Introduction
During the past decades, the incidence of
endometrial cancer has increased in most regions of the world
(1–3). In 2013, there will be ~49,500 new
cases and 8,200 deaths in the United States (4). However, our knowledge of the etiology
of this disease remains poor.
Generally, endometrial cancer is grouped into type I
(accounting for 80% of cases, primarily endometrioid
adenocarcinoma, commonly linked to unopposed exposure to oestrogen,
obesity and hormone receptor positivity) and type II (accounting
for <20% of cases, which consists of serous, clear cell and some
undifferentiated tumors) (5–8). To
date, the main treatments for endometrial cancer are surgery and/or
radiation therapy, and for patients with late stage or distant
metastasis, chemotherapy is also an option (9).
Unlike ovarian and cervical cancer, most endometrial
cancer cases are localized in the uterus body, but lymphatic or
vascular invasion rarely occur (10,11).
Molecularly, endometrial cancer is mainly characterized by
uncontrolled cellular proliferation and unlimited cell cycle
progression, a process in which these cell cycle-related proteins
[such as cyclins and cyclin-dependent kinases (CDKs)] are strongly
involved (9). This phenomenon
suggested it may be a potent approach to inhibit the cell cycle in
endometrial cancer, through targeting these cell cycle-related
factors or their manipulators (12,13).
Recently, the RPRD1B gene was confirmed to be one of them, and can
accelerate cell cycle by upregulating a panel of cyclins and CDKs
(14).
RPRD1B, the regulation of nuclear pre-mRNA domain
containing 1B gene, is a human homolog of the Rtt103 gene (14); it can regulate the binding of RNA
polymerase II to the CCND1 gene (cyclin D1) and prevent degradation
of the CCND1 mRNA (15). Similarly,
RPRD1B also enhances the transcription of many other cell
cycle-related factors (such as CDK2, CDK4, CDK6 and cyclin E)
(15). Given that elevated RPRD1B
was reported in a small panel of endometrial cancer types, in the
present study, we explored its detailed functions in endometrial
cancer (14).
Materials and methods
Tissue collection
Seventy-six endometrial cancer tissues were
collected from patients who underwent surgery in our hospital from
July 2010 to October 2012. The tumor stage and grade were
determined following the criteria of Federation International of
Gynecology and Obstetrics (FIGO, 2009)(16). Fifteen normal endometrium tissues
were obtained as the control. Details given in Table I.
 | Table IRelationship between RPRD1B expression
and clinico-pathological characteristics. |
Table I
Relationship between RPRD1B expression
and clinico-pathological characteristics.
| Variables | Cases | Percentage |
|---|
| Age (years) |
| <55 | 30 | 39.5 |
| ≥55 | 46 | 60.5 |
| Pathological
subtype |
| Endometrioid | 65 | 85.5 |
|
Non-endometrioid | 11 | 14.5 |
| Stagea |
| I | 37 | 56.9 |
| II | 10 | 15.4 |
| III | 15 | 23.1 |
| IV | 3 | 4.6 |
| Gradea |
| I | 26 | 40 |
| II | 21 | 32.3 |
| III | 18 | 27.7 |
| Myometrial
invasionb |
| <1/2 | 23 | 62.7 |
| ≥1/2 | 14 | 37.3 |
| Lymph node
metastasis |
| Negative | 21 | 67.7 |
| Positive | 10 | 32.3 |
| Lymphovascular
space invasion |
| Negative | 52 | 68.4 |
| Positive | 24 | 31.6 |
Immunohistochemical (IHC) staining
The IHC staining was performed as previously
described (14). The primary
antibodies used were: anti-Ki 67 (1:500; Wuhan Boster Biological
Technology, Ltd., Wuhan, China) and anti-cyclin D1 (1:200; Cell
Signaling Technology, Danvers, MA, USA). The staining was
visualized using Histostain-Plus IHC kit (Shanghai Mingrui Biotech
Co., Ltd., Shanghai, China). All slides were scored by two
pathologists following these criteria: 0, none (totally negative
staining); 1, weak (1–25% positive); 2, moderate (26–50% positive);
3, strong (>50% positive).
Cell culture
Six endometrial cancer cell lines were used:
Ishikawa, KLE, RL95-2, HEC-1B, SPEC-2 and AN3CA. All cells were
purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA) and maintained at 37°C in a humidified
atmosphere of 5% CO2. The medium was DMEM/F12+10% fetal
bovine serum (FBS) for KLE, SPEC-2 and RL95-2 and MEM +10% FBS for
Ishikawa, HEC-1B and AN3CA. The medium and the FBS were purchased
from Gibco (Auckland, New Zealand).
RNA isolation and quantitative real-time
PCR
The tissue and cells were homogenized and total RNA
was extracted using TRIzol (Invitrogen, Carlsbad, CA, USA). The
mRNA expression of RPRD1B was measured by quantitative real-time
PCR using SYBR-Green reaction mixture (Takara, Dalian, China). The
primers for RPRD1B were: sense, 5′-ggatgctttttctcatgttgc-3′ and
antisense, 5′-cgccatacacacttcgttctt-3′. The reaction conditions
were: 95°C for 30 sec, 33 cycles of 95°C for 5 sec and 60°C for 32
sec. β-actin was used as the endogenous control. The relative
expression of RPRD1B was calculated by the 2−ΔΔCt
method.
Western blot analysis
Total protein was extracted using the RIPA buffer
(Wuhan Boster Biological Technology) and the concentration was
determined by the BCA assay kit (Thermo Fisher Scientific, New
York, NY, USA). Equal amount (30–50 μg) of protein was separated on
12% SDS-PAGE gel and transferred to the PVDF membrane (GE
Healthcare, Buckinghamshire, UK). The membrane was then incubated
with primary antibodies overnight at 4°C and with secondary
antibodies for a further 1 h at room temperature. The bands were
then developed using an imaging system. The primary antibodies for
cyclin D1, CDK4 and CDK6 were purchased from Cell Signaling
Technology and anti-RPRD1B mouse monoclonal antibody was obtained
from Abgent (San Diego, CA, USA). β-actin was used as the
endogenous control.
MTT assay
Ishikawa cells (3×103) or
5×103 HEC-1B cells/well were seeded into 96-well plates
and incubated overnight. Then, the cells were transfected with
pcDNA3.1-RPRD1B plasmid or siRNA-RPRD1B using
Lipofectamine® 2000 (Invitrogen) for 48 h. Cells
transfected with the scramble siRNA or the empty plasmid were set
as the negative control. Then, 5 μl MTT solution (5 mg/ml; Sigma,
Minneapolis, MN, USA) was added into the medium and incubated at
37°C for a further 1 h. The formazan crystal was dissolved in 100
μl DMSO (Sigma) and the absorbance was measured at 570 nm on a
plate reader. These procedures were repeated in triplicate.
Colony formation assay
Then, 0.2×103 cells/well were plated into
6-well plates and transfected with siRNA-RPRD1B or pcDNA3.1-RPRD1B.
The cells were routinely cultured for two weeks and the colony
number was counted under an inverted microscope. This experiment
was repeated in triplicate.
Cell cycle analysis
Briefly, 2×105 cells/well were seeded
into 6-well plates and incubated overnight. The cells were then
transfected with siRNA-RPRD1B or pcDNA3.1-RPRD1B for 48 h. The
cells were harvested and resuspended in phosphate- buffered saline
(PBS) and fixed in chilled 90% methanol at −20°C for 24 h. The
cells were then resuspended in 1 ml PI staining solution and
analyzed on a FACSCalibur (BD Biosciences, Bedford, MA, USA).
Tumor growth assay
Five to six-week-old female severe combined
immunodeficient mice (purchased from Shanghai SLAC Laboratory
Animal Co., Ltd., Shanghai, China) were used for this assay. To
establish the endometrial cancer model, 1×106 HEC-1B
cells stably transfected with pcDNA3.1-RPRD1B were injected
subcutaneously into the right flank of mice, and the control cells
with empty plasmid were injected into the left flank. One week
after injection, the tumor size was checked and recorded every
other five days. The tumor volume = (length × width2)/2.
Twenty-eight days after injection, all the mice were sacrificed and
the tumor tissue was collected for further analysis.
Alterations of cellular sensitivity to
Raloxifene
Raloxifene was dissolved in DMSO and diluted into
different concentrations (μM): 0.01, 0.1, 1, 10, 20, 40 and 80. The
sensitivity of HEC-1B and Ishikawa cells to Raloxifene was
determined by MTT assay.
Ethics statement
The present study was approved by the Human
Investigation Ethics Committee of the Affiliated Hospital of
Jiangnan University. Written informed consent was obtained from
each patient involved in the present study. The animal research was
carried out following the Guideline for the Care and Use of
Laboratory Animals of China. The protocol was approved by the
Committee on the Ethics of Animal Experiments of Jiangnan
University [Permit Number: JSXK (hu) 2006-0088].
Statistical analysis
The statistical analysis was performed using SPSS
17.0 (SPSS Inc., Chicago, IL, USA). χ2 test or t-test
was used for categorical and quantitative data appropriately.
P<0.05 was considered to indicate a statistically significant
difference.
Results
RPRD1B is frequently overexpressed in
human endometrial cancer
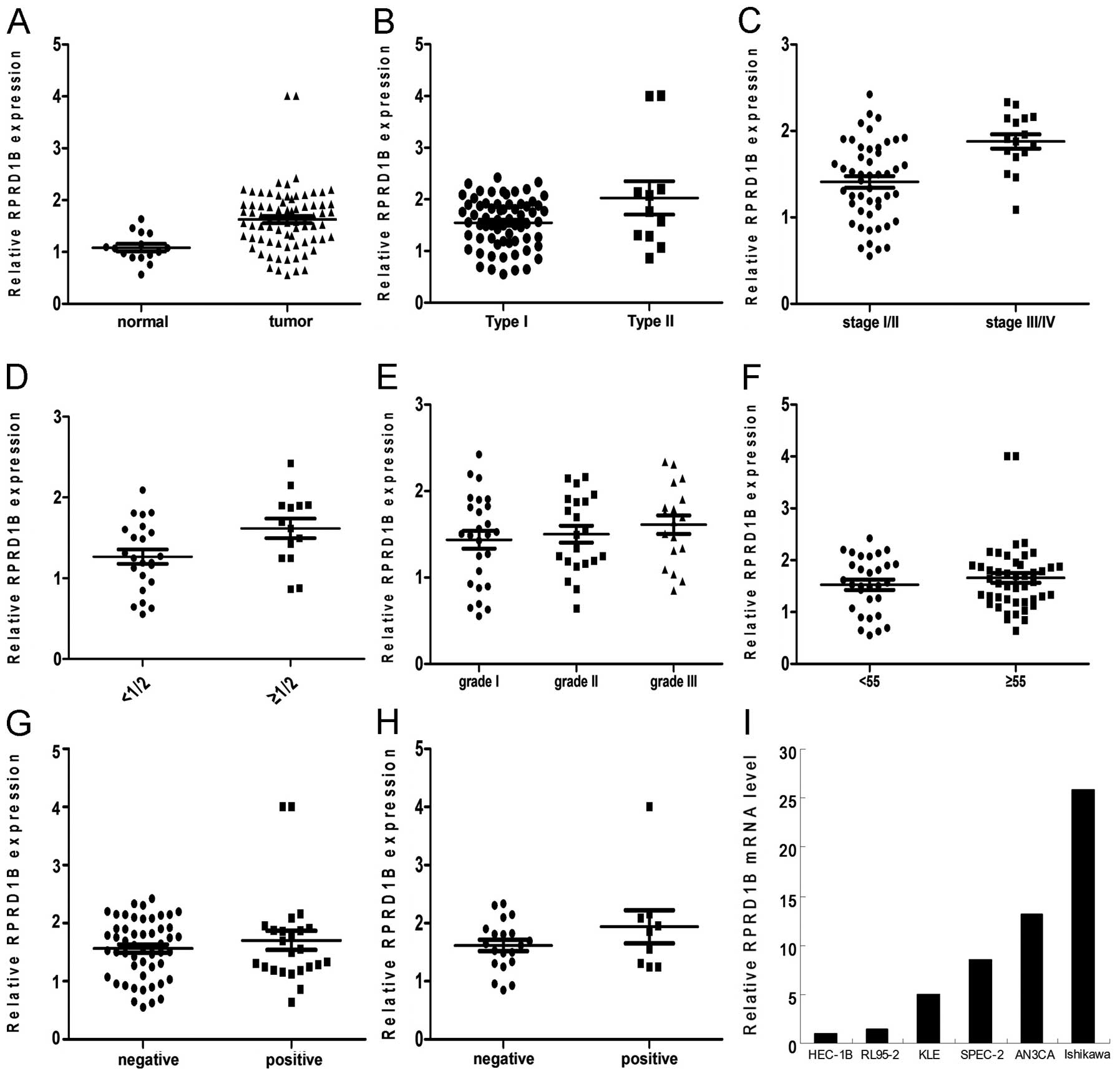
RPRD1B mRNA was significantly overexpressed in
endometrial cancer tissues, compared to the normal endometrium
(Fig. 1A; P=0.0012). By further
analysis, we found that RPRD1B overexpression was correlated with
histology type (Fig. 1B; P=0.0146),
tumor stage (Fig. 1C; P=0.0004) and
depth of myometrial invasion (Fig.
1D; P=0.024), but was not associated with histology grade
(Fig. 1E; P=0.3612), patient age
(Fig. 1F; P=0.4503), lymphovascular
space invasion (LVSI; Fig. 1G;
P=0.3559) or lymph node metastasis (Fig. 1H; P=0.1845).
We then detected RPRD1B mRNA levels in these 6
endometrial cancer cell lines. As shown in Fig. 1I, RPRD1B mRNA was abundant in
Ishikawa and AN3CA cells, but was only slight in HEC-1B and
RL95-2.
Overexpression of RPRD1B promotes
cellular proliferation and accelerates the cell cycle in HEC-1B
cells
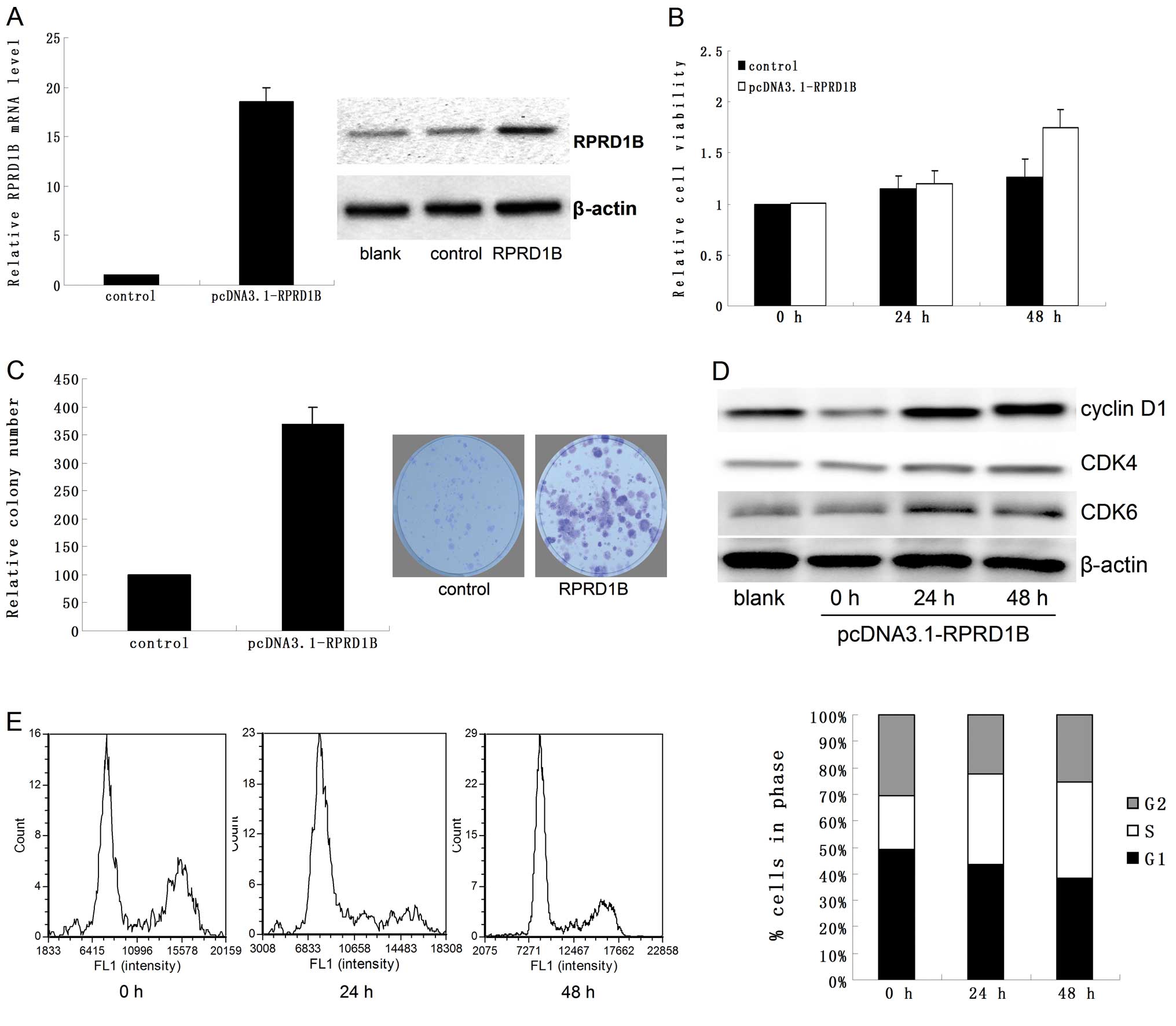
As shown in Fig. 2A,
both the mRNA and protein of RPRD1B were upregulated following
transfection with pcDNA3.1-RPRD1B. Compared to the control, RPRD1B
overexpression promoted cellular proliferation significantly
(Fig. 2B and C; P=0.032 for MTT
assay and P=0.018 for colony formation assay). In addition, RPRD1B
also increased the expression of cyclin D1, CDK4 and CDK6 (Fig. 2D) and accelerated the cell cycle of
HEC-1B cells (Fig. 2E; P=0.018 for
24 h and P=0.007 for 48 h).
Downregulation of RPRD1B suppresses
cellular proliferation and leads to cell cycle arrest in Ishikawa
cells
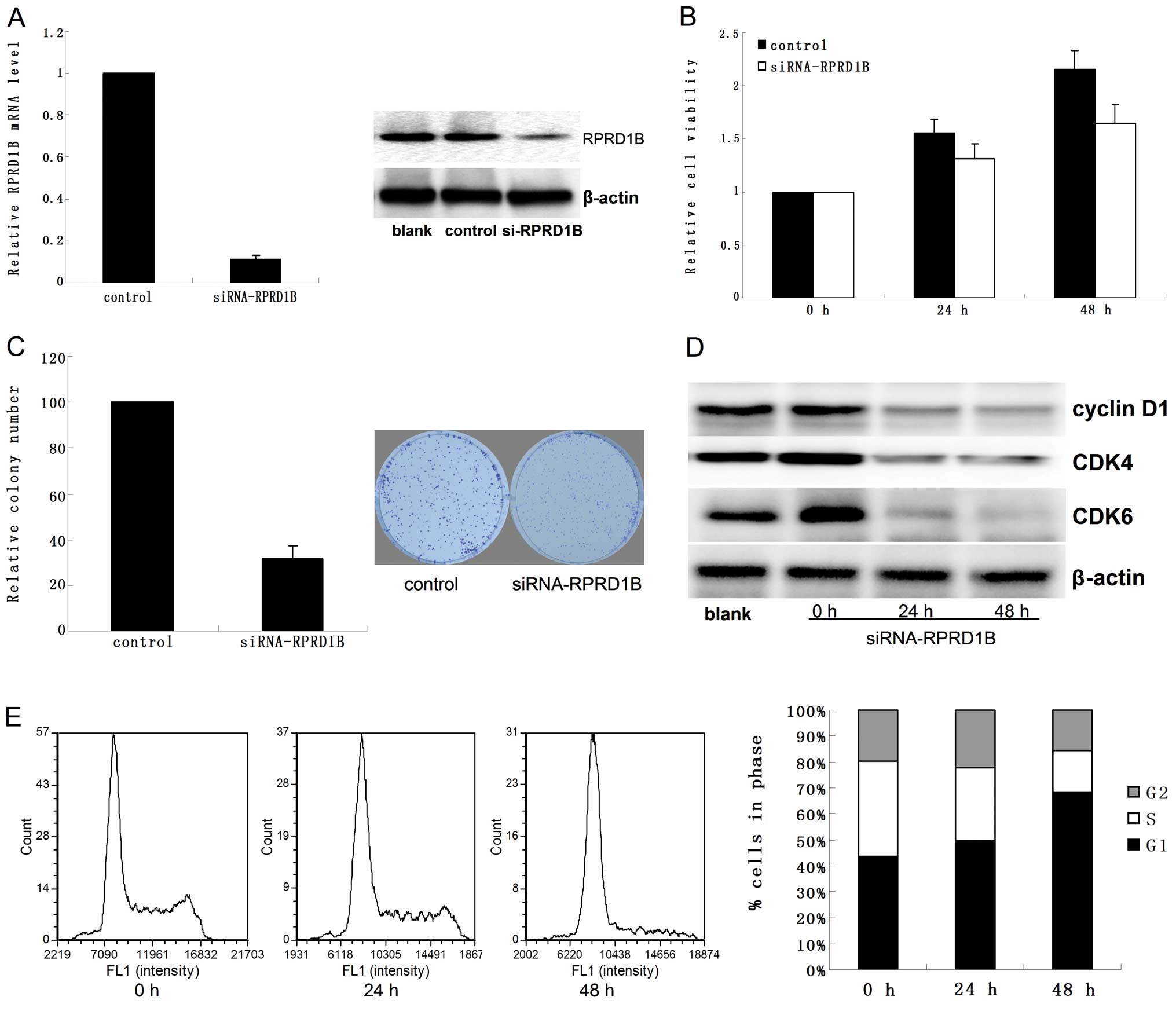
Following treatment with siRNA-RPRD1B for 48 h, both
RPRD1B mRNA and protein were reduced significantly in Ishikawa
cells (Fig. 3A). Loss of RPRD1B
notably suppressed cellular proliferation (Fig. 3B and C; P=0.02 for MTT assay and
P=0.031 for colony formation assay). Flow cytometry analysis showed
that downregulation of RPRD1B inhibited the expression of cyclin
D1, CDK4 and CDK6 (Fig. 3D), and
led to G1 phase arrest at both 24 and 48 h (Fig. 3E; P=0.039 for 24 h and P=0.025 for
48 h).
Overexpression of RPRD1B accelerates
tumor growth in vivo
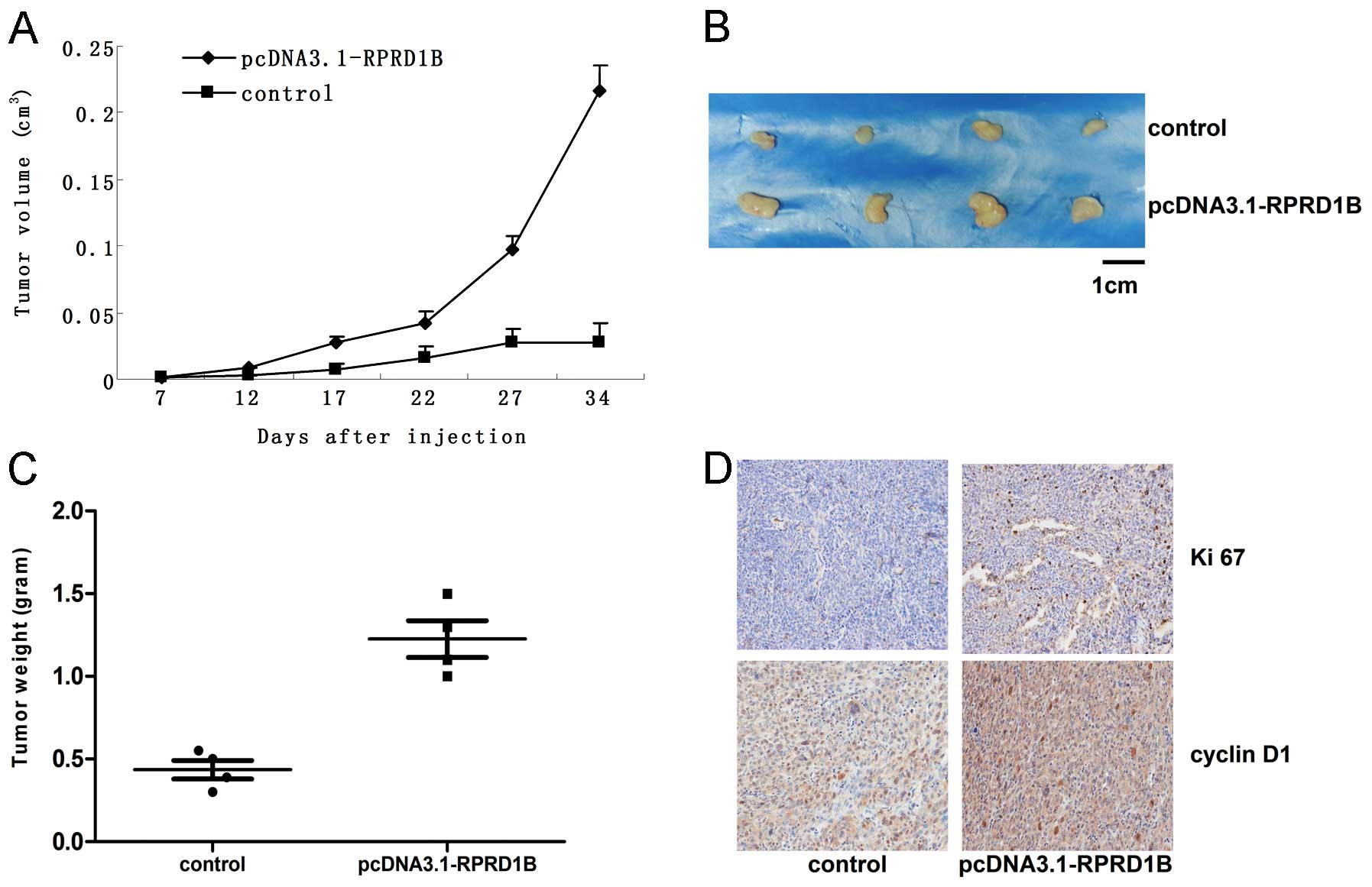
We established the endometrial cancer model by
injecting HEC-1B cells (with or without overexpression of RPRD1B)
into nude mice. According to our results, the xenograft with RPRD1B
overexpression grew much faster than the control (Fig. 4A and B; P=0.0012). We also found the
tumor weight increased significantly following RPRD1B
overexpression (Fig. 4C; P=0.007).
By IHC staining, much higher expression of Ki-67 and cyclin D1 was
detected in the group with over-abundant RPRD1B (Fig. 4D).
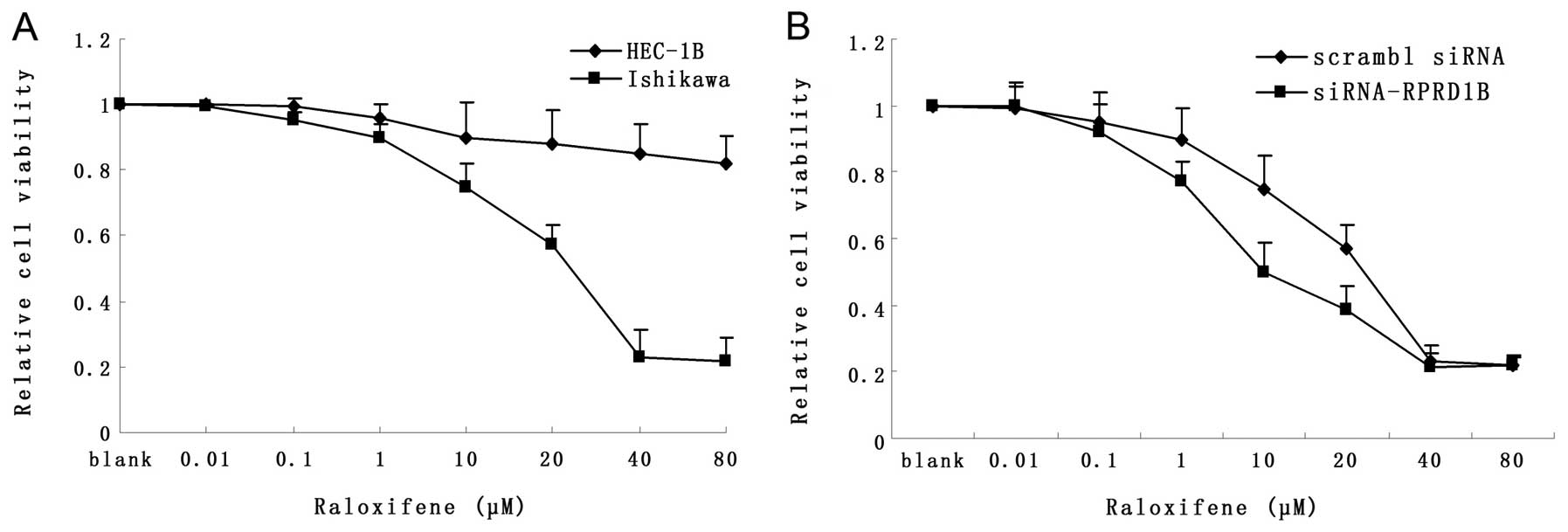
Downregulation of RPRD1B sensitizes
Ishikawa cells to Raloxifene
As shown in Fig. 5A,
the IC50 of Raloxifene was ~25 μM for Ishikawa, but
>80 μM for HEC-1B cells. Loss of RPRD1B increased the
sensitivity of Ishikawa cells to Raloxifene (Fig. 5B; P=0.018); however, RPRD1B
over-expression had no effects on the reactions of HEC-1B to
Raloxifene (data not shown).
Discussion
Due to the high incidence rate and relatively
favorable prognosis, endometrial cancer is now considered a chronic
disease and, hence, preventing the tumor from unscheduled growth
seems to be more appropriate. Dysregulated cell cycle is a common
feature in nearly all types of human cancer (17,18).
To date, the relationship between cell cycle dysregulation and
human cancer have been well documented. In particular, several cell
cycle regulators were confirmed to be critical for the initiation
and progression of endometrial cancer (19–21).
In mammals, the cell cycle is manipulated by the
cyclin-dependent kinases (CDKs), which can be activated by cyclins
(such as cyclin D and E) and can be inhibited by Ink4 and Cip/Kip
inhibitors (13,22,23).
The activities of CDKs and their regulators are often dysregulated
in human tumors owing to genetic or epigenetic changes or
alterations of their upstream signal pathway (24–26).
Recently, RPRD1B, a manipulator of both cyclins and CDKs, was found
overexpressed in endometrial cancer (14). Therefore, we hypothesized that
RPRD1B might be a key factor in endometrial neoplasia and
development.
In the present study, we demonstrated RPRD1B
overexpression in human endometrial cancer tissues, which was
closely related with tumor stage, histology type and depth of
myometrial invasion. In vitro, we explored the effects of
RPRD1B on tumor cells by up- and downregulating its expression in
two endometrial cancer cell lines. In HEC-1B cells, RPRD1B
overexpression promoted cellular growth and accelerated the process
of colony formation, while in Ishikawa cells, loss of RPRD1B
inhibited its growth and colony formation.
As RPRD1B has been reported to be a regulator of
cell cycle-related proteins, we then investigated the cell cycle
status and expression of cyclin D1, CDK4 and CDK6 in the two cell
lines. Compared to the control, RPRD1B overexpression promoted
2-fold more HEC-1B cells into the S phase, and the levels of cyclin
D1, CDK4 and CDK6 were also upregulated. Moreover, RPRD1B
downregulation inhibited the cell cycle and caused significant G1
phase arrest, through suppressing cyclin D1, CDK4 and CDK6. These
findings were consistent with those of a previous study on gastric
carcinoma cells (14).
Furthermore, we established the nude mice model to
explore whether RPRD1B affects the tumor growth in vivo. As
our data showed, HEC-1B cells with abundant RPRD1B grew much faster
than the control group, and showed much stronger staining of Ki-67
(a proliferation index) and cyclin D1.
Considering that selective estrogen receptor
modulators (SERMs) work well in a group of endometrial cancer
patients but soon induced drug resistance in several cases, we then
investigated whether RPRD1B could affect the sensitivity of
Ishikawa and HEC-1B cells to Raloxifene (27,28).
We found that knockdown of RPRD1B could sensitize Ishikawa cells to
Raloxifene, but overexpression of RPRD1B had no effects on HEC-1B
cells. We hypothesized it could be caused by the different ER
status of Ishikawa (ER positive) and HEC-1B (ER negative).
In summary, we demonstrated that RPRD1B was
frequently overexpressed in human endometrial cancer. Both in
vitro and in vivo, overabundant RPRD1B promoted tumor
growth and accelerated cellular cell cycle through upregulating
cyclin D1, CDK4 and CDK6, while knockdown of RPRD1B suppressed
tumor growth and caused cell cycle arrest by decreasing cyclin D1,
CDK4 and CDK6. In addition, knockdown of RPRD1B increased cells
sensitivity to Raloxifene treatment. These findings may aid in the
design of drugs targeting RPRD1B and its partners, which we believe
will be a new strategy for curing this disease.
Acknowledgements
The present study was supported by grants to Jinjin
Yu from Wuxi Science and Technology Bureau (no. CSE01N1113).
References
|
1
|
Wei KR, Chen WQ, Zhang SW, Zheng RS, Wang
YN and Liang ZH: Epidemiology of uterine corpus cancer in some
cancer registering areas of China from 2003–2007. Zhonghua Fu Chan
Ke Za Zhi. 47:445–451. 2013.(In Chinese).
|
|
2
|
Jamison PM, Noone AM, Ries LA, Lee NC and
Edwards BK: Trends in endometrial cancer incidence by race and
histology with a correction for the prevalence of hysterectomy,
SEER 1992 to 2008. Cancer Epidemiol Biomarkers Prev. 22:233–241.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bray F, Dos Santos Silva I, Moller H and
Weiderpass E: Endometrial cancer incidence trends in Europe:
underlying determinants and prospects for prevention. Cancer
Epidemiol Biomarkers Prev. 14:1132–1142. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar
|
|
5
|
Hecht JL and Mutter GL: Molecular and
pathologic aspects of endometrial carcinogenesis. J Clin Oncol.
24:4783–4791. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dedes KJ, Wetterskog D, Ashworth A, Kaye
SB and Reis-Filho JS: Emerging therapeutic targets in endometrial
cancer. Nat Rev Clin Oncol. 8:261–271. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bokhman JV: Two pathogenetic types of
endometrial carcinoma. Gynecol Oncol. 15:10–17. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Boruta DM II, Gehrig PA, Fader AN and
Olawaiye AB: Management of women with uterine papillary serous
cancer: a Society of Gynecologic Oncology (SGO) review. Gynecol
Oncol. 115:142–153. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Amant F, Moerman P, Neven P, Timmerman D,
Van Limbergen E and Vergote I: Endometrial cancer. Lancet.
366:491–505. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Saso S, Chatterjee J, Georgiou E, Ditri
AM, Smith JR and Ghaem-Maghami S: Endometrial cancer. BMJ.
343:d39542011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Del Carmen MG, Boruta DM II and Schorge
JO: Recurrent endometrial cancer. Clin Obstet Gynecol. 54:266–277.
2011.
|
|
12
|
Thanapprapasr D and Thanapprapasr K:
Molecular therapy as a future strategy in endometrial cancer. Asian
Pac J Cancer Prev. 14:3419–3423. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: a changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lu D, Wu Y, Wang Y, et al: CREPT
accelerates tumorigenesis by regulating the transcription of
cell-cycle-related genes. Cancer Cell. 21:92–104. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ni Z, Olsen JB, Guo X, et al: Control of
the RNA polymerase II phosphorylation state in promoter regions by
CTD interaction domain-containing proteins RPRD1A and RPRD1B.
Transcription. 2:237–242. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Creasman W: Revised FIGO staging for
carcinoma of the endometrium. Int J Gynaecol Obstet. 105:1092009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim JK and Diehl JA: Nuclear cyclin D1: an
oncogenic driver in human cancer. J Cell Physiol. 220:292–296.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Massague J: G1 cell-cycle control and
cancer. Nature. 432:298–306. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mitselou A, Ioachim E, Zagorianakou N,
Kitsiou E, Vougiouklakis T and Agnantis NJ: Expression of the
cell-cycle regulatory proteins (cyclins D1 and E) in endometrial
carcinomas: correlations with hormone receptor status,
proliferating indices, tumor suppressor gene products (p53, pRb),
and clinicopathological parameters. Eur J Gynaecol Oncol.
25:719–724. 2004.
|
|
20
|
Semczuk A and Jakowicki JA: Alterations of
pRb1-cyclin D1-cdk4/6-p16(INK4A) pathway in endometrial
carcinogenesis. Cancer Lett. 203:1–12. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Horree N, van Diest PJ, Sie-Go DM and
Heintz AP: The invasive front in endometrial carcinoma: higher
proliferation and associated derailment of cell cycle regulators.
Hum Pathol. 38:1232–1238. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ortega S, Malumbres M and Barbacid M:
Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim
Biophys Acta. 1602:73–87. 2002.PubMed/NCBI
|
|
23
|
Martin A, Odajima J, Hunt SL, et al: Cdk2
is dispensable for cell cycle inhibition and tumor suppression
mediated by p27Kip1 and p21Cip1. Cancer Cell.
7:591–598. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lundberg AS and Weinberg RA: Functional
inactivation of the retinoblastoma protein requires sequential
modification by at least two distinct cyclin-cdk complexes. Mol
Cell Biol. 18:753–761. 1998.
|
|
25
|
Rane SG, Dubus P, Mettus RV, et al: Loss
of Cdk4 expression causes insulin-deficient diabetes and Cdk4
activation results in β-islet cell hyperplasia. Nat Genet.
22:44–52. 1999.PubMed/NCBI
|
|
26
|
Rosu-Myles M, Taylor BJ and Wolff L: Loss
of the tumor suppressor p15Ink4b enhances myeloid progenitor
formation from common myeloid progenitors. Exp Hematol. 35:394–406.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Brzozowski AM, Pike AC, Dauter Z, et al:
Molecular basis of agonism and antagonism in the oestrogen
receptor. Nature. 389:753–758. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Maximov PY, Lee TM and Jordan VC: The
discovery and development of selective estrogen receptor modulators
(SERMs) for clinical practice. Curr Clin Pharmacol. 8:135–155.
2013. View Article : Google Scholar : PubMed/NCBI
|