Introduction
microRNAs (miRNAs) are small, endogenous, non-coding
RNA molecules (19–23 nucleotides) that regulate gene expression
post-transcriptionally by negatively regulating the stability or
translational efficiency of their target mRNAs (1). Aberrant miRNA expression plays diverse
roles in tumorigenesis and tumor development through modulation of
cell proliferation, differentiation, apoptosis, motility and
malignant transformation (2).
Additionally, deregulated miRNAs exhibit oncogenic or tumor
suppressor properties (3). For
example, among other findings, miR-24 and miR-21 have been shown to
influence breast cancer invasion and metastasis (4,5).
Overexpression of miR-187 has been associated with lymph node
metastasis and poor prognosis in breast cancer (6). miR-96 was found to be highly
upregulated in a variety of tumors, including prostate cancer
(7), hepatocellular carcinoma
(8), lung (9), colorectal (10), endometrial (11) and breast cancer (12). Furthermore, overexpression of miR-96
induced cell proliferation in MCF-7 breast cancer cells by
targeting FOXO3a (12). However, to
date, the biological function of miR-96 in breast cancer
carcinogenesis remains largely unknown.
RECK (reversion-inducing cysteine-rich protein with
Kazal motifs), a ubiquitous tumor-suppressor gene, negatively
regulates MMP-9, MMP-2 and MT1-MMP, and has a significant effect on
the regulation of angiogenesis, tumor invasion and metastasis
(13,14). The functional inactivation of RECK
by regulation of its expression has been observed in various types
of solid tumors, including breast cancer (15). Furthermore, RECK was recently
described as a potentially useful prognostic marker for breast
cancer (16). Therefore,
overexpression of RECK should be considered as a therapeutic
approach for breast cancer. Recently, several miRNAs, miR-92a
(17), miR-182 (18), miR-15a (19) and miR-21 (20) were reported to suppress RECK
expression and function as oncogenes. In addition, miR-222 has been
shown to directly silence RECK and promote proliferation in H.
pylori-associated gastric cancer (21). These data highlight the importance
of miRNAs targeting RECK in breast cancer development and provide
insight into the mechanisms underlying tumorigenesis.
In the present study, we reported that miR-96 was
significantly upregulated in breast cancer cells and breast cancer
specimens when compared with that in non-malignant breast
epithelial cells and adjacent normal tissues. Ectopic expression of
miR-96 promoted cellular proliferation, migration and invasion of
MDA-MB-231 cells, at least in part, by targeting RECK. Moreover, we
found that the silencing of RECK by RNA interference mimicked the
oncogenic effects of miR-96. Collectively, the present study
indicates that miR-96 serves as an oncogene in breast cancer and is
a vital regulator of cellular proliferation, migration and
invasion. Targeting miR-96 is a potential novel strategy for the
treatment of human breast cancer.
Materials and methods
Specimens
In the present study, 38 paired breast cancer
specimens and adjacent normal breast tissues were collected from
the Department of General Surgery of the Shanghai Tenth People’s
Hospital. These samples were immediately snap-frozen in liquid
nitrogen. Both tumor and normal tissues were histologically
confirmed by two different experienced pathologists according to
the World Health Organization (WHO) using H&E (hematoxylin and
eosin) staining. No patients received chemotherapy or radiotherapy
prior to surgery.
Cell lines and transfection
The human breast cancer cell lines MDA-MB-231,
MCF-7, MDA-MB-468, MDA-MB-435, T-74D, MDA-MB-453 and non-malignant
breast epithelial cell line MCF-10A were all obtained from the
American Type Culture Collection (ATCC; Manassas, VA, USA). The
breast cancer cells were cultured in Dulbecco’s modified Eagle’s
medium (DMEM) (Gibco, Carlsbad, CA, USA) supplemented with 10%
fetal bovine serum (FBS) (Gibco), penicillin (100 U/ml) and
streptomycin (100 μg/ml) (Enpromise, China). MCF-10A cells were
cultured in Mammary Epithelial Basal Medium (MEBM) (Cambrex). Cells
were incubated at 37°C in a humidified chamber supplemented with 5%
CO2.
miR-96 mimics, inhibitors and their negative control
(NC) were chemosynthesized by Shanghai Genepharma Co., Ltd.
(Shanghai, China). The MDA-MB-231 cells were cultured to ~30–40%
confluence in 6-well plates and were transfected with miR-96 mimics
or miR-96 inhibitors or RECK siRNA (Santa Cruz Biotechnology) at
working concentrations using Lipofectamine 2000 (Invitrogen,
Carlsbad, CA, USA), in accordance with the manufacturer’s
instructions. miR- and siRNA-negative control (NC) were used as
negative controls. After 48 h of incubation, cells were harvested
for further analysis. All transfections were performed in
triplicates.
Quantitative reverse-transcription
polymerase chain reaction (qRT-PCR)
For detection of miR-96 expression, primer design
and qRT-PCR were carried out according to a previously described
method (22). miRNA was isolated
from tissues and cells using the miRcute miRNA Isolation kit
according to the manufacturer’s instructions (Beijing Tianjin,
Beijing, China). The primers for miR-96 were stem-loop RT primer
5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAGCAAA-3′; forward
5′-GCCCGCTTTGGCACTAGCACATT-3′ and reverse 5′-GTGCAGGGTCCGAGGT-3′.
The primers for U6 small nuclear RNA were RT primer
5′-GTCGTATCCAGTGCAGGGTCCGAGGTGCACTGGATACGACAAAATATGG-3′; forward
5′-TGCGGGTGCTCGCTTCGGCAGC-3′ and reverse 5′-CCAGTGCAGG
GTCCGAGGT-3′. cDNA was generated by reverse transcription using the
PrimeScript™ RT-PCR kit in accordance with the manufacturer’s
instructions (Takara, Tokyo, Japan). Real-time PCR was performed on
a 7900HT Fast RT-PCR instrument (Applied Biosystems, Singapore).
The amplification procedure was as follows: 5 min at 95°C, followed
by 40 cycles at 95°C for 30 sec and 65°C for 45 sec.
For detection of RECK mRNA expression, total RNA was
isolated using TRIzol (Invitrogen), and cDNA was generated by
reverse transcription using the PrimeScript RT-PCR kit in
accordance with the manufacturer’s instructions (Takara). Real-time
PCR was performed on a 7900HT Fast RT-PCR instrument using
SYBR-Green and the following primers: RECK,
5′-AACCAAATGTGCCGTGAT-3′ (sense), 5′-ATGGCTTGACAGTATTCTCG-3′
(antisense); β-actin, 5′-CAGAGCCTCGCCTTTGCC-3′ (sense),
5′-GTCGCCCACATAGGAATC-3′ (antisense). The PCR parameters for
relative quantification were as follows: 2 min at 95°C, followed by
40 cycles of 45 sec at 57°C and 45 sec at 72°C. The relative
expression was evaluated following the relative quantification
equation, 2−ΔΔCT (23).
Each sample was tested in triplicate.
Cell proliferation assay
Cell proliferation was determined using an MTT assay
kit (Sigma, Santa Clara, CA, USA) in accordance with the
manufacturer’s instructions. In brief, the transfected cells
(5×103 cells/well) were seeded into 96-well culture
plates (BD Biosciences, Franklin Lakes, NJ, USA) and incubated
overnight at 37°C in 5% CO2. Cell proliferation was
assessed at 24, 48, 72 and 96 h following addition of 0.5 mg/ml MTT
(Sigma) solution. After a 4-h incubation, the medium was replaced
with 100 μl dimethylsulfoxide (DMSO; Sigma) and vortexed for 10
min. The optical density (OD) of each well was measured using a
microplate reader at 490 nm. Each experiment was performed in
triplicate.
Cell migration and invasion assays
For the invasion assay, the miR-96 mimic- or miR-96
inhibitor (100 nmol/l)-transfected MDA-MB-231 cells
(4×104 cells/Transwell) were plated in the top chamber
of Transwells (Millipore) with a Matrigel (2 mg/ml)-coated membrane
containing 8-mm diameter pores in 200 μl serum-free DMEM in
triplicate. Medium containing 10% FBS was added to the lower
chamber. After 48 h of incubation, cells remaining on the upper
membrane surface were removed by cotton swab scrubbing; cells on
the lower surface of the membrane were fixed in 10% formalin at
room temperature for 30 min and stained with 0.5% crystal violet.
The cell number in five random fields (x200) was counted for each
chamber. The stained cells were dissolved in glacial acetic acid,
and solutions were transferred to a 96-well culture plate for
colorimetric reading of OD at 560 nm. The OD value represents the
invasive ability. For the migration assays, the infected cells
(1×104 cells/Transwell) were plated in the top chamber
with no Matrigel. After a 24-h incubation, the number of migrated
cells was counted as described above. Each experiment was carried
out in triplicate.
Luciferase assay
The 3′-UTR segments of the RECK mRNA sequence
containing the predicted miR-96 binding sites were amplified by PCR
in a total volume of 50 μl using the PrimerStar kit (Takara) in
accordance with the manufacturer’s instructions. The primers used
were 5′-AAACTAGCGGCCGCTAGTGCTGCTACTTATATAATTGCCAAAT-3′ (sense) and
5′-CTAGATTTGGCAATTATATAAGTAGCAGCACTAGCGGCCGCTAGTTT-3′ (antisense).
The mutant constructs were generated by mutation. Fragments were
subcloned into the XhoI site in the 3′-UTR of Renilla
luciferase of the psiCHECK-2 reporter vector. MDA-MB-231 cells were
transiently cotransfected in 24-well plates with 0.2 μg
psiCHECK-2/RECK 3′-UTR or psiCHECK-2/RECK 3′-UTR mutant reporter
plasmids and 100 nmol/l miR-96 or miR-NC using Lipofectamine™ 2000
(Invitrogen). After 48 h, firefly and Renilla luciferase
activities were measured by using a Dual Luciferase Assay (Promega,
Madison, WI, USA). The firefly luciferase activity of each sample
was normalized to the Renilla luciferase activity.
Western blot analysis
The protein expression levels were analyzed by
western blot analysis. Cells were lysed in lysis buffer (10 mmol/l
Tris-HCl, pH 7.4, 1% NP-40, 0.1% deoxycholic acid, 0.1% SDS, 150
mmol/l NaCl, 1 mM EDTA and 1% protease inhibitor cocktail) (Sigma).
The protein concentrations were quantified using a BCA protein
assay kit (Pierce). Protein was separated using 8% sodium dodecyl
sulfate polyacrylamide gel electrophoresis and transferred to a
nitrocellulose membrane (Beyotime Institute of Biotechnology,
Jiangsu, China). The membrane was immunoblotted overnight at 4°C
with primary antibodies against RECK (1:1,000 dilution; no. 3433,
Cell Signaling Technology) and β-actin (1:1,000 dilution;
sc-1616-R, Santa Cruz Biotechnology), as a loading control.
Horseradish peroxidase-conjugated secondary antibodies was
incubated with the membrane for 2 h at 37°C after three washes with
TBST. Immunoreactive protein bands were detected with an Odyssey
Scanning system.
Statistical analysis
Data are presented as the means ± standard deviation
(SD) from at least three independent experiments. The t-test
(two-tailed) was used to draw a comparison between groups. P-values
<0.05 were considered to indicate statistically significant
results.
Results
miR-96 is upregulated in human breast
cancer cell lines and clinical specimens
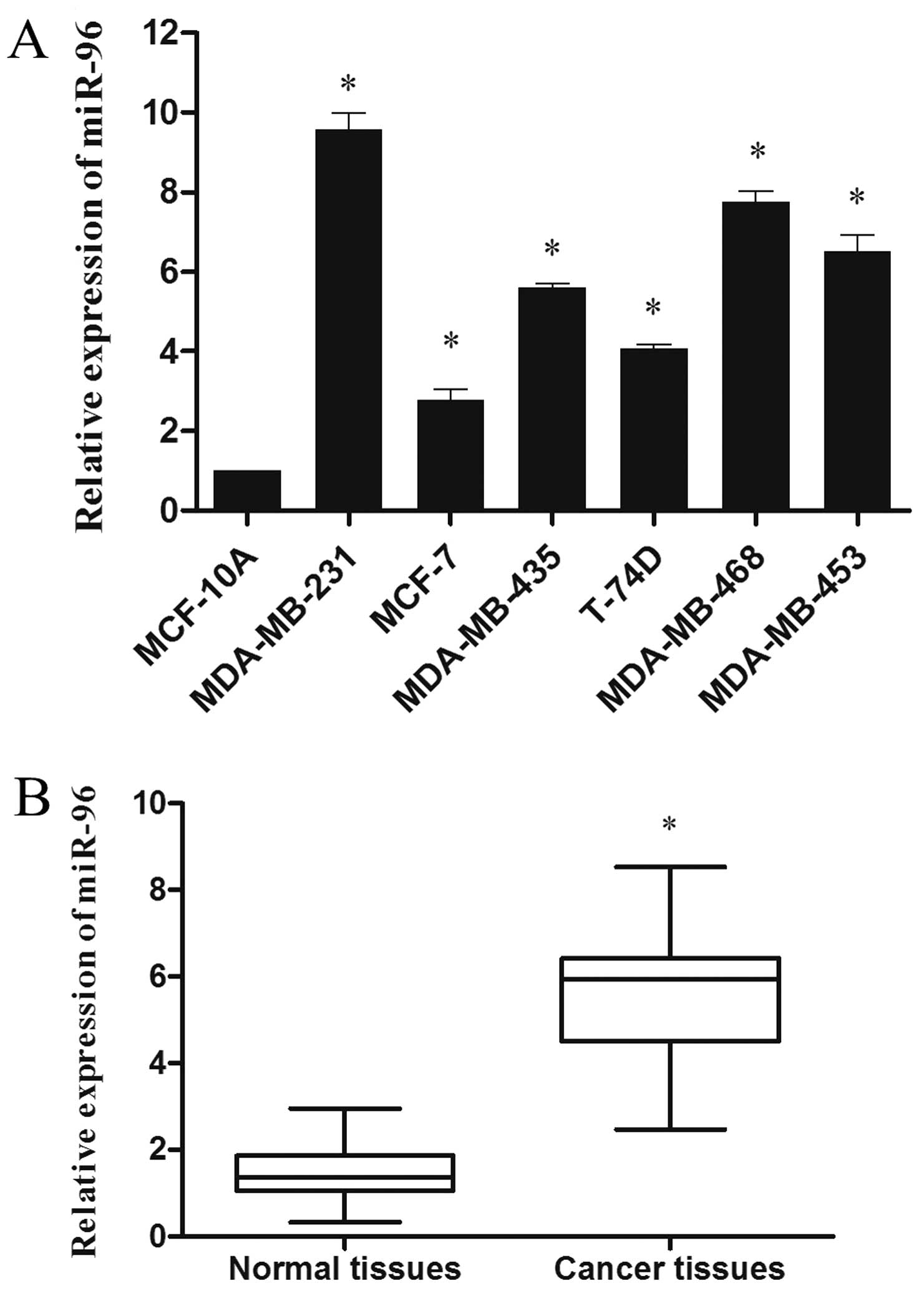
We examined the miR-96 expression in breast cancer
cell lines MDA-MB-231, MCF-7, MDA-MB-435, T-74D, MDA-MB-468 and
MDA-MB-453 as well as breast cancer tissues. As shown in Fig. 1A, all breast cancer cell lines
expressed higher levels of miR-96 when compared with the levels in
the non-malignant breast epithelial cell line MCF-10A. Furthermore,
we compared miR-96 expression profiles between 38 pairs of breast
cancer tissues and matched adjacent normal breast tissues. In
comparison with the adjacent normal breast tissues, miR-96 showed
on average a 3.8-fold higher expression in cancer tissues
(P<0.05; Fig. 1B). These results
indicated that miR-96 is upregulated in breast cancer.
miR-96 promotes MDA-MB-231 cell
proliferation
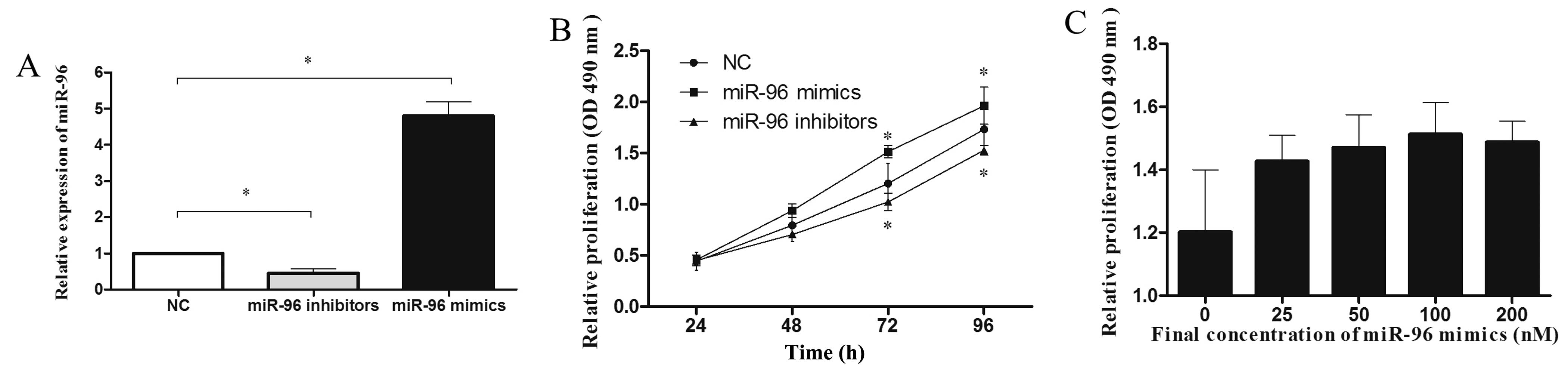
To investigate the effects of miR-96 on breast
cancer cell proliferation. Initially, we assessed miR-96 expression
in MDA-MB-231 cells transfected with miR-96 mimics or inhibitors
(100 nmol/l for 48 h) by qRT-PCR (Fig.
2A). As shown in Fig. 2B,
upregulation of miR-96 significantly increased the growth rate of
MDA-MB-231 cells at 48 h (18%), 72 h (26%) and 96 h (13%) compared
with the negative control (P<0.05). Moreover, MTT assay showed
that suppression of miR-96 markedly inhibited the growth rate of
MDA-MB-231 cells at 48 h (11%), 72 h (14%) and 96 h (12%) as
compared with the control cells (P<0.05). The dose-dependent
effect of miR-96 mimics was also evident in the MTT assay at 72 h
(Fig. 2C). Taken together, these
results revealed that miR-96 promotes the proliferation of
MDA-MB-231 breast cancer cells.
miR-96 promotes MDA-MB-231 cell migration
and invasion
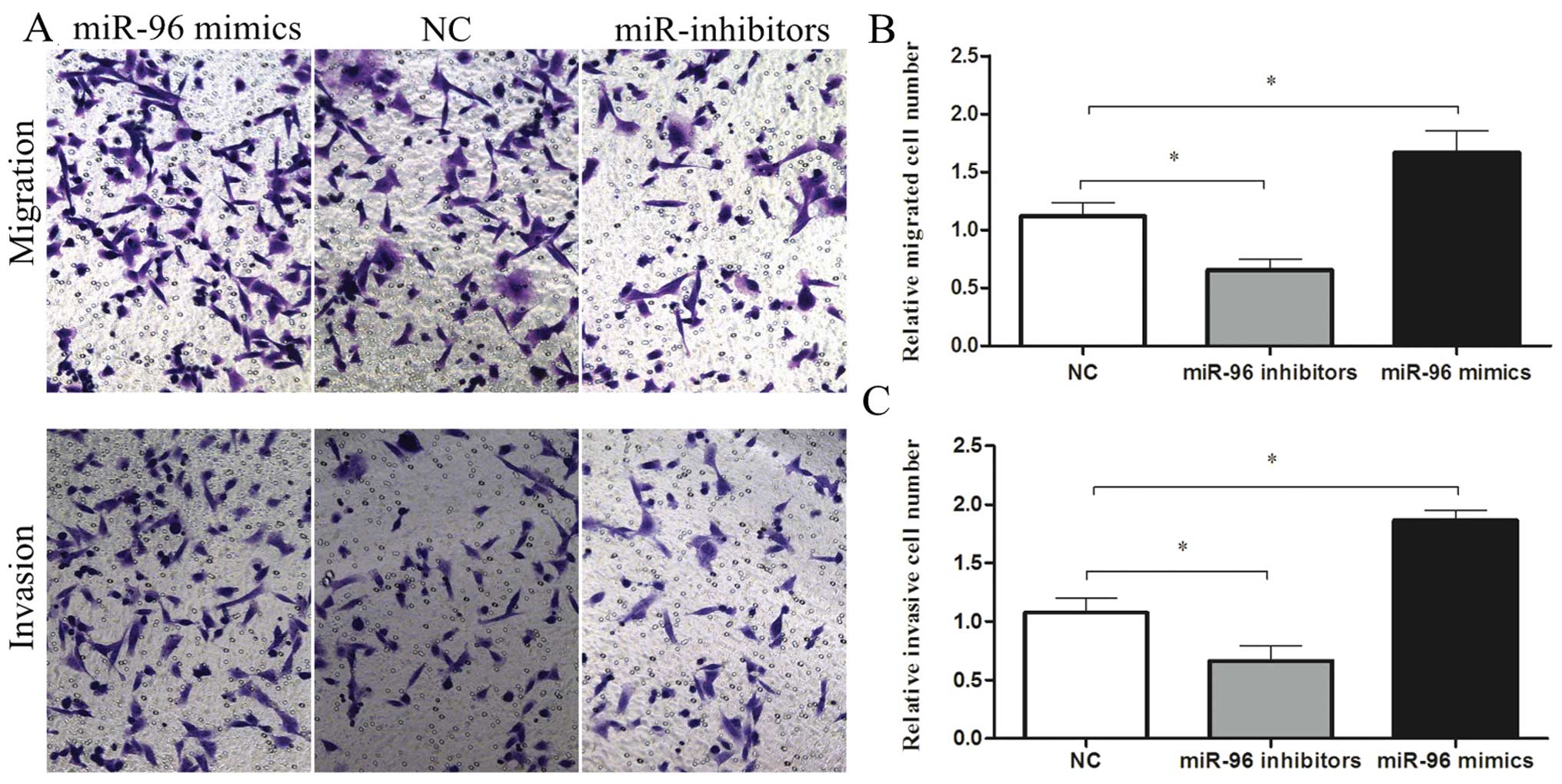
To investigate whether miR-96 affects the migratory
and invasive abilities of the cells, we transfected MDA-MB-231
cells with 100 nmol/l miR-96 mimics or inhibitors and detected
changes in their motility using Transwell assays. Transwell
migration assays performed in the absence of Matrigel showed that
overexpression of miR-96 promoted MDA-MB-231 cell migration by
1.5-fold compared to the control cells. In contrast, MDA-MB-231
cell migratory activity was significantly inhibited when cells were
transfected with the miR-96 inhibitors compared to the control
(0.58-fold; P<0.05) (Fig. 3A and
B). The upregulation of miR-96 significantly promoted the
invasion of MDA-MB-231 cells as measured by Transwell invasion
assays that included 2 mg/ml Matrigel (1.7-fold; P<0.05). In
addition, a decrease in the invasive ability of miR-96
inhibitor-transfected cells when compared with the control
(0.61-fold; P<0.05) indicated that miR-96 also participated in
human breast cancer cell invasion (Fig.
3A and C).
miR-96 downregulates RECK expression by
binding the 3′UTR of RECK
To explore the mechanism by which miR-96 functions
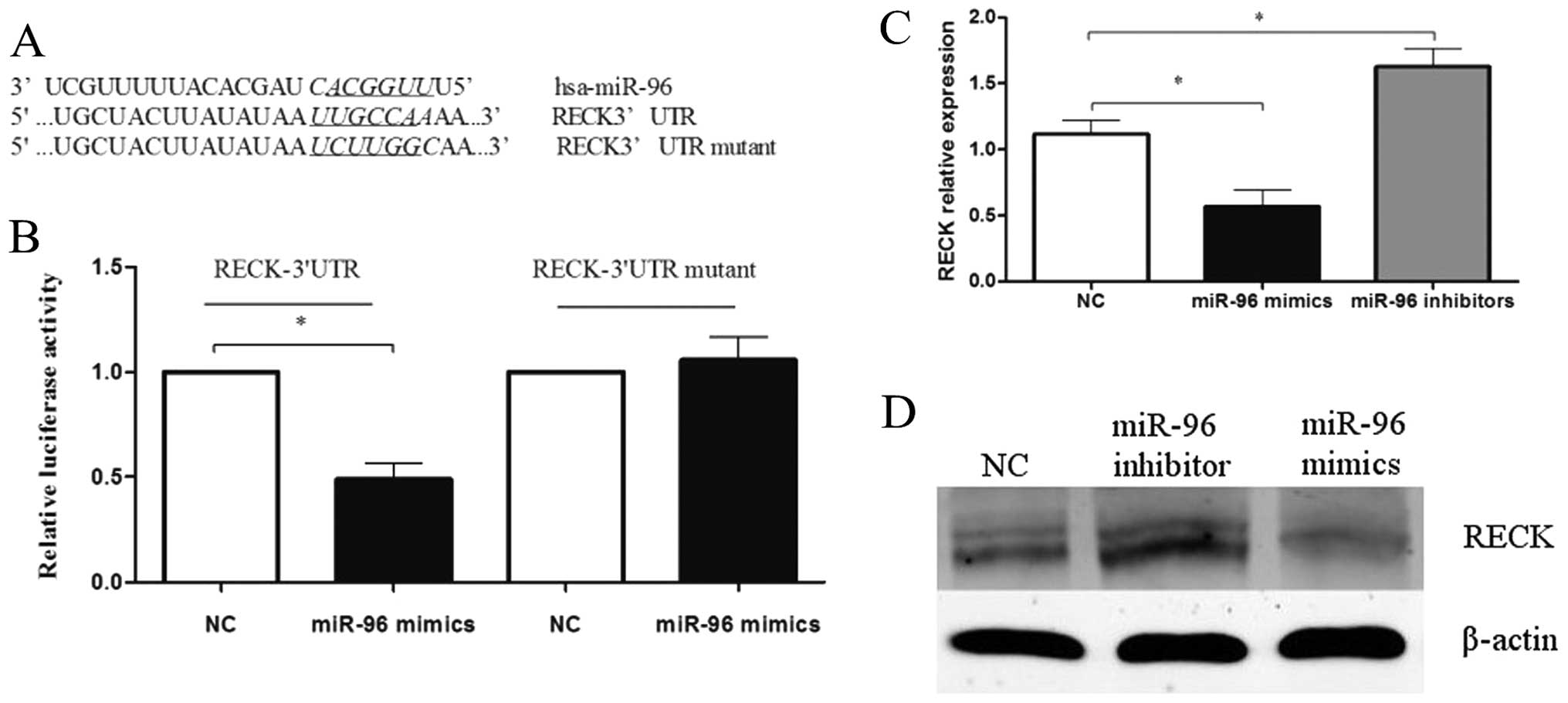
in breast cancer, we searched for putative targets using the
TargetScan database. We identified a binding site for miR-96 in the
3′-UTR of RECK mRNA. To validate whether RECK is a bona fide target
of miR-96, we cloned the 3′-UTR of RECK containing the putative
miR-96 binding site into a luciferase reporter construct, in
addition to a mutated RECK 3′-UTR (Fig.
4A). In comparison with the negative control, miR-96 mimics
(100 nmol/l) significantly decreased the relative luciferase
activity when co-transfected with the psiCHECK-2/RECK 3′-UTR.
However, this effect of miR-96 was abolished following
co-transfection of psiCHECK-2/RECK 3′-UTR mutant and miR-96
(Fig. 4B).
To determine whether miR-96 regulates RECK at both
the mRNA and protein levels, miR-96 inhibitors or mimics (100
nmol/l) were transfected into MDA-MB-231 cells, and the levels of
RECK mRNA and protein were monitored (P<0.05; Fig. 4C and D). qRT-PCR analysis revealed
that inhibition of miR-96 in MDA-MB-231 cells led to increased
expression of endogenous RECK mRNA when compared with the control.
Additionally, western blot analysis showed that RECK protein
expression was clearly upregulated following transfection of
MDA-MB-231 cells with miR-96 inhibitors. In addition, enforced
expression of miR-96 in MDA-MB-231 cells triggered a significant
silencing effect on endogenous RECK expression, both at the mRNA
and protein levels. Together, these data revealed that RECK is a
novel target of miR-96.
Effects of RECK on breast cancer cell
proliferation and invasion
Since overexpression of miR-96 promoted the
proliferation and invasion of MDA-MB-231 breast cancer cells, and
given that RECK is a direct target of miR-96, we hypothesized that
RECK is involved in the miR-96-promotion of proliferation and
invasion by miR-96. The results indicated that protein level of
RECK was decreased in the MDA-MB-231 cells transfected with 200
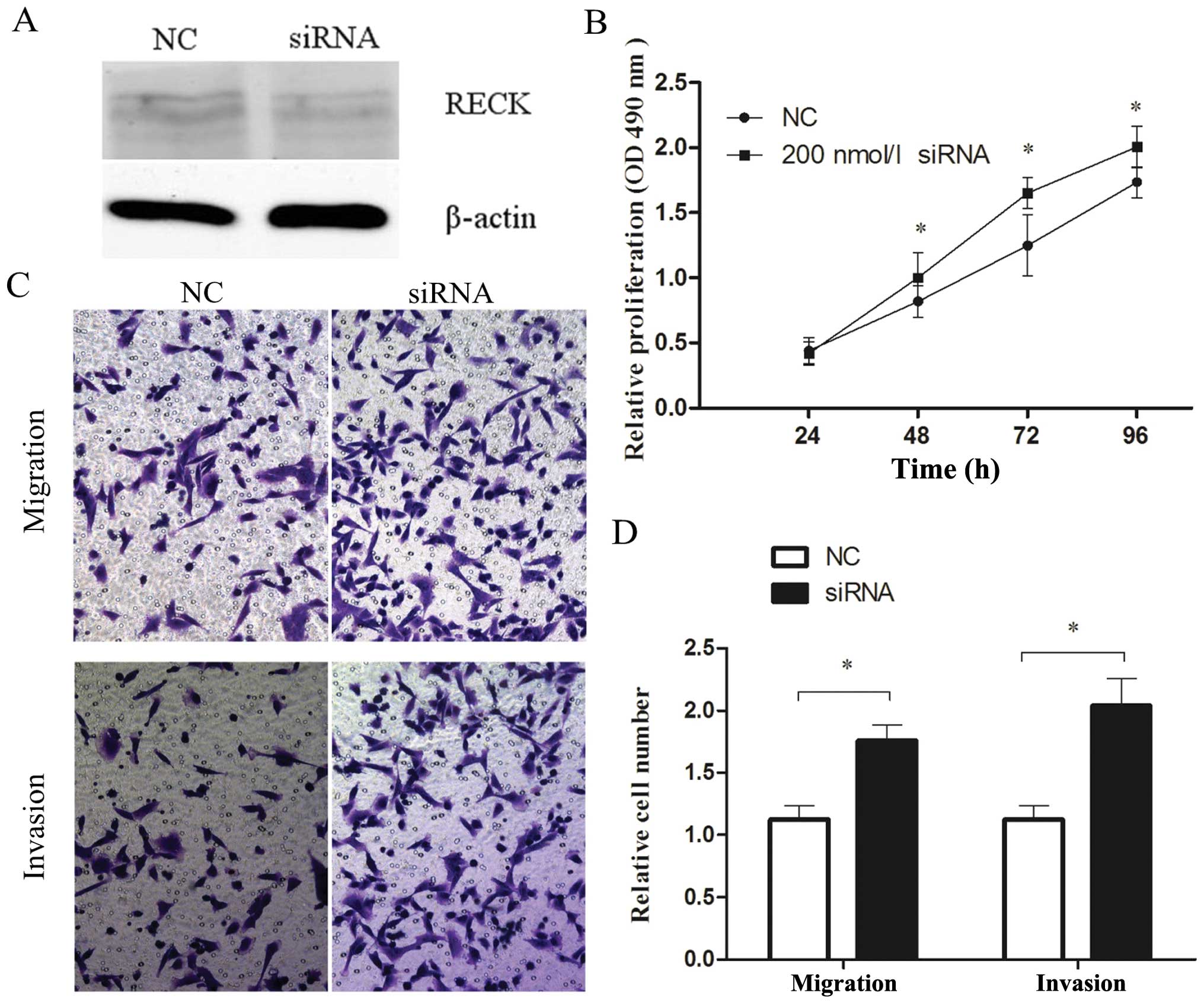
nmol/l RECK siRNA (Fig. 5A).
Furthermore, RECK siRNA transfectants markedly increased cell
proliferation by 22, 32 and 16% at 48, 72 and 96 h, respectively,
compared with the control siRNA (P<0.05; Fig. 5B). In addition, inhibition of RECK
promoted the ability of cells to migrate and invade adjacent
tissues (P<0.05; Fig. 5C and D).
These data indicate that downregulation of RECK expression promotes
cell proliferation, migration and invasion, phenocopying the
overexpression of miR-96 in MDA-MB-231 cells.
Discussion
Growing evidence indicates that miRNAs may
contribute to cancer pathogenesis. Dysregulation of miRNAs is
associated with the initiation and progression of breast cancer,
since they may serve as oncogenes or tumor suppressors (24–26).
In the present study, we performed qRT-PCR to evaluate the
expression pattern of miR-96 in breast cancer tissues and cell
lines. We also investigated the biological impact of miR-96 and
explored the molecular mechanisms by which miR-96 modulates the
behavior of breast cancer cells.
Our results showed that miR-96 was dramatically
upregulated in breast cancer tissues when compared to the
expression in adjacent normal tissues. In addition, we demonstrated
that miR-96 expression was elevated in 6 breast cancer cell lines
when compared with the MCF-10A non-malignant breast epithelial cell
line. Similar findings were reported in many different solid
tumors, including hepatocellular carcinoma (8), lung (9), colorectal (10), endometrial (11), as well as in prostate cancer cell
lines (7). These data indicate that
the dysregulation of miR-96 might be a common occurrence in human
cancer tissues and cell lines. In contrast, two studies reported
the opposite expression pattern of miR-96 in pancreatic cancer,
T-cell lymphoma and lung cancer (27,28).
Therefore, the expression profiles of miR-96 may be tissue- and
cell type-specific. Further research is warranted to investigate
the functional role and detailed mechanism of miR-96 in breast
cancer.
To assess the role of miR-96 in breast cancer, we
investigated the gain-or-loss of function effects of miR-96 on
various aspects of breast cancer biology. The present study
demonstrated that targeted knockdown of miR-96 expression by miR-96
inhibitors in MDA-MB-231 cells led to significant inhibition of
cellular proliferation, migration and invasion. Conversely,
transfection of miR-96 mimics into MDA-MB-231 cells induced
corresponding malignant tumor cell behaviors. Previous studies
demonstrated that suppression of miR-96 expression might also
inhibit clonogenicity and invasion in hepatocellular carcinoma
cells (8,29). Taken together, these results
indicate that dysregulated expression of miR-96 may function as an
oncogene in multiple cancers.
Identification of putative miRNA targets is critical
for understanding their pleiotropic functions in tumorigenesis
(30). In endometrial cancer,
overexpression of miR-96 induced impaired apoptotic responses by
directly repressing FOXO1 expression (11). A recent study demonstrated that
miR-96 promoted cell proliferation via targeting FOXO3a,
p27Kip1 and p21Cip1 in breast cancer
(12). In the present study, we
identified RECK as a direct target of miR-96 in MDA-MB-231 cells.
Furthermore, endogenous RECK expression, both at the mRNA and
protein levels, was decreased in MDA-MB-231 cells transfected with
miR-96 mimics, but increased in MDA-MB-231 cells transfected with
miR-96 inhibitors. Together, these data indicate that miR-96
directly interacts with RECK mRNA and suppresses RECK protein
expression. Additionally, silencing of RECK expression by siRNA in
MDA-MB-231 cells significantly promoted cellular proliferation,
migration and invasion, consistent with the results of ectopic
miR-96 expression in the same cells. These findings support the
hypothesis that RECK is a new target of miR-96.
In summary, our findings demonstrate that miR-96 is
upregulated in breast cancer tissues and cell lines, and is able to
promote cellular proliferation, migration and invasion via direct
regulation of the expression of RECK, implying that miR-96 can
serve as a potential therapeutic target for breast cancer.
Acknowledgements
This research was made possible with financial
support from the National Natural Sciences Foundation of China, for
the project 81272240, and the Shanghai Science Committee Foundation
(to L.F.) (no. STCSM 10411964700).
References
|
1
|
O’Hara SP, Mott JL, Splinter PL, Gores GJ
and LaRusso NF: MicroRNAs: key modulators of post-transcriptional
gene expression. Gastroenterology. 136:17–25. 2009.
|
|
2
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bartel DP: MicroRNAs: target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Du WW, Fang L, Li M, et al: MicroRNA
miR-24 enhances tumor invasion and metastasis by targeting PTPN9
and PTPRF to promote EGF signaling. J Cell Sci. 126:1440–1453.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhu S, Wu H, Wu F, Nie D, Sheng S and Mo
YY: MicroRNA-21 targets tumor suppressor genes in invasion and
metastasis. Cell Res. 18:350–359. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mulrane L, Madden SF, Brennan DJ, et al:
miR-187 is an independent prognostic factor in breast cancer and
confers increased invasive potential in vitro. Clin Cancer
Res. 18:6702–6713. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Haflidadottir BS, Larne O, Martin M,
Persson M, Edsjo A, Bjartell A and Ceder Y: Upregulation of miR-96
enhances cellular proliferation of prostate cancer cells through
FOXO1. PLoS One. 8:e724002013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xu D, He X, Chang Y, Xu C, Jiang X, Sun S
and Lin J: Inhibition of miR-96 expression reduces cell
proliferation and clonogenicity of HepG2 hepatoma cells. Oncol Rep.
29:653–661. 2013.PubMed/NCBI
|
|
9
|
Zhu W, Liu X, He J, Chen D, Hunag Y and
Zhang YK: Overexpression of members of the microRNA-183 family is a
risk factor for lung cancer: a case control study. BMC Cancer.
11:3932011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bandres E, Cubedo E, Agirre X, et al:
Identification by real-time PCR of 13 mature microRNAs
differentially expressed in colorectal cancer and non-tumoral
tissues. Mol Cancer. 5:292006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Myatt SS, Wang J, Monteiro LJ, et al:
Definition of microRNAs that repress expression of the tumor
suppressor gene FOXO1 in endometrial cancer. Cancer Res.
70:367–377. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lin H, Dai T, Xiong H, et al: Unregulated
miR-96 induces cell proliferation in human breast cancer by
downregulating transcriptional factor FOXO3a. PLoS One.
5:e157972010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Takahashi C, Sheng Z, Horan TP, et al:
Regulation of matrix metalloproteinase-9 and inhibition of tumor
invasion by the membrane-anchored glycoprotein RECK. Proc Natl Acad
Sci USA. 95:13221–13226. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Oh J, Takahashi R, Kondo S, et al: The
membrane-anchored MMP inhibitor RECK is a key regulator of
extracellular matrix integrity and angiogenesis. Cell. 107:789–800.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Noda M and Takahashi C: Recklessness as a
hallmark of aggressive cancer. Cancer Sci. 98:1659–1665. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang Y, Cheng S, Zhang G, et al: Low
expression of RECK indicates a shorter survival for patients with
invasive breast cancer. Cancer Sci. 103:1084–1089. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lin HY, Chiang CH and Hung WC: STAT3
upregulates miR-92a to inhibit RECK expression and to promote
invasiveness of lung cancer cells. Br J Cancer. 109:731–738. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chiang CH, Hou MF and Hung WC:
Up-regulation of miR-182 by β-catenin in breast cancer increases
tumorigenicity and invasiveness by targeting the matrix
metalloproteinase inhibitor RECK. Biochim Biophys Acta.
1830:3067–3076. 2013.
|
|
19
|
Xin C, Buhe B, Hongting L, et al:
MicroRNA-15a promotes neuroblastoma migration by targeting
reversion-inducing cysteine-rich protein with Kazal motifs (RECK)
and regulating matrix metalloproteinase-9 expression. FEBS J.
280:855–866. 2013.PubMed/NCBI
|
|
20
|
Gabriely G, Wurdinger T, Kesari S, Esau
CC, Burchard J, Linsley PS and Krichevsky AM: MicroRNA 21 promotes
glioma invasion by targeting matrix metalloproteinase regulators.
Mol Cell Biol. 28:5369–5380. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li N, Tang B, Zhu ED, et al: Increased
miR-222 in H. pylori-associated gastric cancer correlated
with tumor progression by promoting cancer cell proliferation and
targeting RECK. FEBS Lett. 586:722–728. 2012.PubMed/NCBI
|
|
22
|
Chen C, Ridzon DA, Broomer AJ, et al:
Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic
Acids Res. 33:e1792005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
|
|
24
|
Iorio MV, Casalini P, Tagliabue E, Menard
S and Croce CM: MicroRNA profiling as a tool to understand
prognosis, therapy response and resistance in breast cancer. Eur J
Cancer. 44:2753–2759. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Luo Q, Li X, Li J, et al: MiR-15a is
underexpressed and inhibits the cell cycle by targeting CCNE1 in
breast cancer. Int J Oncol. 43:1212–1218. 2013.PubMed/NCBI
|
|
26
|
Li J, Kong X, Zhang J, Luo Q, Li X and
Fang L: MiRNA-26b inhibits proliferation by targeting PTGS2 in
breast cancer. Cancer Cell Int. 13:72013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu S, Lu Z, Liu C, et al: miRNA-96
suppresses KRAS and functions as a tumor suppressor gene in
pancreatic cancer. Cancer Res. 70:6015–6025. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vishwamitra D, Li Y, Wilson D, et al:
MicroRNA 96 is a post-transcriptional suppressor of anaplastic
lymphoma kinase expression. Am J Pathol. 180:1772–1780. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen RX, Xia YH, Xue TC and Ye SL:
Suppression of microRNA-96 expression inhibits the invasion of
hepatocellular carcinoma cells. Mol Med Rep. 5:800–804.
2012.PubMed/NCBI
|
|
30
|
Calin GA, Liu CG, Sevignani C, et al:
MicroRNA profiling reveals distinct signatures in B cell chronic
lymphocytic leukemias. Proc Natl Acad Sci USA. 101:11755–11760.
2004. View Article : Google Scholar : PubMed/NCBI
|