Introduction
Malignant pleural mesothelioma (MPM) is an
aggressive cancer arising from the surface serosal cells of pleural
cavities. MPM is associated with exposure to asbestos fibers. MPM
is highly resistant to conventional chemotherapy and radiotherapy,
and has an extremely poor prognosis; median survival is 4–13 months
for untreated patients (1) and 6–18
months for treated patients (2,3).
Although platinum-based chemotherapy is used widely, it has a
modest therapeutic effect. Hence, identification of new agents that
enhance its therapeutic effect is warranted.
The mammalian target of rapamycin (mTOR) is a
289-kDa serine-threonine kinase that was identified as a target
molecule of rapamycin (a macrolide fungicide isolated from the
bacteria Streptomyces hygroscopicus). mTOR is a member of
the large phosphatidylinositol 3-kinase (PI3K)-related kinase
(PIKK) family of protein kinases, and is located downstream of the
PI3K/Akt signaling pathway. Growth factors such as insulin-like
growth factor (IGF), epidermal growth factor (EGF) and vascular
endothelial growth factor (VEGF) regulate mTOR signals through the
PI3K/Akt signaling pathway (4).
Rapamycin inhibits mTOR by binding to its immunophilin,
FK506-binding protein 12 (FKBP12), and blocks progression from the
G1 phase to the S phase of the cell cycle and induces
the apoptosis of tumor cells (5).
mTOR also plays a key role in angiogenesis. mTOR increases the
translation of hypoxia-inducible factor 1 (HIF-1)/HIF-2. A major
role of the HIF transcription factors is to upregulate
transcription of mitogenic growth factors such as VEGF which lead
to angiogenesis (6).
Temsirolimus, a water-soluble dihydroester analog of
rapamycin, has been approved for renal cell carcinoma and breast
cancer, based on the results of a randomized phase III study
(7) and a phase II study (8), respectively. Recent studies have
demonstrated that mTOR inhibitors are useful for the treatment of
MPM (9,10). Several studies have suggested that
temsirolimus inhibits angiogenesis through inhibition of
HIF-1-dependent VEGF production in breast cancer and multiple
myeloma (6,11).
In the present study, we evaluated the benefits of
temsirolimus against MPM cell lines and the efficacy of combination
therapy with cisplatin or pemetrexed (which are presently used as
therapeutic agents for MPM) and assessed the antiangiogenic effect
of temsirolimus against MPM in vitro.
Materials and methods
Cell lines and culture conditions
The MPM cell lines H290 and Y-meso14 as well as the
human mesothelial cell line Met-5A were kindly provided by Dr
Sekido (Aichi Cancer Center Research Institute, Nagoya, Japan). The
MPM cell lines H226 and MSTO-211H as well as the human lung
fibroblast cell lines MRC-5 and IMR-90 were purchased from the
American Type Culture Collection (ATCC; Manassas, VA, USA). Cells
were maintained in specific media during incubation at 37°C in a
humidified atmosphere of 5% CO2 in air. Four human MPM
cell lines and MeT-5A were cultured in RPMI-1640 medium
supplemented with 10% fetal bovine serum (FBS). MRC-5 and IMR-90
were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10%
FBS. Each medium contained 100 U/ml of penicillin and 0.1 mg/ml of
streptomycin.
Drugs
Temsirolimus was obtained from Pfizer (New York, NY,
USA), cisplatin was from Nippon Kayaku (Tokyo, Japan), and
pemetrexed was from Eli Lilly (Tokyo, Japan). Temsirolimus was
stored as a 1-mM solution in dimethyl sulfoxide (DMSO) at −20°C,
cisplatin was stored as an undiluted solution (0.5 mg/ml) at −20°C,
and pemetrexed was stored as a solution of concentration 1 mg/ml in
physiological (0.9%) saline at −20°C.
Cell viability and proliferation
assays
Cell viability was measured by the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT)
dye reduction method (12). In
brief, cells were seeded onto 96-well plates (2,000 cells/well) in
100 μl RPMI-1640 or DMEM with 10% FBS for 24 h. Afterward, the
cells were exposed either to temsirolimus alone (at concentrations
ranging from 0 to 10 μM), cisplatin (0–10 μg/ml) or pemetrexed
(0–10 μg/ml) for 72 h at 37°C with 5% CO2 or
combinations of the agents. In addition, 50 μl of a stock MTT
solution (2 mg/ml) was added to all of the wells, and the cells
were incubated for 2 h at 37°C with 5% CO2. The media
containing the MTT solution were removed and the dark-blue crystals
dissolved by adding 100 μl DMSO. Absorbance was measured at a
wavelength of 570 nm with a microplate reader. Cell proliferation
was determined by cell counting. Cells were seeded onto 6-well
plates (10,000 cells/well) and incubated for the indicated periods.
After cells were harvested, the cell number was counted.
Determination of protein levels of VEGF
and platelet-derived growth factor-AA (PDGF-AA)
Culture supernatants were evaluated. For culture
supernatants, tumor cells were seeded into 6-well plates at
2×105 cells/well in 2 ml of RPMI-1640 medium with 10%
FBS and exposed to temsirolimus (0–1 μM) for 48 h. The supernatants
were harvested, and the levels of VEGF and PDGF-AA were determined
using enzyme-linked immunosorbent assay (ELISA) kits (R&D
Systems, Minneapolis, MN, USA) according to the manufacturer’s
instructions. The lower limit of detection was 31.2 pg/ml for VEGF
and 31.2 pg/ml for PDGF-AA.
Antibodies and western blot analyses
Tumor cells were incubated in 10 ml of RPMI-1640
buffer with 10% FBS in temsirolimus (0–1 μM) for 1 h. The total
protein level was measured using a bicinchoninic acid (BCA) protein
assay kit (Pierce Biotechnology, Rockford, IL, USA). For western
blot analyses, 40 μg of total protein was resolved by sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE)
(Bio-Rad Laboratories, Waltham, MA, USA). The proteins were
transferred onto polyvinylidene difluoride membranes (Bio-Rad
Laboratories). The following primary antibodies were used:
anti-phospho-S6 ribosomal protein; anti-S6 ribosomal protein, or
anti-cleaved caspase 3 (Asp175) antibodies (1:1,000 dilution; Cell
Signaling Technology, Beverly, MA, USA) and anti-G3PDH polyclonal
antibody (1:1,000 dilution; Trevigen, El Paso, TX, USA). Membranes
were incubated for 1 h at room temperature with species-specific
horseradish peroxidase-conjugated secondary antibodies.
Immunoreactive bands were visualized with SuperSignal West Dura
Extended Duration Substrate Enhanced Chemiluminescent Substrate
(Pierce Biotechnology). Each experiment was carried out
independently in triplicate.
Immunohistochemical analyses
Five tumor specimens were obtained from 5 MPM
patients, all of whom provided written informed consent, at the
Kanazawa University Hospital. The present study was approved by the
Institutional Review Boards of the Kanazawa University.
Paraffin sections were autoclaved and immunostained
with the following primary antibodies: mTOR (mTOR/FRAP; rabbit
monoclonal; 1:350 dilution; Epitomics, San Francisco, CA, USA),
anti-phospho-mTOR [mTOR/FRAP phospho (pS2448); rabbit monoclonal;
1:100 dilution; Epitomics], anti-VEGF (rabbit polyclonal; 1:100
dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA), and
anti-PDGF-AA (rabbit polyclonal; 1:200 dilution; EMD Millipore
Corp., Waltham, MA, USA). Antibodies were visualized by a ChemMate
EnVision/peroxidase complex kit (Dako, Tokyo, Japan). The
‘immunohistochemical expression level’ was scored by a three-tier
system: negative expression (score 0), <10%; low expression
(score 1), ≥10% but <50%; high expression (score 2), ≥50% tumor
cells with ‘significant’ staining (13).
Statistical analyses
Statistical analyses were carried out using GraphPad
Prism ver 5.02. The statistical significance of differences between
control and test values was analyzed using Student’s t-test,
Mann-Whitney U test, and one-way analysis of variance (ANOVA) with
Bonferroni’s post hoc test, where applicable. Differences at
P<0.05 were deemed significant.
Results
Effects of temsirolimus, cisplatin or
pemetrexed on MPM cell lines
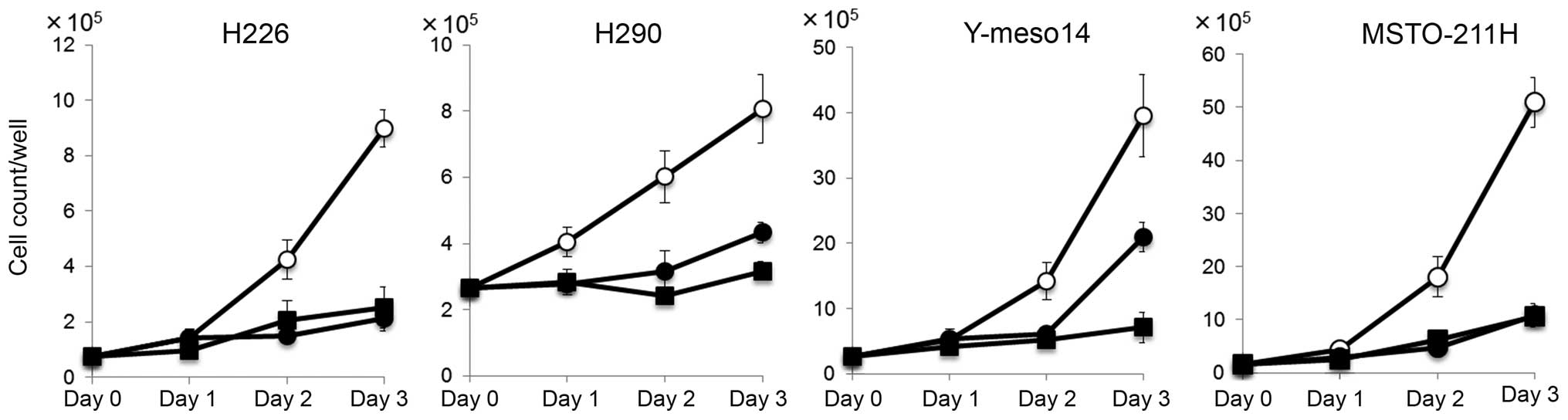
Temsirolimus treatment for 72 h inhibited the
viability of all MPM cell lines in a concentration-dependent manner
(Fig. 1A). The half-maximal
inhibitory concentration (IC50) of temsirolimus in H226,
H290, Y-meso14 and MSTO-211H cells was ~2, 0.5, 0.5 and 0.2 μM,
respectively. Temsirolimus also inhibited the cell numbers of all
MPM cell lines tested (Fig. 2),
indicating that temsirolimus suppressed cell proliferation of the
MPM cell lines. Cisplatin also inhibited the viability of all MPM
cell lines. The IC50 of cisplatin in H226, H290,
Y-meso14 and MSTO-211H cells was ~2, 0.5, 0.7 and 0.4 μg/ml,
respectively. Pemetrexed moderately inhibited the viability of MPM
cell lines when compared with the effect of the other two drugs
except for MSTO-211H cells. The IC50 of pemetrexed in
MSTO-211H cells was ~0.04 μg/ml, whereas it was >10 μg/ml in the
other three cell lines.
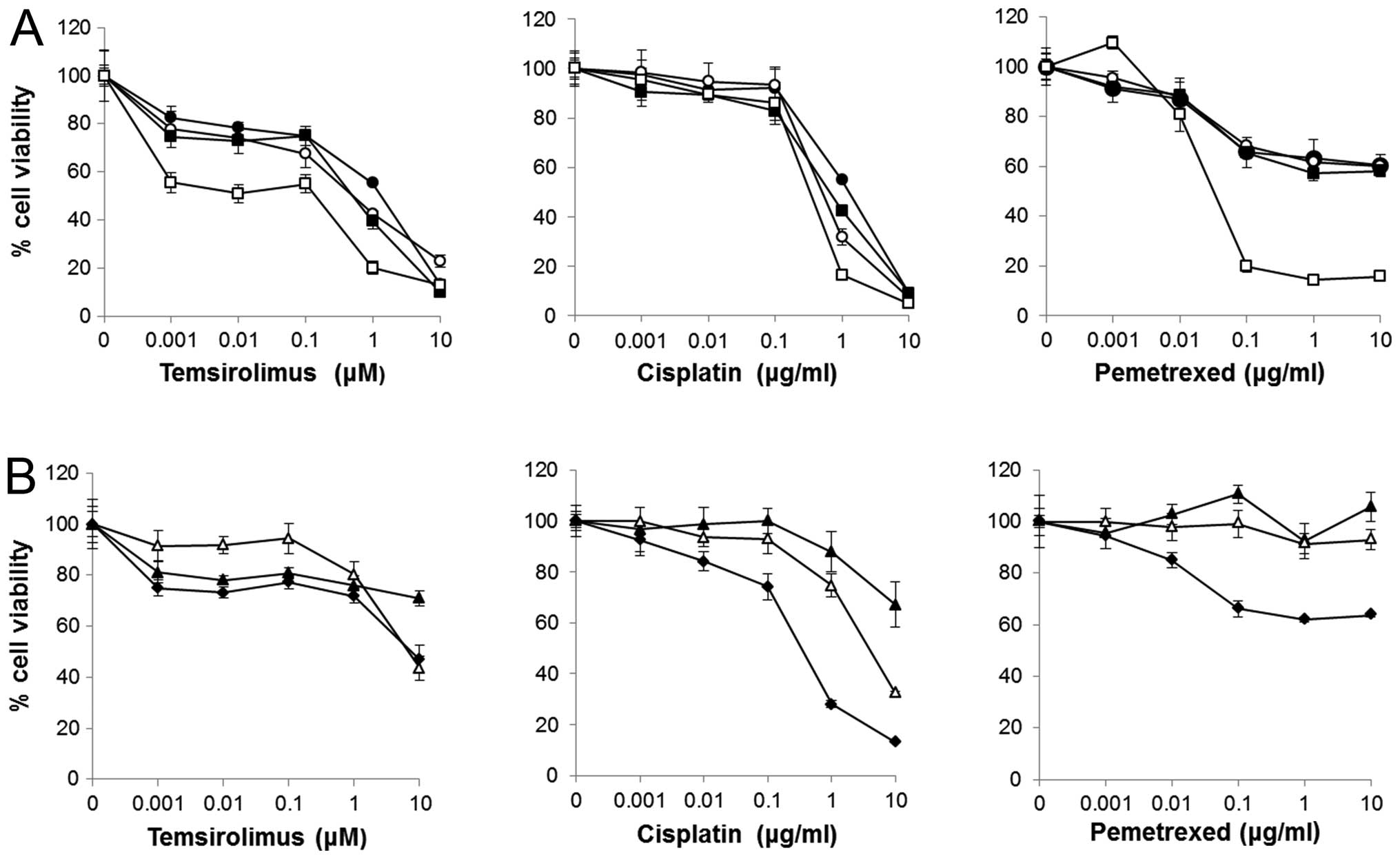 | Figure 1Effect of temsirolimus, cisplatin, and
pemetrexed on the viability of MPM cell lines and non-neoplastic
cells. (A) H226 (●), H290 (○), Y-meso14 (■) and MSTO-211H (□)
(2×103/well) cells were incubated for 72 h with
temsirolimus (0–10 μM), cisplatin (0–10 μg/ml) or pemetrexed (0–10
μg/ml). (B) MRC-5 (▲), IMR-90 (△) and Met-5A (◆)
(2×103/well) cells were incubated for 72 h with
temsirolimus (0–10 μM), cisplatin (0–10 μg/ml), or pemetrexed (0–10
μg/ml). Points, mean of quintet cultures; bars, SD. |
Effect of temsirolimus, cisplatin or
pemetrexed on MRC-5, IMR-90 and Met-5A as ‘non-neoplastic
cells’
Temsirolimus-induced inhibition of the viability of
non-neoplastic cells was less compared with that of the MPM cell
lines (Fig. 1B). Cisplatin
displayed strong inhibition of the viability of Met-5A cells, but
less inhibition of the other cell lines. Pemetrexed showed slight
inhibition of the viability of Met-5A cells, but not MRC-5 or
IMR-90 cells.
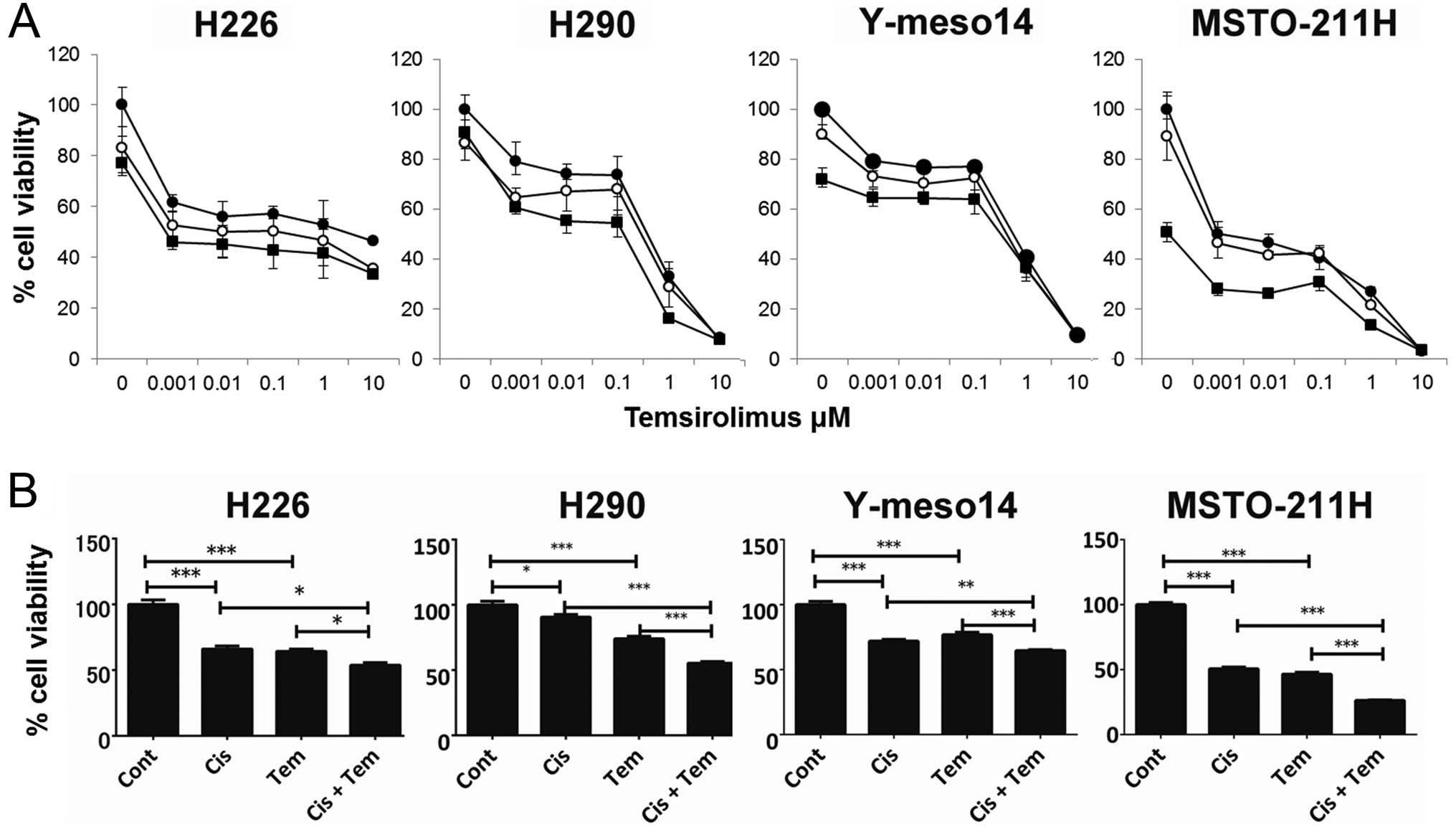
Effects of a combined treatment of
temsirolimus and cisplatin or the combined treatment of
temsirolimus and pemetrexed on the viability of MPM cell lines
To examine the effect of temsirolimus combined with
cisplatin or pemetrexed on MPM cell lines, cells were cultured with
combinations of temsirolimus (0–10 μM) and cisplatin (0.1 and 0.3
μg/ml) or pemetrexed (0.1 and 0.3 μg/ml) for 72 h. MSTO-211H cells
demonstrated high sensitivity to pemetrexed, thus the MSTO-211H
cell line was excluded from combination treatment with temsirolimus
and pemetrexed. The dose-response curves of the combination
treatment are depicted in Fig. 3A.
Combination treatment with temsirolimus and cisplatin inhibited the
viability of all MPM cell lines more effectively than temsirolimus
alone. When the effect of the combination treatment of 0.01 μM
temsirolimus and 0.3 μg/ml cisplatin was compared with 0.01 μM
temsirolimus alone or 0.3 μg/ml cisplatin alone, the combination
treatment was significantly more effective wheb compared with each
drug alone in all MPM cell lines (Fig.
3B). There was no significant difference between the
combination treatment of 0.01 μM temsirolimus and 0.3 μg/ml
pemetrexed with pemetrexed alone in the H226 cells or with
temsirolimus alone in the H290 cells (data not shown). Compared
with each drug alone, the combination of temsirolimus and
pemetrexed was more effective only in the Y-meso14 cells.
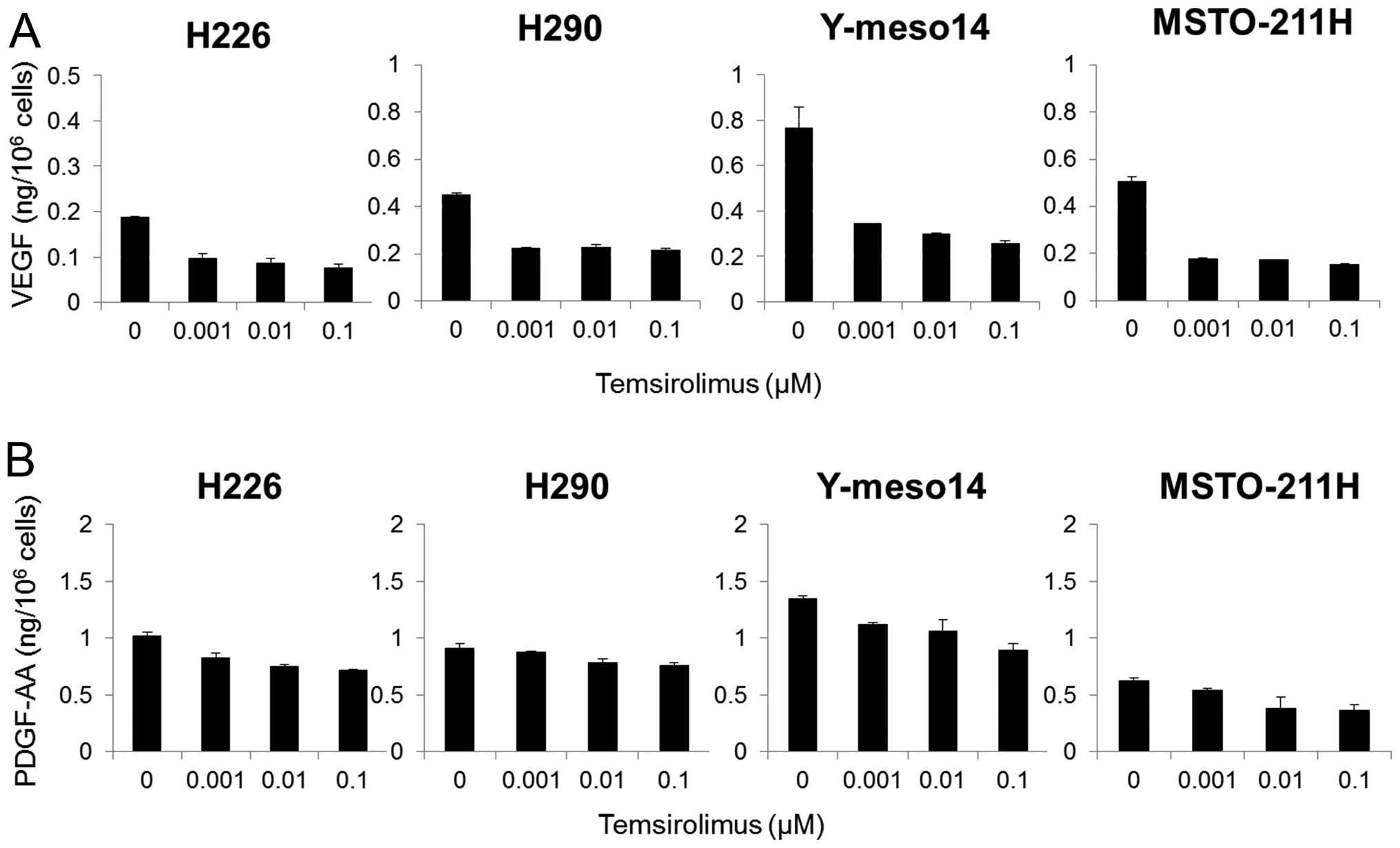
Effect of temsirolimus on proangiogenic
cytokine production in the MPM cell lines
To examine the effect of temsirolimus on the release
of proangiogenic cytokines in the MPM cell lines, tumor cells were
exposed to temsirolimus (0–1 μM) and the levels of VEGF and PDGF-AA
in the supernatants were measured by ELISA. Temsirolimus strongly
inhibited VEGF production (Fig.
4A), and moderately inhibited the production of PDGF-AA in all
four MPM cell lines (Fig. 4B).
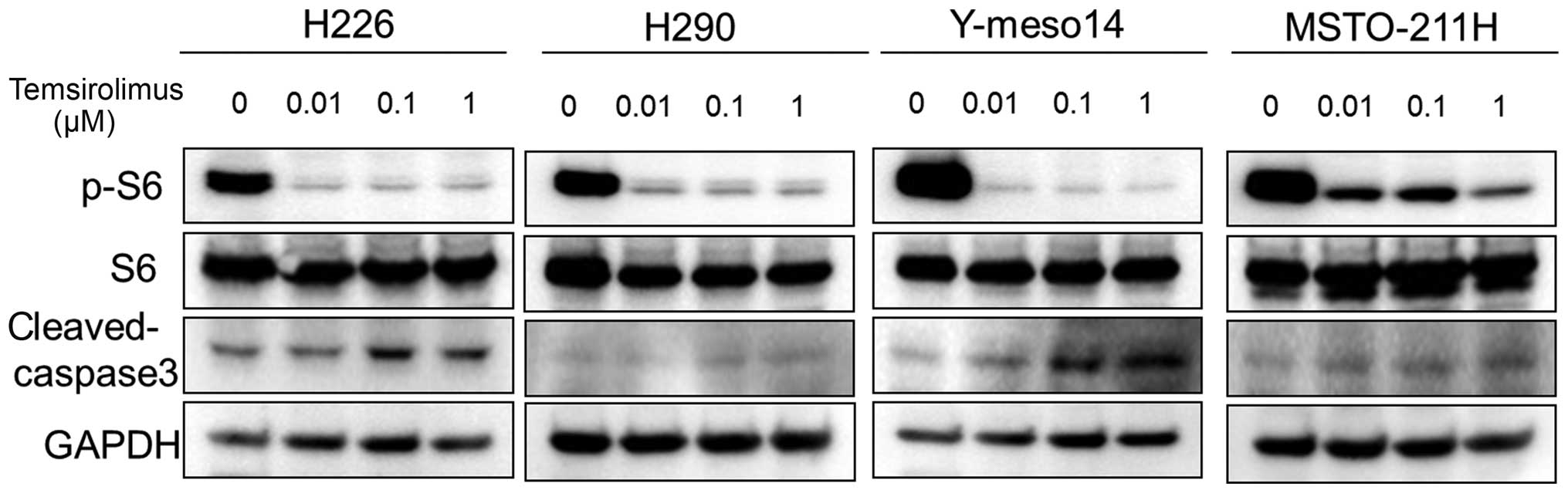
Effect of temsirolimus on the downstream
proteins of mTOR signaling
Western blot analyses showed that temsirolimus
strongly inhibited the phosphorylation of S6 ribosomal protein (a
kinase regulated by mTOR). The level of cleaved caspase-3 (a known
marker of apoptosis) increased in a dose-dependent manner in the
H226 and Y-meso14 cells, but not in the MSTO-211H and H290 cells,
following treatment with temsirolimus (Fig. 5).
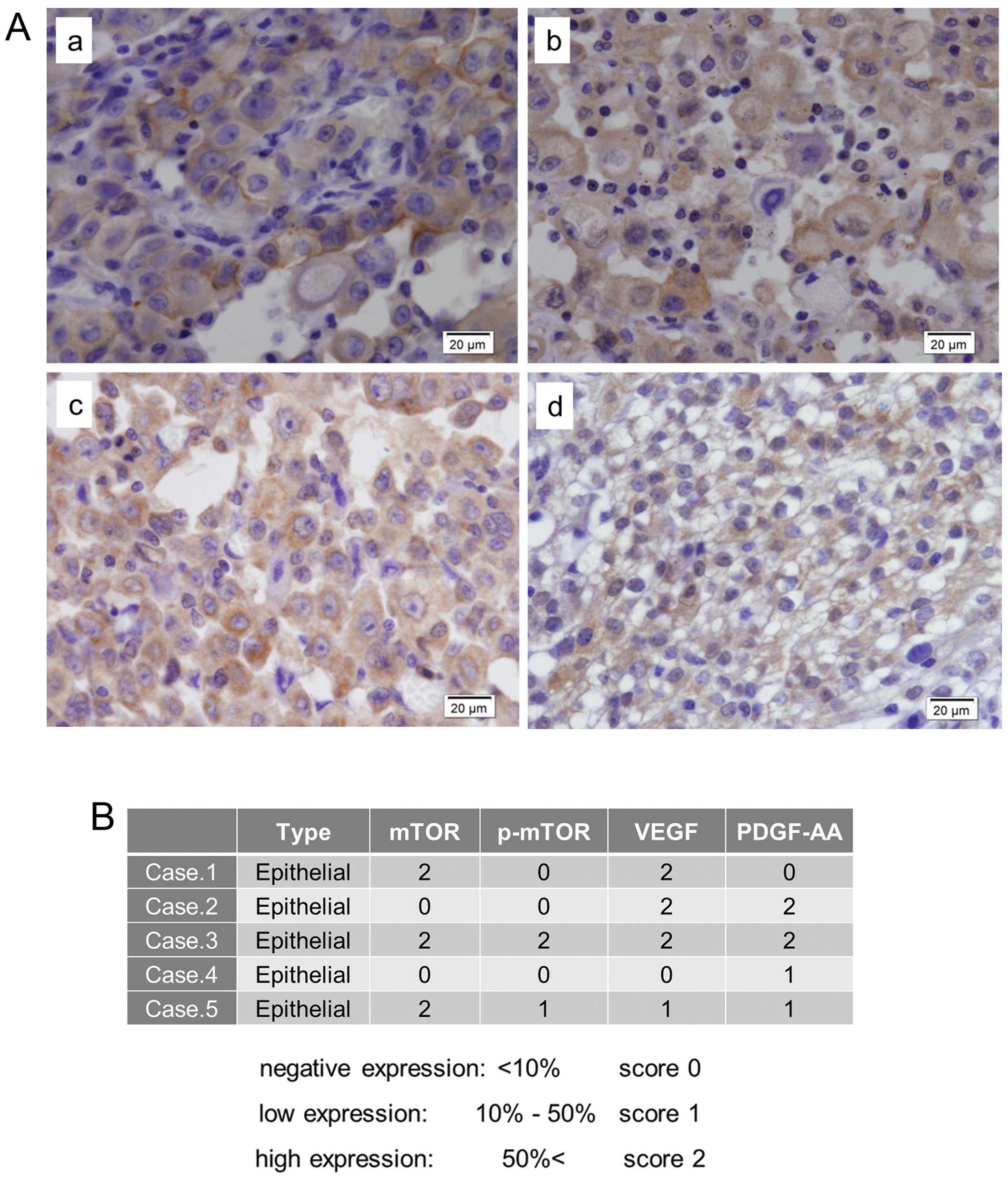
Immunohistochemical expression of mTOR,
p-mTOR, VEGF and PDGF-AA in MPM tissues
mTOR immunohistochemical staining exhibited an
appreciable expression and cytoplasmic staining pattern in three of
five MPM tissues (Fig. 6A–a and B).
p-mTOR immunohistochemical staining also demonstrated a cytoplasmic
pattern, and was observed in two of five MPM tissues (Fig. 6A–b and B). VEGF and PDGF-AA were
expressed in the cytoplasm of numerous cells and in the nuclei of
some cells in four of five MPM tissues (Fig. 6A–c, d and B).
Discussion
In the present study, we demonstrated that
temsirolimus inhibited the proliferation of MPM cells and that
combined treatment with temsirolimus and cisplatin (a standard
chemotherapeutic agent for MPM) resulted in greater cell death in
the MPM cell lines.
Temsirolimus is a water-soluble dihydroester analog
of rapamycin and is being developed for the treatment of cancer.
Temsirolimus was approved by the US Food and Drug Administration
(FDA) for the treatment of advanced renal cell carcinoma (RCC) in
2007 as efficacy and safety were demonstrated at a second interim
analysis of a phase III, multicenter, international, randomized and
open-label study (7). In addition
to RCC, several clinical trials were conducted in diverse types of
carcinoma, such as breast cancer (8) and glioblastoma (14).
Several reports have focused on the effect of mTOR
inhibitors on MPM. Hartman et al (9) demonstrated that sirolimus (previously
known as rapamycin) inhibited the proliferation of MPM cell lines,
and that a combined treatment of sirolimus and cisplatin led to
greater cell death in MPM cell lines. Hoda et al (10) reported that temsirolimus attenuated
MPM cell growth in vitro and tumor formation in vivo,
and it synergized with cisplatin against MPM models in vitro
and in vivo.
mTOR is a protein kinase located downstream of the
PI3K/Akt signaling pathway, which has an important role in the
survival and proliferation of cells. Temsirolimus gains function by
binding to FKBP12 and this complex inhibits mTOR kinase activity,
consequently inhibiting phosphorylation of the 40S ribosomal
protein S6 kinase (p70s6k) and the eukaryotic initiation factor
4E-binding protein-1 (4E-BP1) (15). Inhibition of these elements results
in a decrease in protein synthesis and translation of specific mRNA
species, and interferes with the progression from the G1
phase to S phase of the cell cycle (5). Temsirolimus also inhibits the
translation of HIF-1/HIF-2, which upregulate the transcription of
mitogenic growth factors such as VEGF and PDGF (6). In western blot analysis,
phosphorylation of mTOR was observed in MPM specimens and
temsirolimus strongly inhibited the phosphorylation of p70s6k
(which is located downstream of mTOR) in all MPM cell lines in a
dose-dependent manner. These data suggest that temsirolimus
treatment inhibits the proliferation of tumor cells by inhibiting
the phosphorylation of mTOR by inhibiting the activity of mTOR
kinase. Temsirolimus treatment led to an increase in the level of
cleaved caspase-3 (considered to be a marker of apoptosis) in H226
and Y-meso14 cells, but did not lead to an increase in the H290 and
MSTO-211H cells. On the other hand, MTT assay showed that MSTO-211H
cells had the most sensitivity to temsirolimus treatment. These
data indicate that, in MSTO-211H and H290 cells, temsirolimus
treatment did not induce apoptosis, but strongly obstructed cell
proliferation. As expected, temsirolimus inhibited the cell count
of all MPM cell lines tested. In contrast, Schedel et al
(16) reported that temsirolimus
significantly reduced cell viability and induced apoptosis and cell
cycle arrest in bladder cancer and head and neck squamous cell
carcinoma.
MPM is highly resistant to conventional
chemotherapy. Cisplatin is one of the most effective
chemotherapeutic agents against MPM but, if used alone, its
response rate against MPM has been shown to be only 16.7%, with a
median survival time of 9.3 months (17). When used in combination with the
multitargeted antifolate agent pemetrexed, the response rate
increased significantly to 41.3% and the median survival time was
significantly prolonged to 12.1 months; however, the difference was
only 2.8 months. Therefore, the identification of new agents is
warranted. In the present study, we evaluated the therapeutic
efficacy of the combined treatment of temsirolimus and cisplatin in
MPM cell lines. Combination treatment with temsirolimus and
cisplatin inhibited the viability of all MPM cell lines more
effectively than treatment with temsirolimus alone. Hence, a
synergistic or additive effect for the combination treatment of
temsirolimus and cisplatin can be hypothesized, in accordance with
another report (10).
Rapidly proliferating tumors require an efficient
blood supply to meet their nutritional needs; therefore,
angiogenesis is essential for the growth and metastasis of tumors
(18). VEGF is one of the most
potent stimulators of angiogenesis, and high expression of VEGF is
associated with intratumoral microvessel density and a poor
prognosis in various types of carcinomas (19,20).
PDGF has also been shown to support growth, angiogenesis and stroma
recruitment in various tumor types (21,22).
Del Bufalo et al (6)
reported that temsirolimus may potently inhibit angiogenesis by
multiple mechanisms: indirectly through transcriptional inhibition
of hypoxia-stimulated, HIF-1α-dependent VEGF production as well as
directly through inhibition of the endothelial cell functions
involved in neoangiogenesis (e.g., growth factor-stimulated
proliferation and morphogenesis). Other authors have reported that
an mTOR inhibitor inhibits tumor growth by an antiangiogenic effect
or by tumor-specific thrombosis associated with a decrease in VEGF
production (23). In accordance
with our previous studies (24,25),
tumor progression of Y-meso14 and EHMES-10 (human MPM cell lines),
which expressed high levels of VEGF, was inhibited by bevacizumab
(an anti-VEGF antibody) or E7080 (a multi-tyrosine kinase
inhibitor) in vivo. In contrast, the tumor progression of
MSTO-211H cells (which expressed low levels of VEGF) was not
inhibited by treatment with bevacizumab. In the present study, we
showed that temsirolimus strongly inhibited the production of VEGF
in all four MPM cell lines tested. Temsirolimus is expected to
inhibit the growth of VEGF-high-producing tumors (e.g., Y-meso14),
due not only to an anticancer effect but also an antiangiogenic
effect in vivo.
In summary, our results suggest that the mTOR
inhibitor temsirolimus inhibits the proliferation of MPM cells, and
strengthens the effect of platinum-based chemotherapy. The
treatment effect resulted in dephosphorylation of the downstream
proteins of the PI3K/Akt signaling pathway. In addition, the
antiangiogenic effects may contribute substantially to the
antitumor activity of tumors producing high levels of VEGF, such as
in Y-meso 14 cells. Temsirolimus may be beneficial for a subset of
MPM patients with high mTOR expression. We intend to investigate
the effect of temsirolimus treatment in a MPM model in
vivo.
References
|
1
|
Ong ST and Vogelzang NJ: Chemotherapy in
malignant pleural mesothelioma. A review. J Clin Oncol.
14:1007–1017. 1996.PubMed/NCBI
|
|
2
|
Antman KH: Natural history and
epidemiology of malignant mesothelioma. Chest. 103:373S–376S. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Aisner J: Current approach to malignant
mesothelioma of the pleura. Chest. 107:332S–344S. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schmelzle T and Hall MN: TOR, a central
controller of cell growth. Cell. 103:253–262. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Grewe M, Gansauge F, Schmid RM, Adler G
and Seufferlein T: Regulation of cell growth and cyclin D1
expression by the constitutively active FRAP-p70s6K pathway in
human pancreatic cancer cells. Cancer Res. 59:3581–3587.
1999.PubMed/NCBI
|
|
6
|
Del Bufalo D, Ciuffreda L, Trisciuoglio D,
et al: Antiangiogenic potential of the mammalian target of
rapamycin inhibitor temsirolimus. Cancer Res. 66:5549–5554.
2006.PubMed/NCBI
|
|
7
|
Hudes G, Carducci M, Tomczak P, et al:
Global ARCC Trial. Temsirolimus, interferon alfa, or both for
advanced renal-cell carcinoma. N Engl J Med. 356:2271–2281. 2007.
View Article : Google Scholar
|
|
8
|
Chan S, Scheulen ME, Johnston S, et al:
Phase II study of temsirolimus (CCI-779), a novel inhibitor of
mTOR, in heavily pretreated patients with locally advanced or
metastatic breast cancer. J Clin Oncol. 23:5314–5322. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hartman ML, Esposito JM, Yeap BY and
Sugarbaker DJ: Combined treatment with cisplatin and sirolimus to
enhance cell death in human mesothelioma. J Thorac Cardiovasc Surg.
5:1233–1240. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hoda MA, Mohamed A, Ghanim B, et al:
Temsirolimus inhibits malignant pleural mesothelioma growth in
vitro and in vivo: synergism with chemotherapy. J Thorac Oncol.
6:852–863. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Frost P, Moatamed F, Hoang B, et al: In
vivo antitumor effects of the mTOR inhibitor CCI-779 against human
multiple myeloma cells in a xenograft model. Blood. 15:4181–4187.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Green LM, Reade JL and Ware CF: Rapid
colorimetric assay for cell viability: application to the
quantitation of cytotoxic and growth inhibitory lymphokines. J
Immunol Methods. 70:257–268. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dobashi Y, Suzuki S, Sato E, Hamada Y,
Yanagawa T and Ooi A: EGFR-dependent and independent activation of
Akt/mTOR cascade in bone and soft tissue tumors. Mod Pathol.
22:1328–1340. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Galanis E, Buckner JC, Maurer MJ, et al:
Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma
multiforme: a North Central Cancer Treatment Group Study. J Clin
Oncol. 23:5294–5304. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jefferies HB, Fumagalli S, Dennis PB,
Reinhard C, Pearson RB and Thomas G: Rapamycin suppresses 5′TOP
mRNA translation through inhibition of p70s6k. EMBO J.
16:3693–3704. 1997.
|
|
16
|
Schedel F, Pries R, Thode B, et al: mTOR
inhibitors show promising in vitro activity in bladder
cancer and head and neck squamous cell carcinoma. Oncol Rep.
25:763–768. 2011.
|
|
17
|
Vogelzang NJ, Rusthoven JJ, Symanowski J,
et al: Phase III study of pemetrexed in combination with cisplatin
versus cisplatin alone in patients with malignant pleural
mesothelioma. J Clin Oncol. 21:2636–2644. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fidler IJ and Ellis LM: The implications
of angiogenesis for the biology and therapy of cancer metastasis.
Cell. 79:185–188. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Niedergethmann M, Hildenbrand R, Wostbrock
B, et al: High expression of vascular endothelial growth factor
predicts early recurrence and poor prognosis after curative
resection for ductal adenocarcinoma of the pancreas. Pancreas.
25:122–129. 2002. View Article : Google Scholar
|
|
20
|
Takahashi Y, Kitadai Y, Bucana CD, Cleary
KR and Ellis LM: Expression of vascular endothelial growth factor
and its receptor, KDR, correlates with vascularity, metastasis, and
proliferation of human colon cancer. Cancer Res. 55:3964–3968.
1995.PubMed/NCBI
|
|
21
|
Ostman A and Heldin CH: PDGF receptors as
targets in tumor treatment. Adv Cancer Res. 97:247–274. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Andrae J, Gallini R and Betsholtz C: Role
of platelet-derived growth factors in physiology and medicine.
Genes Dev. 22:1276–1312. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guba M, Yezhelyev M, Eichhorn ME, et al:
Rapamycin induces tumor-specific thrombosis via tissue factor in
the presence of VEGF. Blood. 105:4463–4469. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li Q, Yano S, Ogino H, et al: The
therapeutic efficacy of anti-vascular endothelial growth factor
antibody, bevacizumab, and pemetrexed against orthotopically
implanted human pleural mesothelioma cells in severe combined
immunodeficient mice. Clin Cancer Res. 13:5918–5925. 2007.
View Article : Google Scholar
|
|
25
|
Ikuta K, Yano S, Trung VT, et al: E7080, a
multi-tyrosine kinase inhibitor, suppresses the progression of
malignant pleural mesothelioma with different proangiogenic
cytokine production profiles. Clin Cancer Res. 15:7229–7237. 2009.
View Article : Google Scholar
|