Introduction
pp32 (ANP32A) consists of N-terminal leucine-rich
repeats (LRR) and a C-terminal acidic domain (1,2).
pp32r1 (ANP32C) and pp32r2 (ANP32D) are members of the pp32 family,
and genes of this family are located on different chromosomes from
that of the pp32 gene (3). Although
pp32 has been reported to suppress cellular transformation induced
by multiple oncogenes (2), pp32r1
and pp32r2 are the cause of prostate and breast adenocarcinoma, can
transform rodent cells, and produce tumors in nude mice (3,4). pp32
has been shown to enhance apoptotic activity through the
stimulation of caspase, which is thought to be required for the
tumor suppressive activity of pp32 (5). In cancer cells, phosphorylated
retinoblastoma protein forms a complex with pp32 to inhibit
pp32-mediated apoptosis, whereas pp32r1 and pp32r2 fail to interact
with retinoblastoma protein (6).
However, the oncogenic mechanism mediated by pp32r1 and pp32r2 is
not fully understood.
pp32 binds to many important proteins such as
ataxin-1 (7), Granzyme A (8), the phosphorylated form of Rb (6) and HuR (9) to modulate their functions. pp32 is
involved in the stabilization of the AU-rich element (ARE)
containing mRNA through interaction with the RNA-binding protein
HuR (9,10). HuR is a member of the embryonic
lethal abnormal vision (ELAV) family of RNA-binding proteins
(11). It can shuttle between the
nucleus and the cytoplasm, and the stabilization of ARE-mRNA by HuR
is believed to be linked to its localization in the cytoplasm
(12,13). HuR is abundant in the cytoplasm in
many types of cancer cells, and cytoplasmic HuR expression has been
implicated in the malignancy of several types of carcinomas such as
colon cancers (14,15).
In a previous study, we identified pp32 as an
associated protein of the adenovirus oncogene product E4orf6. It
became clear that E4orf6 exported ARE-mRNA from the nucleus to the
cytoplasm of cells by interacting with pp32 and HuR protein
(16). Moreover, we elucidated that
stabilized ARE-mRNA such as c-fos and c-myc mRNA can
contribute to the transformation of cells (17). Cytoplasmic expression of HuR and
ARE-mRNA was also abundant in oral cancer cells, which are not
caused by virus genes (18), and
HuR knockdown downregulated a malignant phenotype such as the
invasive activity of oral cancer cells (19). Since ARE-mRNA was found to be
exported and stabilized in numerous cancer cells as well (13), the export and stabilization of
ARE-mRNA are thought to be general oncogenic mechanisms. To
understand the mechanism by which cytoplasmic HuR expression is
upregulated in cancer cells, we focused on HuR decay mediated by
the pp32 family.
It has been shown that when the HuR-pp32 complex is
exported to the cytoplasm by lethal stress such as staurosporine
(STS), HuR is cleaved by caspase-3 and -7, and the released pp32
activates apoptosomes to induce the apoptosis of cells (20). However, no evidence concerning the
effects of pp32r1 on HuR regulation has been shown. In this study,
we examined the effect of pp32r1 on HuR degradation. The expression
of pp32r1 in cancer cells was higher than that in normal cells.
Unlike pp32, pp32r1 was localized to the cytoplasm of cells. pp32r1
inhibited the cleavage of HuR in cells treated with lethal stress.
pp32r1 bound to HuR, and HuR survived in the cytoplasm of cancer
cells when they expressed pp32r1. These findings suggest that
pp32r1 binds to HuR to control its decay, and the survival of HuR
contributes to the transformation of cells.
Materials and methods
Cell culture, transfection, stress
treatment and cell fractionation
HEK293 (human embryonic kidney cells transformed by
the adenovirus E1 gene), SAS (oral cancer), HeLa (cervical cancer),
HT1080 (fibrosarcoma), MRC5 (human lung fibroblast), HGF (human
gingival fibroblast) and BJ (human foreskin fibroblast) cell lines
were obtained from the American Type Culture Collection (ATCC;
Manassas, VA, USA) and were cultured at 37°C and 5% CO2
atmosphere in Dulbecco’s modified Eagle’s medium (DMEM; Sigma)
containing 10% fetal bovine serum (FBS) (HyClone) with antibiotics
(Sigma). Cells were transfected with pcDNA3-FLAG-pp32,
pCMV2-FLAG-pp32r1 and each empty plasmid (pcDNA3 and pCMV2-FLAG)
using Hilymax (Dojindo) according to the manufacturer’s protocol.
For the lethal stress experiments, cells were treated with 1 μM
staurosporine (Sigma). In order to separate cells into cytoplasmic
and nuclear fractions, cells were resuspended in a fractionating
buffer (10 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1.5 mM
MgCl2, 0.5% Nonidet P-40, protease inhibitor cocktail),
followed by vigorous shaking for 5 min and centrifugation at 12,000
rpm for 30 sec as previously described (21). The supernatant was used as the
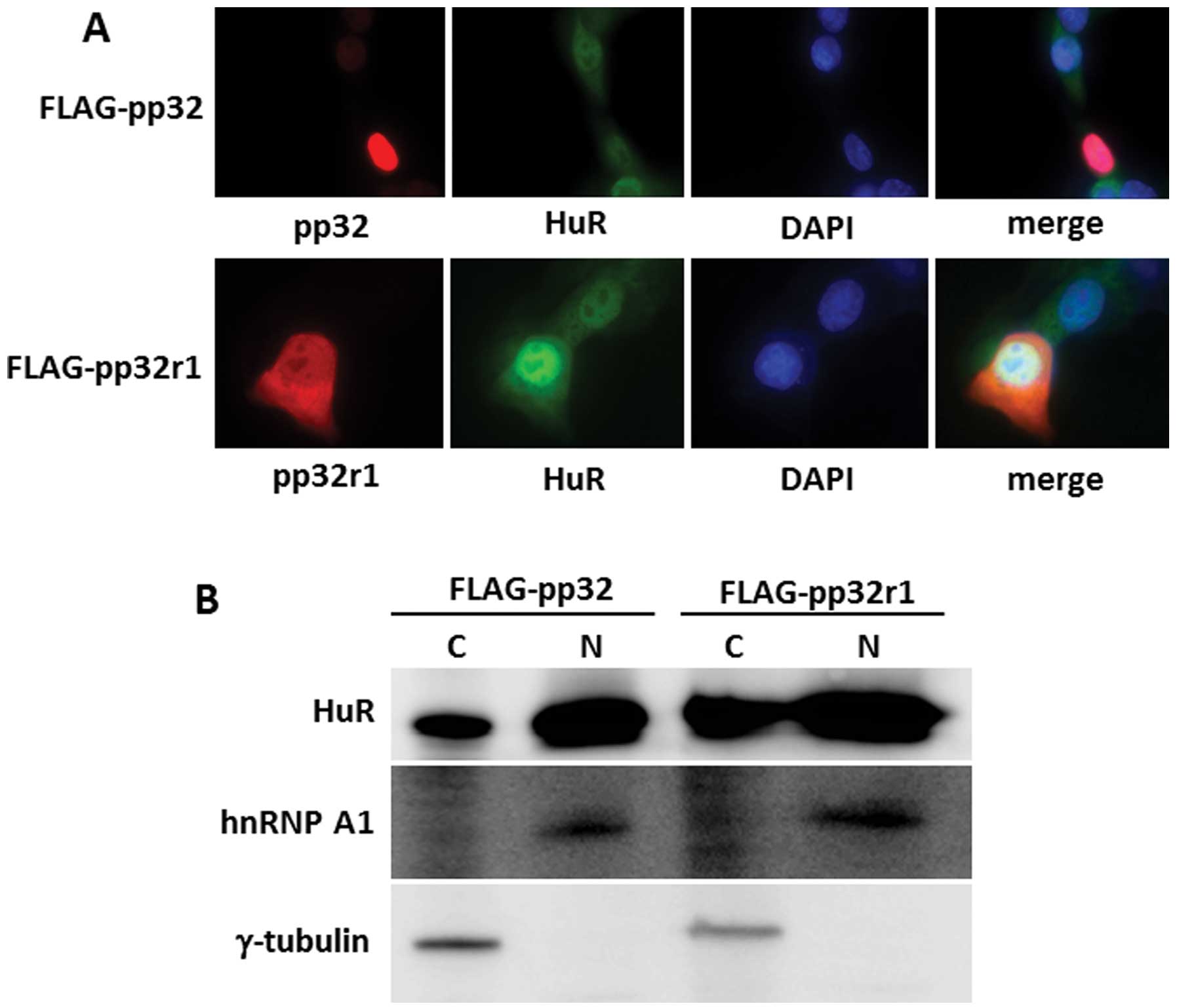
cytoplasmic fraction. To estimate the accuracy of cell
fractionation, cytoplasmic (γ-tubulin) and nuclear (hnRNP A1)
proteins were detected by western blotting.
Protein analysis
Western blotting was performed as previously
described (22). Antibodies used in
this study were specific to HuR, pp32, caspase-3, hnRNP A1 (Santa
Cruz), and pp32r1, β-actin, γ-tubulin (Sigma). The secondary
antibody was horseradish peroxidase-conjugated IgG (Jackson
ImmunoResearch Laboratories). Immunoprecipitation was performed as
previously described (23). SAS
cells were lysed in RIPA buffer [25 mM Tris-HCl (pH 7.6), 150 mM
NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS] and proteins were
immunoprecipitated using anti-pp32 and anti-pp32r1 antibodies.
Immunofluorescence
pp32- and pp32r1-expressing cells plated on
coverslips in 35-mm dishes were fixed with 4% paraformaldehyde,
permeabilized with 0.5% Triton X-100 (Sigma), and incubated with
antibodies as follows: mouse monoclonal anti-HuR antibody, goat
monoclonal anti-pp32 antibody, rabbit monoclonal anti-pp32r1
antibody. FITC-conjugated anti-mouse, rhodamine-conjugated
anti-goat or anti-rabbit antibodies were used as secondary
antibody. Cells were observed using an IX71 inverted microscope
(Olympus).
Soft-agar colony formation assay
pp32-overexpressing or pp32r1-overexpressing cells
(3×104) were plated per 60-mm culture dish in 3 ml of
DMEM containing 10% FBS and 0.36% agar on a layer of 5 ml of the
same medium containing 0.75% agar. Three weeks after plating, the
colonies were stained with 0.04% crystal violet-2% ethanol in
PBS.
Results
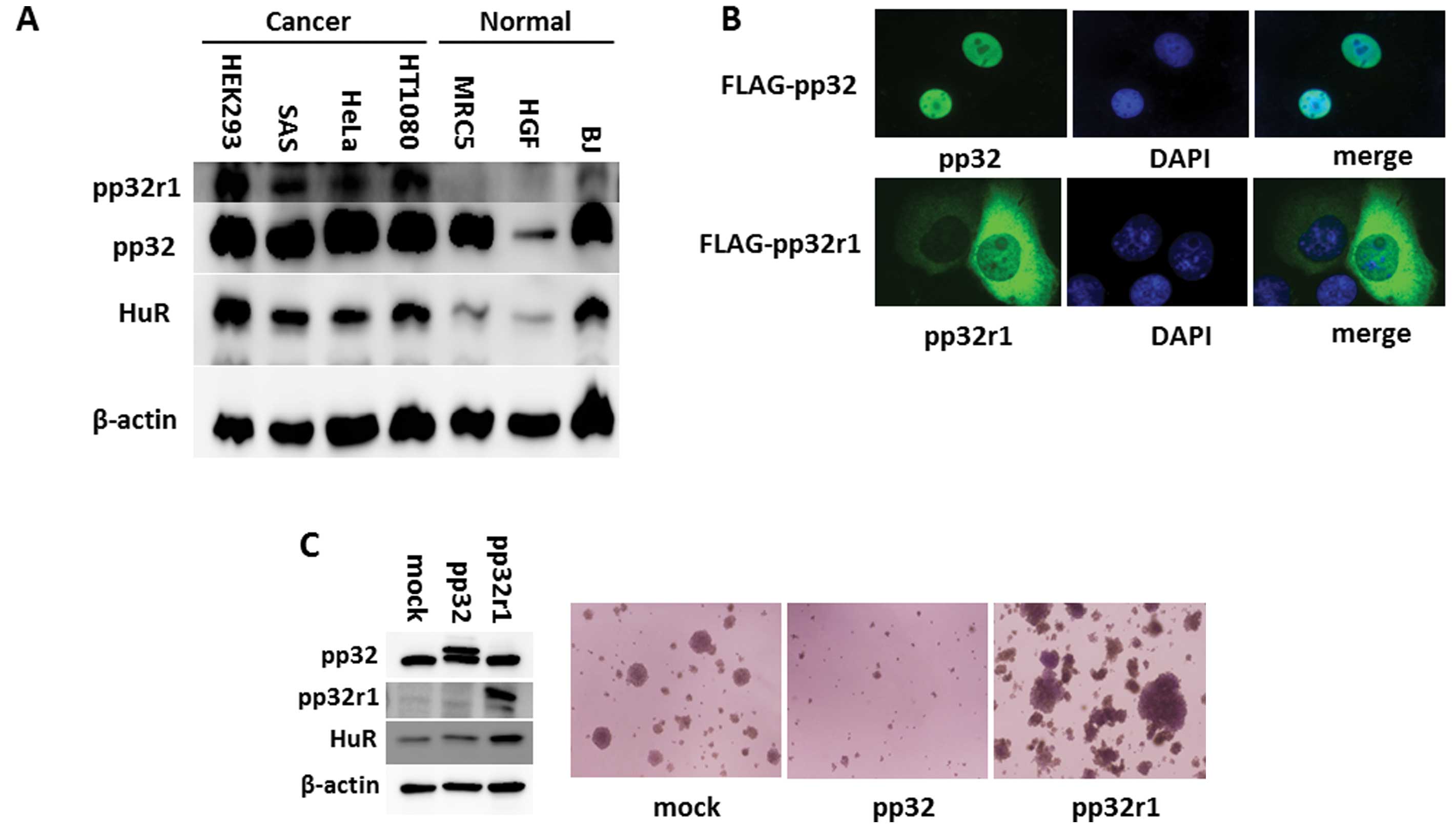
Expression, localization and function of
pp32r1 in cancer cells
Since it has been demonstrated that pp32r1 can
transform cells and that expression of pp32r1 and pp32r2 has been
observed in breast and prostate cancer tissues but not in benign
tissues (3,4), we determined the expression of the
pp32r1 protein in several cancer cell lines: HEK293 (human
embryonic kidney cells transformed by the adenovirus E1 gene), SAS
(oral cancer), HeLa (cervical cancer), and HT1080 (fibrosarcoma).
As shown in Fig. 1A, pp32r1 was
expressed in these cancer cells, whereas only slight expression was
evident in the normal MRC5 (human lung fibroblast), HGF (human
gingival fibroblast) and BJ (human foreskin fibroblast) cells. In
contrast, there was only slight difference in pp32 expression
between the cancer cells and the normal cells except HGF cells.
To observe the localization of pp32r1, we used an
antibody for pp32r1; however, endogenous pp32r1 slightly reacted
with it. We then transfected FLAG-tagged pp32 or pp32r1 into SAS
oral cancer cells, and the localization of each protein was
observed by immunofluorescence using the same antibody. pp32 was
mainly expressed in the nucleus, while pp32r1 was localized in the
cytoplasm, although it existed both in the nucleus and cytoplasm
(Fig. 1B). These results indicate
that pp32r1 is abundantly expressed in the cancer cells and is
localized in the cytoplasm of the cells.
As pp32r1-induced transformed cells produced tumors
in nude mice (3), we examined the
anchorage-independent growth activity of pp32r1-expressing cancer
cells by the soft-agar colony formation assay. A FLAG-tagged pp32
or pp32r1 expression construct was introduced into HeLa cells that
were then cultured in soft-agar. Numerous colonies were formed by
cells with the control plasmid. Cells expressing pp32 were unable
to form colonies relative to the control cells, while
pp32r1-expressing cells formed large colonies in the soft-agar
(Fig. 1C). These results suggest
that pp32r1 enhanced the anchorage-independent growth activity of
HeLa cells. Taken together these results indicate that pp32 and
pp32r1 have totally different properties.
HuR decay in pp32r1-expressing cells upon
a lethal stress condition
Upon a lethal stress condition such as staurosporine
(STS), HuR and pp32 translocate together to the cytoplasm, and HuR
is cleaved into HuR-CP1 and -CP2 by activated caspase-3 and -7. The
released pp32 activates apoptosomes to induce the apoptosis of
cells (20). In order to understand
the effect of pp32r1 on the cleavage of HuR, SAS cells were
transfected with a FLAG-pp32 or FLAG-pp32r1 expression vector and
cells were treated with STS. The decay and localization of HuR and
expression of pp32 and pp32r1 were observed. In the pp32-expressing
cells, HuR was localized only in the nucleus, whereas HuR was
expressed throughout the cytoplasm of cells expressing pp32r1
(Fig. 2A; arrowheads).
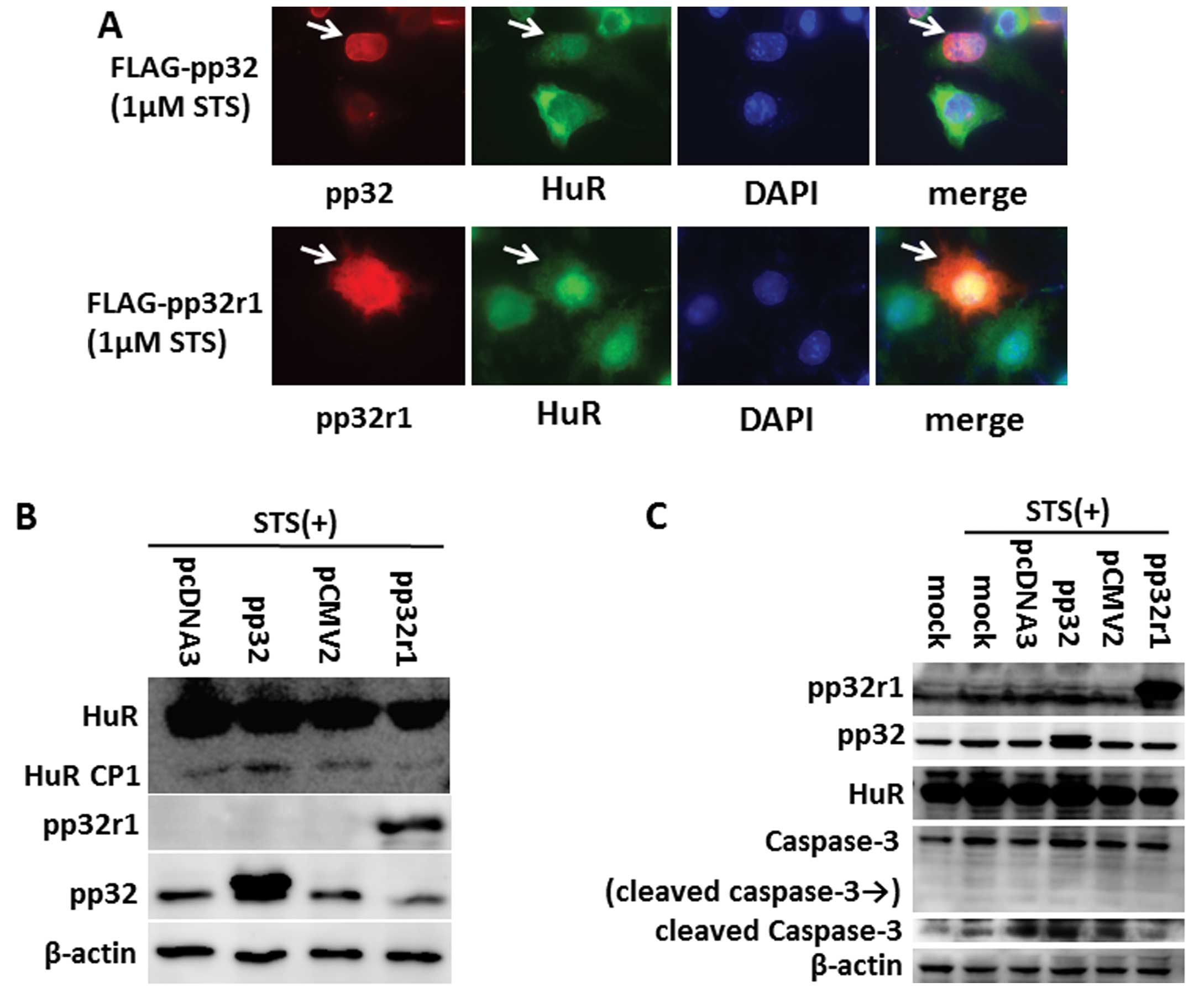 | Figure 2pp32r1 does not allow the cleavage of
HuR in the cytoplasm upon a lethal stress condition. (A) FLAG-pp32
and FLAG-pp32r1 were introduced into SAS cells, and cells were
treated with 1 μm staurosporine (STS). The localization of HuR,
pp32 and pp32r1 was observed as described in Fig. 1B. DAPI-stained nuclei are shown.
Arrowheads indicate pp32- and pp32r1-expressing cells. (B)
FLAG-pp32, FLAG-pp32r1 and their empty vectors (pcDNA3 and
pCMV2-FLAG) were transfected into SAS cells, and the expression of
HuR-CP1 in each cell line was analyzed by western blotting after
treatment with STS. pp32r1, pp32 and β-actin expression is also
indicated. (C) To examine the cleavage of caspase-3, the same
transfected cells were treated with STS, and expression of pp32r1,
pp32, HuR, caspase-3 and β-actin was analyzed using western
blotting. |
We analyzed the cleaved HuR fragment HuR-CP1
expressed in the same cells using western blot analysis. As shown
in Fig. 2B, the amount of HuR-CP1
in pp32r1-expressing cells was decreased compared to that of pp32
expressing cells. These results indicate that HuR cleavage in the
cytoplasm of cells under a stress condition was inhibited by the
expression of pp32r1.
In a previous study, pp32 was found to activate
apoptosome-mediated caspase in the cytoplasm to induce apoptosis,
while pp32r1 did not (5). To
confirm caspase activity in cells expressing pp32r1, we examined
the cleavage of caspase-3 in SAS cells bearing pp32 or pp32r1
plasmids. When a pp32 expression vector was transfected into cells,
cleaved caspase-3 was increased by STS stimulation. However, the
band declined with pp32r1 expression when compared to cells with
the control empty vector even when cells were treated with STS
(Fig. 2C). These results indicate
that pp32r1 failed to activate caspase in STS-treated cells, which
is the reason why HuR protein survived in the cytoplasm of the
cancer cells.
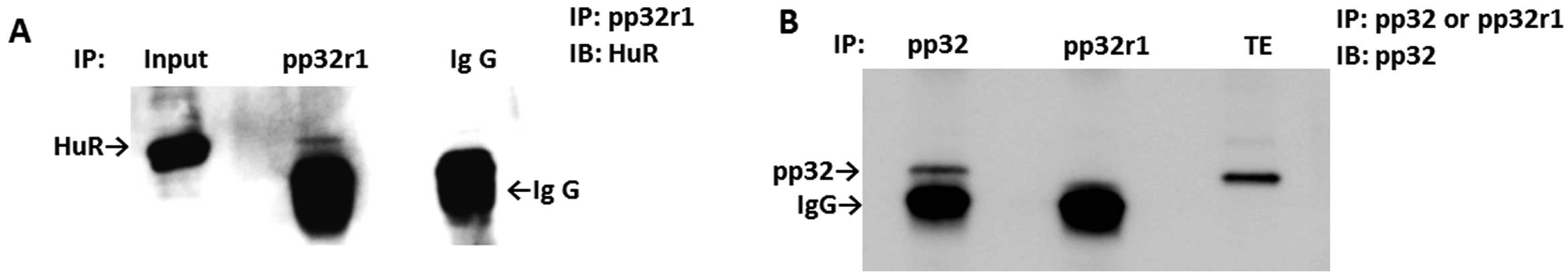
pp32r1 binds to HuR
Since pp32 is known to be an associated protein of
HuR (9), we examined whether or not
pp32r1 binds to HuR. To examine the endogenous interactions of
these proteins, the lysate of SAS cells was immunoprecipitated with
the antibody for pp32r1, followed by western blotting with the
anti-HuR antibody. As the HuR band appeared in samples of pp32r1
immunoprecipitation, HuR was co-precipitated with pp32r1 (Fig. 3A). To eliminate the possibility that
pp32r1 indirectly binds to HuR mediated by pp32, we examined the
binding of pp32r1 with pp32. pp32 was not co-precipitated with
pp32r1 (Fig. 3B), indicating that
pp32r1 was not able to bind to pp32. These results indicate that
pp32r1 interacts with HuR similar to the case of pp32 and that
binding is not mediated by pp32.
HuR is not degraded in cancer cells
expressing pp32r1
As shown in previous studies (3,4) and in
Fig. 1A, the expression level of
pp32r1 in cancer cells is generally higher than that of normal
cells. Furthermore, as pp32r1 was found to bind to HuR (Fig. 3A), HuR was hypothesized to bind
predominantly to pp32r1 in cancer cells which inhibits the cleavage
of HuR similar to that of the stress condition (Fig. 2). To confirm this hypothesis, we
observed HuR cleavage mediated by pp32 and pp32r1 in cancer cells.
FLAG-pp32 or FLAG-pp32r1 expression constructs were introduced into
SAS cells, and the amount of HuR in the nucleus and cytoplasm of
these cells was observed. pp32 expression diminished the expression
of HuR in the cytoplasm of SAS cells relative to that in the
non-transfected control cells. In contrast, HuR existed throughout
the cytoplasm of cells expressing pp32r1 similar to the case of the
control cells (Fig. 4A). HuR decay
in the cytoplasm was then estimated by western blotting. As shown
in Fig. 4B, the amount of HuR in
the cytoplasm of pp32-expressing SAS cells was obviously less than
cytoplasmic HuR expressed in cells with pp32r1. These results
suggest that HuR is stabilized in the cytoplasm of cancer cells
when they express pp32r1.
Discussion
In the present study, we compared several properties
of pp32 and pp32r1. pp32r1 was abundantly expressed in cancer cells
whereas its expression was extremely low in normal cells. In
contrast, there was only slight difference in pp32 expression.
pp32r1 was expressed in the cytoplasm and nucleus of cancer cells,
but pp32 existed only in the nucleus. pp32r1 enhanced the
anchorage-independent growth properties of cancer cells, whereas
pp32 inhibited this ability. pp32 induced the cleavage of HuR in
the cytoplasm of cells in a lethal stress condition; however,
pp32r1 did not induce it. We confirmed that the binding of pp32r1
with HuR was not mediated by pp32. Additionally, in cancer cells,
HuR survived in the cytoplasm of cells expressing pp32r1. Taken
together, these findings suggest that when pp32r1 is as abundant in
cells as it is in cancer cells, pp32r1 will preferentially
associate with HuR and the cytoplasmic expression of HuR may
increase through attenuation of caspase-mediated decay.
In a previous study, pp32 was shown to induce
apoptosis through the stimulation of Apaf-1-mediated caspase
activity. This activity was thought to be required for the tumor
suppressive activity of pp32 (5).
pp32r1 did not stimulate caspase activity as confirmed in the
present study (Fig. 2C).
Furthermore, pp32r1 was not able to inhibit the caspase activity of
pp32 as caspase activity was not attenuated by adding pp32r1 in an
in vitro caspase assay (5).
These findings indicate that pp32r1 failed to antagonize the
apoptotic activity of pp32 by a competitive manner and that the
oncogenic activity of pp32r1 is not implicated in the
anti-apoptotic activity. Although pp32r1 and its mutant have been
shown to dysregulate the cell cycle (24), the molecular mechanism of the
oncogenic activity of pp32r1 remains unclear. In the present study,
we demonstrated that pp32r1 directly bound to HuR and the cleavage
of HuR was inhibited in the cytoplasm of cancer cells where pp32r1
expression was abundantly. We propose that our findings provide one
of the explanations for the oncogenic mechanism of pp32r1.
Moreover, these data suggest that pp32r1 does not compete with pp32
in cells undergoing apoptosis, but compete with it for HuR
stabilization.
We reported that the level of HuR expression was
upregulated in the cytoplasm of several types of cancer cells such
as oral cancer (18) and
viral-mediated transformed cancer (16). Furthermore, the cytoplasmic
expression of HuR is evident in a number of human cancers and has
been implicated in the malignancy of these carcinomas (13,18).
In such cancer cells, several ARE-mRNAs were stabilized in
synchrony with cytoplasmic HuR protein levels since HuR binds to
ARE and is able to stabilize ARE-mRNA. Although we have no evidence
to indicate the relationship between HuR and pp32r1 in such cancer
cells, we suggest that pp32r1 inhibits the decay of HuR in the
cytoplasm of these cells and that HuR stabilizes ARE-mRNA to exert
its oncogenic activity.
Acknowledgements
We thank the members of our laboratories for the
helpful discussion and support. We also thank Takeshi Kuroshima for
his technical advice on immunofluorescence. This work was supported
by a Grant-in-Aid for Scientific Research from the Ministry of
Education, Science and Culture of Japan.
References
|
1
|
Matsuoka K, Taoka M, Satozawa N, et al: A
nuclear factor containing the leucine-rich repeats expressed in
murine cerebellar neurons. Proc Natl Acad Sci USA. 91:9670–9674.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen TH, Brody JR, Romantsev FE, et al:
Structure of pp32, an acidic nuclear protein which inhibits
oncogene-induced formation of transformed foci. Mol Biol Cell.
12:2045–2056. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kadkol SS, Brody JR, Pevsner J, Bai J and
Pasternack GR: Modulation of oncogenic potential by alternative
gene use in human prostate cancer. Nat Med. 5:275–279. 1999.
View Article : Google Scholar
|
|
4
|
Kadkol SS, El Naga GA, Brody JR, et al:
Expression of pp32 gene family members in breast cancer. Breast
Cancer Res Treat. 68:65–73. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pan W, da Graca LS, Shao Y, Yin Q, Wu H
and Jiang X: PHAPI/pp32 suppresses tumorigenesis by stimulating
apoptosis. J Biol Chem. 284:6946–6954. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Adegbola O and Pasternack G:
Phosphorylated retinoblastoma protein complexes with pp32 and
inhibits pp32-mediated apoptosis. J Biol Chem. 280:15497–15502.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Matilla A, Koshy BT, Cummings CJ, Isobe T,
Orr HT and Zoghbi HY: The cerebellar leucine-rich acidic nuclear
protein interacts with ataxin-1. Nature. 389:974–978. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Beresford PJ, Kam CM, Powers JC and
Lieberman J: Recombinant human granzyme A binds to two putative
HLA-associated proteins and cleaves one of them. Proc Natl Acad Sci
USA. 94:9285–9290. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brennan CM, Gallouzi IE and Steitz JA:
Protein ligands to HuR modulate its interaction with target mRNA
in vivo. J Cell Biol. 151:1–14. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gallouzi IE, Brennan CM and Steitz JA:
Protein ligands mediate the CRM1-dependent export of HuR in
response to heat shock. RNA. 7:1348–1361. 2001. View Article : Google Scholar
|
|
11
|
Ma WJ, Cheng S, Campbell C, Wright A and
Furneaux H: Cloning and characterization of HuR, a ubiquitously
expressed Elav-like protein. J Biol Chem. 99:8144–8151.
1996.PubMed/NCBI
|
|
12
|
Hinman MN and Lou H: Diverse molecular
functions of Hu proteins. Cell Mol Life Sci. 65:3168–3181. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lopez de Silanes I, Lal A and Gorospe M:
HuR: post-transcriptional paths to malignancy. RNA Biol. 2:11–13.
2005.PubMed/NCBI
|
|
14
|
López de Silanes I, Fan J, Yang X, et al:
Role of the RNA-binding protein HuR in colon carcinogenesis.
Oncogene. 22:7146–7154. 2003.PubMed/NCBI
|
|
15
|
Srikantan S and Gorospe M: HuR function in
disease. Front Biosci. 17:189–205. 2012. View Article : Google Scholar
|
|
16
|
Higashino F, Aoyagi M, Takahashi A, et al:
Adenovirus E4orf6 targets pp32/LANP to control the fate of
ARE-containing mRNA by perturbing the CRM1-dependent mechanism. J
Cell Biol. 170:15–20. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kuroshima T, Aoyagi M, Yasuda M, et al:
Viral-mediated stabilization of AU-rich element containing mRNA
contributes to cell transformation. Oncogene. 30:2912–2920. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hasegawa H, Kakuguchi W, Kuroshima T, et
al: HuR is exported to the cytoplasm in oral cancer cells in a
different manner from that of normal cells. Br J Cancer.
100:1943–1948. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kakuguchi W, Kitamura T, Kuroshima T, et
al: HuR knockdown changes the oncogenic potential of oral cancer
cells. Mol Cancer Res. 8:520–528. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mazroui R, Di Marco S, Clair E, et al:
Caspase-mediated cleavage of HuR in the cytoplasm contributes to
pp32/PHAP-I regulation of apoptosis. J Cell Biol. 180:113–127.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Weigel S and Dobbelstein M: The nuclear
export signal within the E4orf6 protein of adenovirus type 5
supports virus replication and cytoplasmic accumulation of viral
mRNA. J Virol. 74:764–772. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Aoyagi M, Higashino F, Yasuda M, et al:
Nuclear export of the adenovirus E4orf6 protein is necessary for
its ability to antagonize the apoptotic activity of the BH3-only
proteins. Oncogene. 22:6919–6927. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Higashino F, Pipas JM and Shenk T:
Adenovirus E4orf6 oncoprotein modulates the function of the
p53-related protein, p73. Proc Natl Acad Sci USA. 95:15683–15687.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Buddaseth S, Göttmann W, Blasczyk R, et
al: Dysregulation of cell cycle caused by overexpression of the
oncogene pp32r1 (ANP32C) and the Tyr>His mutant pp32r1Y140H.
Biochim Biophys Acta. 1833:1212–1221. 2013. View Article : Google Scholar : PubMed/NCBI
|