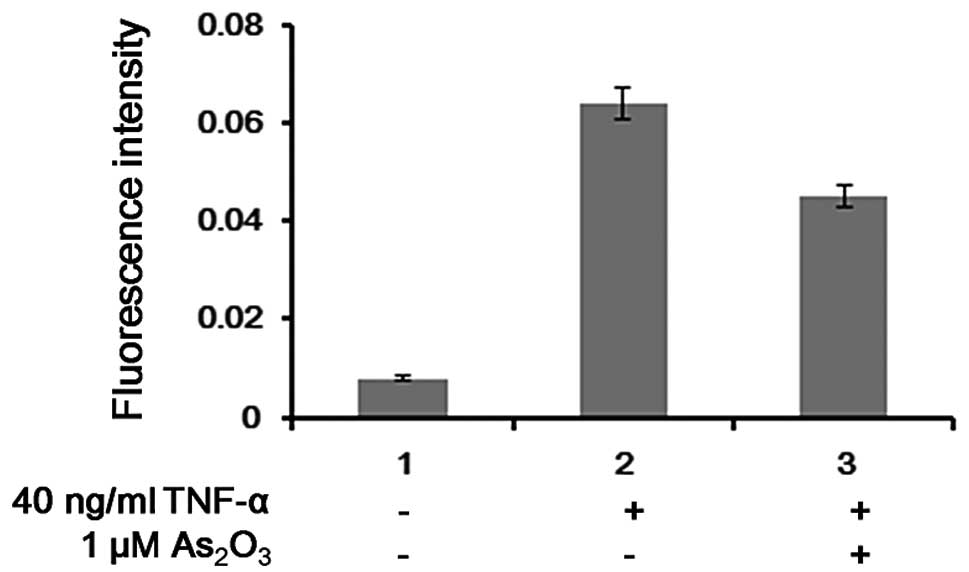
TNF-α for 24 h. We found that without TNF-α stimulation, low
luciferase activity was observed, and the activity of luciferase
increased as the concentration of TNF-α increased. The activity of
luciferase was increased up to 9-fold with 40 ng/ml TNF-α, and it
was suppressed by ~25% following treatment with 1 μM
As2O3 (Fig.
4).
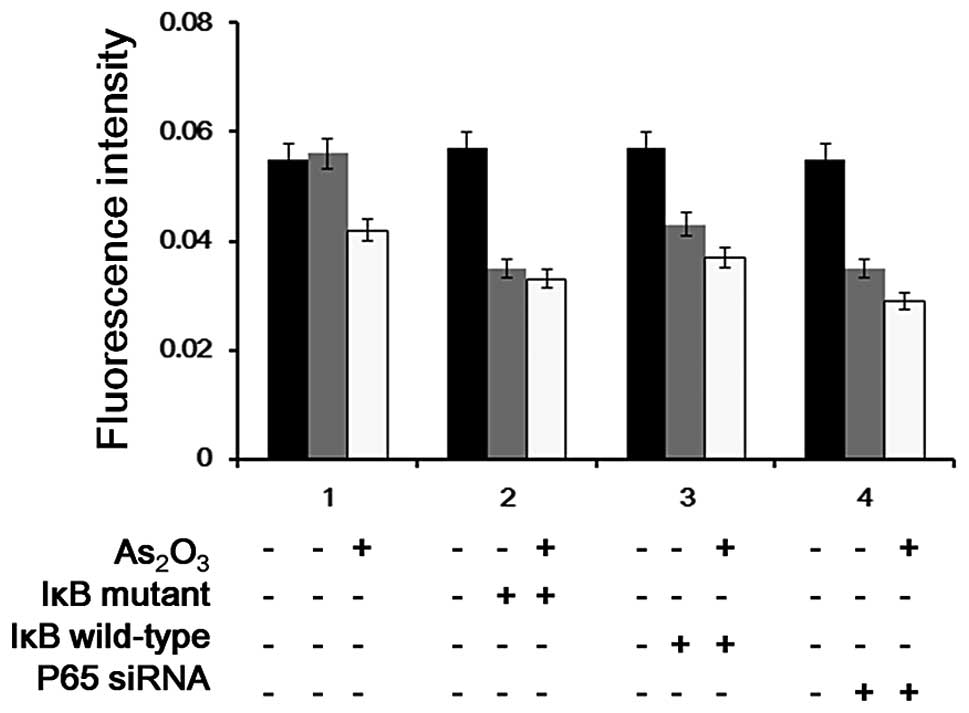
Influence of the NF-κB response element
in the suppression of MDR1 promoter activity following
As2O3 treatment
The activity of the MDR1 promoter induced by TNF-α
was inhibited by As2O3. This inhibition was
significantly reduced after the cells were transfected with p65
siRNA (Figs. 5 and 6). The results indicated that
As2O3 partially inhibited the TNF-α
induced-activity of the MDR1 promoter via NF-κB.
Discussion
Human P-gp is present not only in tumor cells, but
also in normal tissues including the kidney, liver, adrenal glands
and the pregnant uterus (9). The
human MDR1 gene is located at 7p21-21.1; containing 28 exons
(10). The upstream region of the
MDR1 gene promoter lacks the TATA box commonly found in many
protein-coding genes; however, it does contain a reverse CCAAT and
GC-rich region that can bind with NF-Y and Sp family transcription
factors, which recruit histone acetyltransferase to initiate
histone acetylation, chromatin remodeling, thus, activating the
MDR1 promoter. Moreover, the promoter contains binding sites for
lymphoid enhancer factor/T cell transcription factor, heat shock
factor, early growth response factor 1 and myocyte enhancer factor
1 (11). Research has demonstrated
that cell stress such as DNA damage and various activation signals
(including TNF-α and NF-κB) can activate the MDR1 gene and induce
its expression (12–15). Bentires-Alj et al (7) found that the MDR1 promoter contained a
binding sequence (GGGGCTTTCC) for NF-κB. In the present study, we
verified this specific binding using EMSA, and demonstrated that
TNF-α activates the transcription of the MDR1 promoter using a
luciferase reporter system.
In the present study, we used the K562/D and 293T
cell lines as our models. K562/D cells possess a high level of P-gp
expression; therefore, the function of the external input pump of
the cells leads to low efficiency of cell transfection. Thus, in
the present study, we used 293T cells when undertaking the
transfection experiments. After transfection of the reporter gene,
the weak signal was enhanced by NF-κB expression induced by TNF-α,
and this enhancement was significantly reduced again after
treatment with As2O3. These results
demonstrated that As2O3 inhibited activation
of the MDR1 promoter induced by TNF-α.
In our previous research and in studies by other
authors, we found that As2O3 directly
affected the expression level of P-gp in K562/D cells (unpublished
data) (1);
As2O3 was observed to significantly inhibit
the expression and function of P-gp by flow cytometry. These
studies demonstrated that As2O3 could
regulate the expression of MDR1. Further research on the regulatory
mechanisms of MDR1 will facilitate the study of the reversal of
drug resistance.
Expression of MDR1 is affected by many
tumor-associated proteins, such as mutant ras isoforms, and c-raf
can directly bind with the Sp-1 motif to increase expression of
MDR1. SXR/PXR, steroid receptors, and a number of carcinogens can
also directly bind to the MDR1 promoter and activate its
expression. As previously demonstrated, mutant p53 specifically
stimulated the MDR1 promoter and wild-type p53 exerted specific
repression (16–18). These results imply that the MDR1
gene activated during tumor progression is associated with many
factors. As this area of study has developed, researchers have
recognized P-gp as a target for cancer therapy.
As2O3 regulates the
activity of the MDR1 promoter via the NF-κB pathway
In order to further analyze the inhibitory effects
of As2O3 on the activation of the MDR1
promoter, we examined IκB, phosphorylated IκB and the expression of
NF-κB in K562/D cells treated with or without
As2O3.
The NF-κB family consists of five correlative
transcription factors (19,20): i) NF-κB1, including p50 and p105;
ii) NF-κB2, including p52 and p100; iii) NF-κB3, also known as
Rel-A or p65, generally considered to play an important role in the
transcriptional activation of NF-κB (20); iv) Rel-B; and v) Rel (also known as
c-Rel). In the cytoplasm, NF-κB is in an inactive form and binds to
the inhibitor of IκBα. Under the stimulus of certain factors, it is
often activated by a classical pathway, in which two conserved
serine residues (Ser32/36) of IκBα are phosphorylated by the IκB
kinase (IKK) complex. Phosphorylation of IκB leads to rapid
ubiquitination, followed by a conformational change, which is
recognized and degraded by a catalytic ATP-dependent 26S protease.
Thus, the suppression of NF-κB is reversed, and it is translocated
into the nucleus where it exerts its function of transcriptional
activation (22).
Our results demonstrated that phosphorylation of IκB
and nuclear localization of NF-κB were decreased, while expression
of IκB was increased in the As2O3-treated
K562/D cells when compared with the control group. The activity
increased with the increasing concentration of
As2O3. We hypothesized that inhibition of
phosphorylase activity caused by As2O3
resulted in the reduction of phosphorylation of IκB and degradation
of IκB proteins. Thus, IκB proteins bound to NF-κB outside of the
nucleus, and further reduced NF-κB translocation into the
nucleus.
In the present study, we detected an influence on
the promoter activity, resulting from the alteration in the binding
of MDR1 and NF-κB. The results demonstrated that the MDR1
promoter-driven reporter gene, which contained NF-κB sites, had a
high level of transcriptional activity following stimulation with
TNF-α, and As2O3 inhibited the activity of
transcription promoted by TNF-α. This inhibition was also enhanced
by transfection with siRNA targeting p65 or an expression vector
containing IκB. Hence, the NF-κB response element played an
important role in the transcriptional activation of the MDR1
promoter. Moreover, the inhibition of activity was significantly
reduced in the group transfected with the IκB mutant vector
compared with the wild-type vector. These results further
demonstrated that As2O3 inhibited the
phosphorylation of IκB in the NF-κB pathway to regulate MDR1.
NF-κB has antiapoptotic functions. It can be
activated by cytotoxic drugs, and once activated it can induce the
expression of antiapoptotic genes (23–25),
leading to the resistance to chemotherapy (15,26–28).
P-gp has been demonstrated to possess antiapoptotic
functions in stem cells (29–31).
As2O3 inhibits P-gp expression via the NF-κB
pathway, and plays a dual role in the reversal of tumor resistance.
Studies continue to demonstrate the special effects that
As2O3 has on relapsed/refractory tumors. In
the present study using As2O3-treated
wild-type K562/S cells and drug-resistant K562/D cells, we found
that the cell lethal concentration of the former was higher than
that of the latter, which corroborated the collateral sensitivity
theory of multi-drug-resistant cells (32–34).
The finding that resistant cells may be ultra-sensitive to
unconventional drugs (such as As2O3) adds
significant value to the study of cancer treatment.
In summary, the present study demonstrated that the
+561 – +571 region of the MDR1 promoter is the response element of
NF-κB. As2O3 regulated the binding of NF-κB
through the phosphorylation of IκB, and consequently led to
NF-κB-mediated transcriptional repression of the MDR1 promoter,
which is one of the mechanisms of the reversal of the drug
resistance by As2O3. Furthermore, the effect
of As2O3 on the apoptosis of resistant cells
may involve more complex mechanisms, which remain to be elucidated.
Finally, As2O3 has been clinically used as an
anticancer drug, and when its ability to reverse resistance is
further developed and utilized, As2O3 may
become an option for the first-line treatment of cancer.
In conclusion, we identified that
As2O3 reversed the P-gp-induced
drug-resistance of leukemia cells through the NF-κB pathway. A
specific binding sequence for NF-κB was identified in the upstream
region of the MDR1 gene, which NF-κB can use to affect MDR1 gene
expression. Additionally, As2O3 was observed
to inhibit NF-κB binding to this sequence.
In leukemia cells, As2O3 may
inhibit the activity of phosphorylase to inhibit IκB
phosphorylation, thereby inhibiting NF-κB activity and MDR1 gene
expression, leading to the reversal of drug resistance.
References
|
1
|
Wang T, Ma LM, Zhang HP, Wang HW, Yang LH
and Qiao ZH: The effect of arsenic trioxide
(As2O3) combined with BSO on K562/ADM cells
and its mechanisms. Zhonghua Xue Ye Xue Za Zhi. 28:438–443.
2007.(In Chinese).
|
|
2
|
Mathas S, Lietz A, Janz M, et al:
Inhibition of NF-κB essentially contributes to arsenic-induced
apoptosis. Blood. 102:1028–1034. 2003.
|
|
3
|
Thomas H and Coley HM: Overcoming
multidrug resistance in cancer: an update on the clinical strategy
of inhibiting p-glycoprotein. Cancer Control. 10:159–165.
2003.PubMed/NCBI
|
|
4
|
Lee HR, Cheong HJ, Kim SJ, Lee NS, Park HS
and Won JH: Sulindac enhances arsenic trioxide-mediated apoptosis
by inhibition of NF-κB in HCT116 colon cancer cells. Oncol Rep.
20:41–47. 2008.PubMed/NCBI
|
|
5
|
Mathieu J and Besancon F: Arsenic trioxide
represses NF-κB activation and increases apoptosis in ATRA-treated
APL cells. Ann NY Acad Sci. 1090:203–208. 2006.
|
|
6
|
Park MJ, Lee JY, Kwak HJ, et al: Arsenic
trioxide (As2O3) inhibits invasion of HT1080
human fibrosarcoma cells: role of nuclear factor-κB and reactive
oxygen species. J Cell Biochem. 95:955–969. 2005.PubMed/NCBI
|
|
7
|
Bentires-Alj M, Barbu V, Fillet M, et al:
NF-κB transcription factor induces drug resistance through MDR1
expression in cancer cells. Oncogene. 22:90–97. 2003.
|
|
8
|
Kuo MT, Liu Z, Wei Y, et al: Induction of
human MDR1 gene expression by 2-acetylaminofluorene is
mediated by effectors of the phosphoinositide 3-kinase pathway that
activate NF-κB signaling. Oncogene. 21:1945–1954. 2002.
|
|
9
|
Tanigawara Y: Role of P-glycoprotein in
drug disposition. Ther Drug Monit. 22:137–140. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Callen DF, Baker E, Simmers RN, Seshadri R
and Roninson IB: Localization of the human multiple drug resistance
gene, MDR1, to 7q21.1. Hum Genet. 77:142–144. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Scotto KW and Johnson RA: Transcription of
the multidrug resistance gene MDR1: a therapeutic target. Mol
Interv. 1:117–125. 2001.PubMed/NCBI
|
|
12
|
Hirsch-Ernst KI, Ziemann C, Schmitz-Salue
C, Foth H and Kahl GF: Modulation of P-glycoprotein and
mdr1b mRNA expression by growth factors in primary rat
hepatocyte culture. Biochem Biophys Res Commun. 215:179–185.
1995.
|
|
13
|
Hirsch-Ernst KI, Ziemann C, Foth H, Kozian
D, Schmitz-Salue C and Kahl GF: Induction of mdr1b mRNA and
P-glycoprotein expression by tumor necrosis factor alpha in primary
rat hepatocyte cultures. J Cell Physiol. 176:506–515. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fardel O, Lecureur V, Daval S, Corlu A and
Guillouzo A: Up-regulation of P-glycoprotein expression in rat
liver cells by acute doxorubicin treatment. Eur J Biochem.
246:186–192. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cheng Q, Lee HH, Li Y, Parks TP and Cheng
G: Upregulation of Bcl-x and Bfl-1 as a potential mechanism of
chemoresistance, which can be overcome by NF-κB inhibition.
Oncogene. 19:4936–4940. 2000.PubMed/NCBI
|
|
16
|
Chin KV, Ueda K, Pastan I and Gottesman
MM: Modulation of activity of the promoter of the human MDR1 gene
by Ras and p53. Science. 255:459–462. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Synold TW, Dussault I and Forman BM: The
orphan nuclear receptor SXR coordinately regulates drug metabolism
and efflux. Nat Med. 7:584–590. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bush JA and Li G: Regulation of the
Mdr1 isoforms in a p53-deficient mouse model.
Carcinogenesis. 23:1603–1607. 2002.
|
|
19
|
Sen R and Baltimore D: Inducibility of κ
immunoglobulin enhancer-binding protein NF-κB by a
posttranslational mechanism. Cell. 47:921–928. 1986.
|
|
20
|
Lin L, DeMartino GN and Greene WC:
Cotranslational biogenesis of NF-κB p50 by the 26S proteasome.
Cell. 92:819–828. 1998.
|
|
21
|
Schmitz ML and Baeuerle PA: The p65
subunit is responsible for the strong transcription activating
potential of NF-kappa B. EMBO J. 10:3805–3817. 1991.PubMed/NCBI
|
|
22
|
Quivy V and Van Lint C: Regulation at
multiple levels of NF-κB-mediated transactivation by protein
acetylation. Biochem Pharmacol. 68:1221–1229. 2004.
|
|
23
|
Wu M, Lee H, Bellas RE, et al: Inhibition
of NF-kappaB/Rel induces apoptosis of murine B cells. EMBO J.
15:4682–4690. 1996.PubMed/NCBI
|
|
24
|
Ryan KM, Ernst MK, Rice NR and Vousden KH:
Role of NF-κB in p53-mediated programmed cell death. Nature.
404:892–897. 2000.
|
|
25
|
Pham LV, Tamayo AT, Yoshimura LC, et al: A
CD40 signalosome anchored in lipid rafts leads to constitutive
activation of NF-κB and autonomous cell growth in B cell lymphomas.
Immunity. 16:37–50. 2002.PubMed/NCBI
|
|
26
|
Ueda M, Kokura S, Imamoto E, et al:
Blocking of NF-κB activation enhances the tumor necrosis factor
α-induced apoptosis of a human gastric cancer cell line. Cancer
Lett. 193:177–182. 2003.
|
|
27
|
Wang CY, Mayo MW, Korneluk RG, Goeddel DV
and Baldwin AS Jr: NF-κB antiapoptosis: induction of TRAF1 and
TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation.
Science. 281:1680–1683. 1998.
|
|
28
|
Thevenod F, Friedmann JM, Katsen AD and
Hauser IA: Up-regulation of multidrug resistance P-glycoprotein via
nuclear factor-κB activation protects kidney proximal tubule cells
from cadmium- and reactive oxygen species-induced apoptosis. J Biol
Chem. 275:1887–1896. 2000.
|
|
29
|
Pallis M, Turzanski J, Higashi Y and
Russell N: P-glycoprotein in acute myeloid leukaemia: therapeutic
implications of its association with both a multidrug-resistant and
an apoptosis-resistant phenotype. Leuk Lymphoma. 43:1221–1228.
2002. View Article : Google Scholar
|
|
30
|
Ruefli AA, Tainton KM, Darcy PK, Smyth MJ
and Johnstone RW: P-glycoprotein inhibits caspase-8 activation but
not formation of the death inducing signal complex (disc) following
Fas ligation. Cell Death Differ. 9:1266–1272. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Turzanski J, Grundy M, Shang S, Russell N
and Pallis M: P-glycoprotein is implicated in the inhibition of
ceramide-induced apoptosis in TF-1 acute myeloid leukemia cells by
modulation of the glucosylceramide synthase pathway. Exp Hematol.
33:62–72. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schuurhuis GJ, Pinedo HM, Broxterman HJ,
van Kalken CK, Kuiper CM and Lankelma J: Differential sensitivity
of multi-drug-resistant and -sensitive cells to
resistance-modifying agents and the relation with reversal of
anthracycline resistance. Int J Cancer. 46:330–336. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ludwig JA, Szakacs G, Martin SE, et al:
Selective toxicity of NSC73306 in MDR1-positive cells as a new
strategy to circumvent multidrug resistance in cancer. Cancer Res.
66:4808–4815. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hall MD, Handley MD and Gottesman MM: Is
resistance useless? Multidrug resistance and collateral
sensitivity. Trends Pharmacol Sci. 30:546–556. 2009. View Article : Google Scholar : PubMed/NCBI
|