Introduction
The Wilms tumor 1 (WT1) gene has, among others, an
essential role in early development and differentiation of the
urinary tract, particularly the kidneys (1,2).
Mutations of WT1 were first described in Wilms tumors (WTs) of
patients with Wilms tumor, aniridia, genitourinary anomalies, and
mental retardation (WAGR syndrome) (3), and the function of WT1 is believed to
be that of a tumor suppressor gene (TSG). Several studies have
shown a crucial role of WT1 in other types of cancer and syndromes
such as acute myeloic leukemia (AML), Denys-Drash syndrome, and
Frasier syndrome. While genetic alterations, such as mutations,
deletions and/or truncations of WT1, are consistently detectable in
those syndromes, only 20–30% of previously described sporadic WTs
are positive for WT1 anomalies (4).
Therefore, while playing a key role in early
development, recent studies have shown that WT1 not only functions
as a TSG, but also as an oncogene (1,5,6). Two
predominant splice isoforms of WT1 have been described to be
responsible for these different functions; the first is
characterized by omitting exon 5 and thus 17 amino acids of the WT1
protein, which are inserted between the proline-rich amino terminus
and the zinc finger domains (7).
The second results from alternative splicing of exon 9 that
produces an insertion of the three amino acids, lysine, threonine
and serine (KTS) between zinc fingers 3 and 4. While the functional
impact of exon 5 splicing remains unknown, the KTS isoforms have
different affinity to the DNA (5,8–10).
These isoforms are expressed in a stable ratio of 2:1 (KTS+/KTS−)
in healthy tissues (8).
However, (mis-)regulation of WT1 in pathological
conditions is only partially understood. One well-known regulatory
mechanism in WT is the methylation dependent (in-) activation of
imprinted genes (11). Some
imprinted genes are regulated by methylation of regulatory regions
inside of CpG-rich sites which leads to a subsequent inability of
the CCCTC-binding factor (CTCF) protein to bind to its designated
site (12).
CTCF is a ubiquitously expressed and highly
conserved 11-zinc finger transcription factor, which is known to
have diverse regulatory functions all over the human genome.
Depending on the locus, CTCF can be a promoter associated
transcriptional repressor or activator. Moreover, CTCF is involved
in long range processes such as chromatin looping, chromatin
insulation, enhancer blocking, establishing interchromosomal
contacts and especially at imprinted loci, protection against
ectopic de novo methylation (13,14).
WT1 is expressed from both alleles in human kidney;
however, it can also be imprinted in human uterus or fetal brain
(15). Although the WT1 promoter
seems to be unmethylated in WTs (16), previous studies suggest an
epigenetic regulation for the WT1 antisense transcript and an
alternative transcript of WT1 (AWT1), which is related to a CTCF
binding site (17).
The aim of the present study was to analyze the
methylation pattern at regulatory elements of the WT1 gene in a
cohort of WT patients and to compare its influence on gene
expression and KTS splice variants.
Materials and methods
Patients
Forty-four native WT specimens were examined from
patients undergoing surgical tumor resection in our department. The
median age at time of surgery was 34.5 months (range, 2 months-17
years) with a gender ratio of 1:1.5 (f:m). Thirty-seven patients
(82%) underwent neoadjuvant chemotherapy according to the
International Society of Pediatric Oncology (SIOP) protocol
(18). Eleven patients (24%) were
found to have bilateral WT. The control group (n=11) consisted of
renal tissue from the healthy part of the resected specimen after
tumor nephrectomy. These controls are labeled as ‘control’
throughout the text. The median age of the control group was 39
months (range, 3 months-5 years) with a gender ratio of 1:2.7
(f:m). A trained pathologist performed histological classification
of the samples. The study was approved by the local Ethics
Committee of the Ludwig-Maximilians University, Munich. Written
consent was obtained from all parents.
Search for candidate CTCF binding
sites
We searched for CTCF binding sites in the
surrounding of WT1 by investigating the following criteria: i)
similarity to the consensus of the CTCF core sequence (19 bp) from
the JASPAR® database according to position weight matrix
(PWM); ii) confirmation of the putative CTCF binding site through
ChIP-sec data sets included in the Ensemble® regulatory
database (human regulatory built 11); iii) localization in a
CpG-rich area. Following these criteria, we found one candidate
site which has a PWM-score of 0.93, a confirmed CTCF binding in 24
ChIP-seq data sets in 11 different human cell lines, and is located
in an area with a CpG ratio >60%.
Real-time reverse transcription-PCR
(RT-PCR)
TRI reagent® was used for isolation of
total RNA from native samples. Total RNA was depleted from DNA and
subsequently purified using DNase set and RNeasy Mini kit,
respectively (Qiagen, Hilden, Germany). Reverse transcription of
total RNA was performed using random hexamers (Roche Diagnostics,
Penzberg, Germany) and SuperScript II reverse transcriptase
(Invitrogen, Carlsbad, CA, USA). Intron-spanning primers were
designed for the human gene WT1 using Primer Express®
v2.0 (Applied Biosystems, Foster City, CA, USA) based on the
sequence information contained in the Ensembl database. Primers
specific for all transcripts are listed in Table I. PCR amplifications were carried
out with 40 ng of cDNA, 500 nM forward and reverse primer and iTaq
SYBR-Green Supermix (Bio-Rad Laboratories, Hercules, CA, USA) in a
final volume of 20 μl. PCR reactions were run for 40 cycles
consisting of 15 sec denaturation at 95°C, primer annealing for 15
sec at 55°C, and extension for 30 sec at 72°C on a Mastercycler
Realplex2 cycler (Eppendorf, Hamburg, Germany). All PCR
reactions were prepared in doublets and standardized to the
reference gene TATA box-binding protein (TBP). Level of expression
was calculated according to the mathematical model of Pfaffl
(19).
 | Table IPrimers used for gene expression, DNA
methylation and splice variant analyses. |
Table I
Primers used for gene expression, DNA
methylation and splice variant analyses.
| Gene expression via
RT-PCR | Sequence
(5′->3′) |
|---|
| WT1 fw |
CGAGAGCCAGCCCGCTA |
| WT1 rv |
TCGAAGGTGACCGTGCTGTA |
| TBP fw |
GCCCGAAACGCCGAATAT |
| TBP rv |
CCGTGGTTCGTGGCTCTCT |
|
| Methylation analysis
via pyrosequencing | |
|
| WT1 f1 |
GAAAGGTTATTAGGTATTGTGTTAAGG |
| WT1 r1 |
ATACCACTAAATATCCTCACATATACAC |
| WT1 s1 |
AAAGTATTTGTTTTTTATATTGAG |
|
| Splice variant
analysis via pyrosequencing | |
|
| WT1_KTS_F |
TCCGACCACCTGAAGACC |
| WT1_KTS_R |
ACAACTTGGCCACCGACAG |
| WT1_KTS_Pyro |
ACACCAGGACTCATACAG |
Quantification of CTCF binding site
methylation using Pyrosequencing®
Genomic DNA was extracted from native samples using
standard procedures and converted using the EpiTect Bisulfite
kit® (Qiagen) according to the manufacturer’s manual.
Pyrosequencing primers were designed with PyroMark Assay Design
Software 2.0 and are provided in Table
I. PCR was carried out using 50 ng DNA, 500 nM forward and
reverse primer, and Maxima HotStart Taq (Fermentas, Glen Burnie,
MD, USA) in a final volume of 15 μl. PCR reactions were run for 45
cycles consisting of 20 sec denaturation at 95°C, primer annealing
for 20 sec at 58.3°C, and extension for 35 sec at 72°C on a
Mastercycler Realplex2 cycler (Eppendorf). PCR products
were sequenced on a Qiagen PyroMark™ Q24 instrument using PyroMark
Gold reagents and PyroMark Q24 Vacuum Prep Workstation (Qiagen) as
recommended by the manufacturer. Quantitative analysis of
methylation was accomplished using the Pyro Q-CpG Software
(Qiagen).
Limits for hyper-respective hypomethylation were
considered as two-fold the standard deviation (± 2SD).
Analysis of WT1 mRNA KTS+/KTS− splice
variants by Pyrosequencing
Pyrosequencing primers were designed to flank the
KTS+/KTS− splicing event of exon 9 using PyroMark Assay Design
Software 2.0 (Table I). PCR results
in two alternative DNA amplification products, which were
subsequently sequenced as described above to determine their
relative proportion. The KTS+/KTS− ratio was calculated according
to Méreau et al (20).
Statistical analysis
Statistical analysis was performed with
SPSS® v19.0. An explorative analysis was made without
corrections of p-values for multiple testing. Student’s t-test was
used for comparison of gene expression data and methylation status.
Graphs and figures were created using Graphpad Prism version 6.
Results
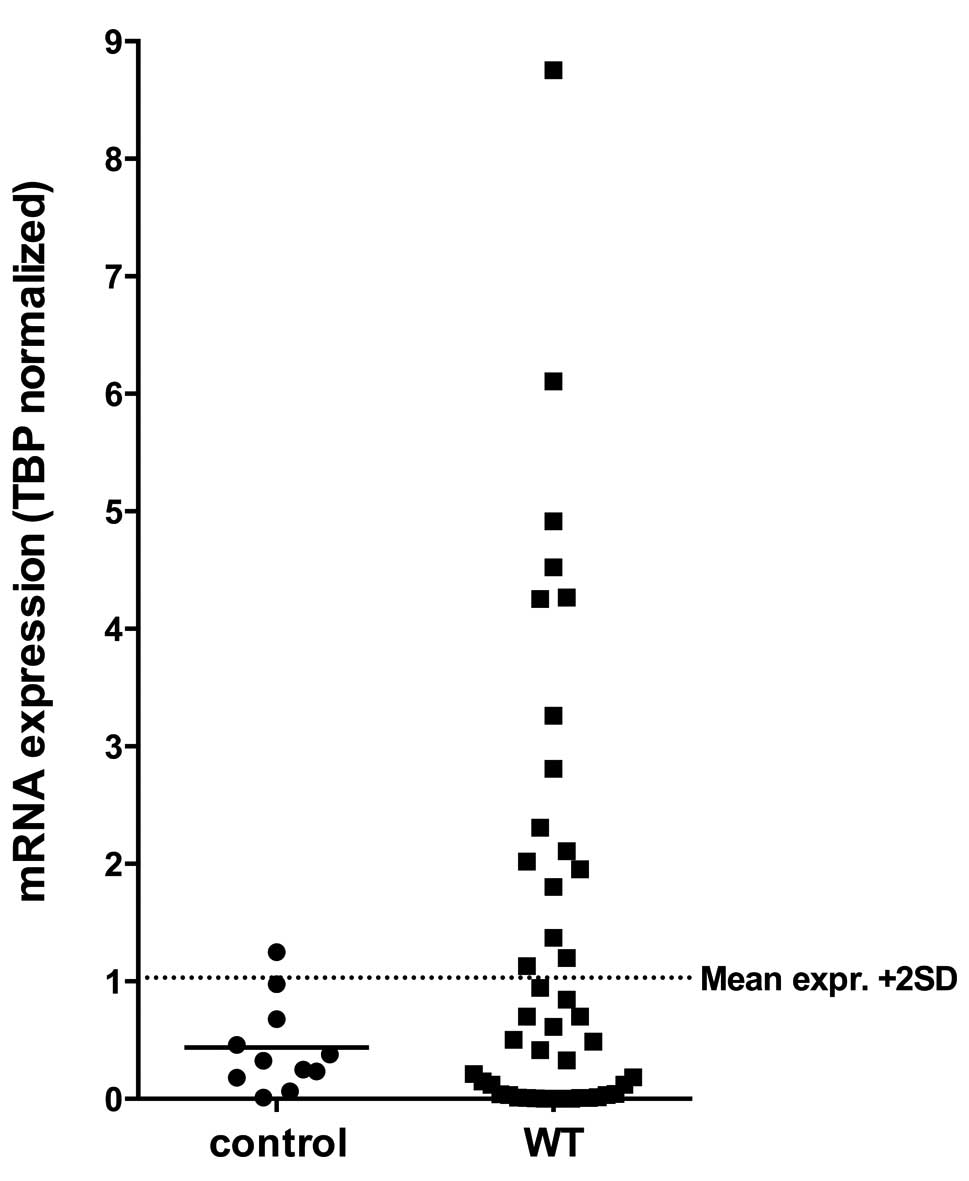
WT1 mRNA expression is upregulated in
WT
First, we investigated the transcriptional activity
of WT1 in a collection of 44 WTs and compared it to renal control
samples. Statistical analysis revealed that there is no difference
of mean WT1 expression between tumors and controls (p=0.64).
However, 16 (36.4%) WT cases showed a much higher WT1 expression
the mean expression + 2 SD of controls (Fig. 1).
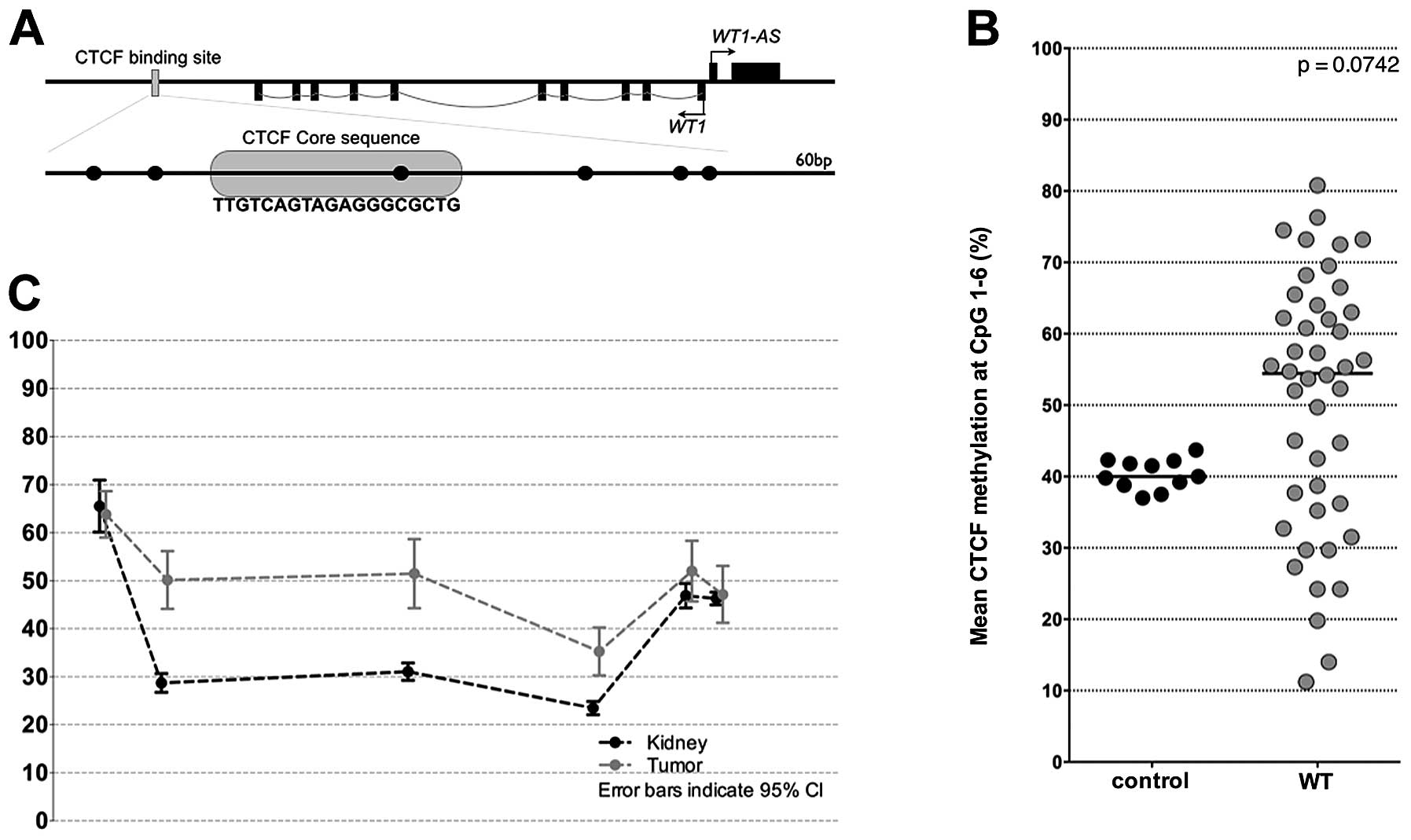
DNA hypermethylation of the CTCF binding
site in WT
To analyze the methylation status at the CTCF
binding site, we designed primers that cover a 60 bp DNA segment
containing 6 CpGs (Fig. 2A).
Investigating the degree of DNA methylation we revealed a mean
methylation rate of 54.5% for all tumors, whereas that of controls
was 40.0% (Fig. 2B). While DNA
methylation level of all tested controls showed only slight
variations (range from 37.0 to 43.7%), DNA methylation of tumors
varied extremely (range from 11.0 to 80.8%).
Notably, mean methylation at the individual CpGs
differed considerably, with a range from 65.5% at CpG1 to 23.5% at
CpG4 in controls (Fig. 2C).
However, differences in DNA methylation between tumors and controls
was observed only at CpGs 2–4 (Fig.
2C), which span the core CTCF-binding site.
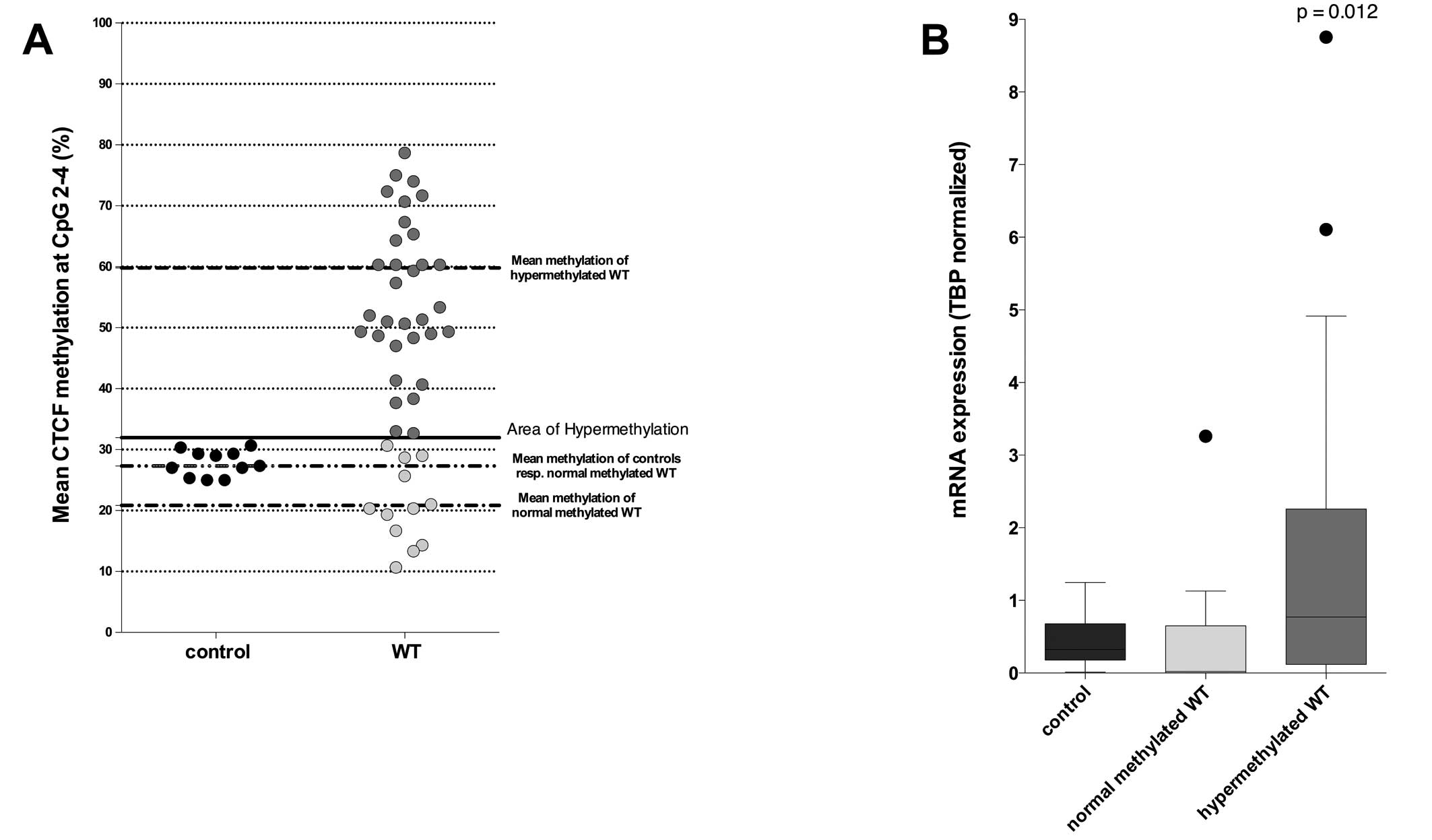
Considering only the three differentially methylated
CpGs, we divided our samples into two groups; one contained tumors
with a normal CTCF methylation, and the other contained
hypermethylated tumors, using 31.95% methylation as a cut-off value
(mean methylation of the CpGs 2–4 in controls + 2SD). The normal
methylated group contained 12 (27.3%) samples with a mean
methylation of 27.2% for CpG 2–4. The second group contained 32
(72.7%) samples, which we considered as hypermethylated. The mean
methylation rate for these tumors was 59.8% (Fig. 3A).
Hypermethylated tumors showed significant
overexpression of WT1 compared to normal methylated tumors
(p=0.012) (Fig. 3B). There was a
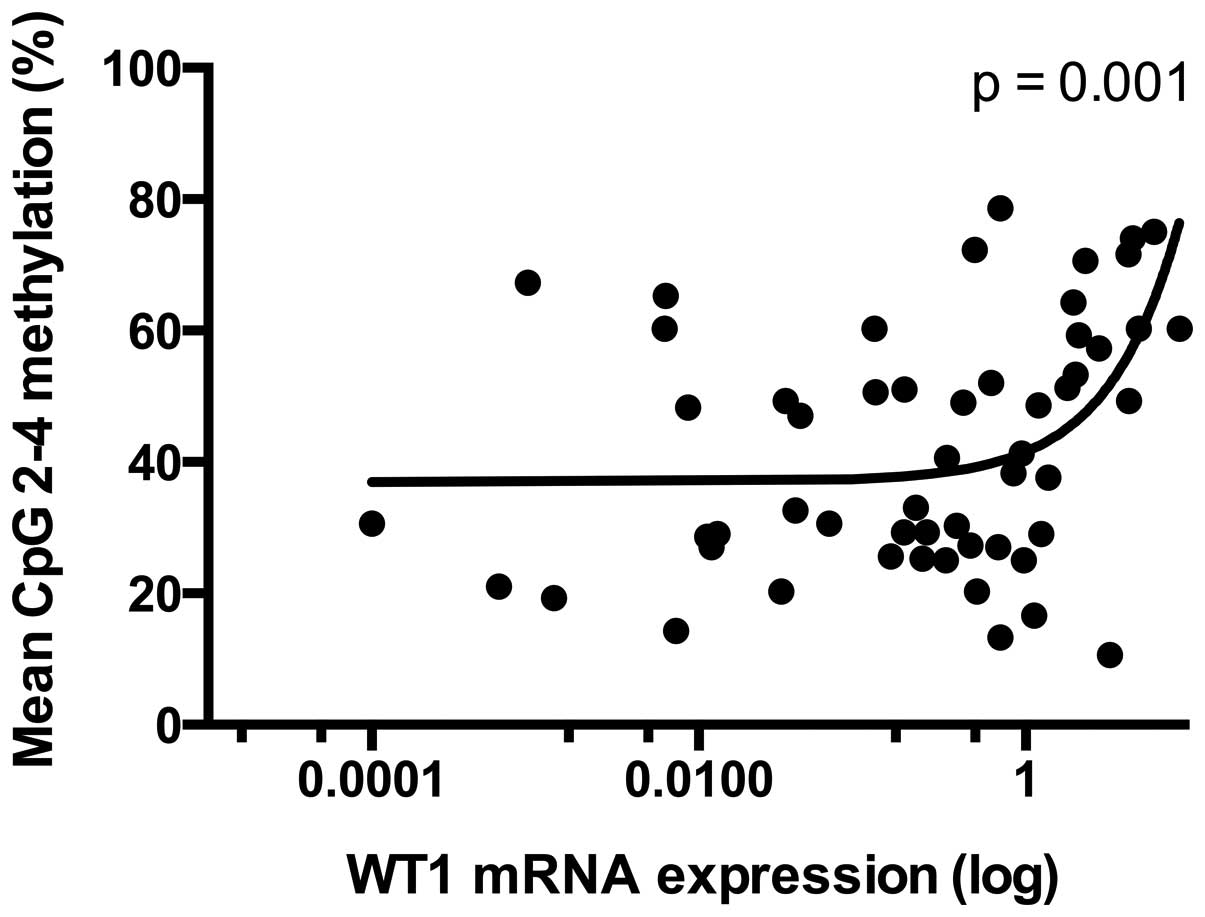
relevant correlation (r=0.435) between mean methylation of the CpGs
2–4 and the mRNA expression of WT1 (p=0.001) (Fig. 4).
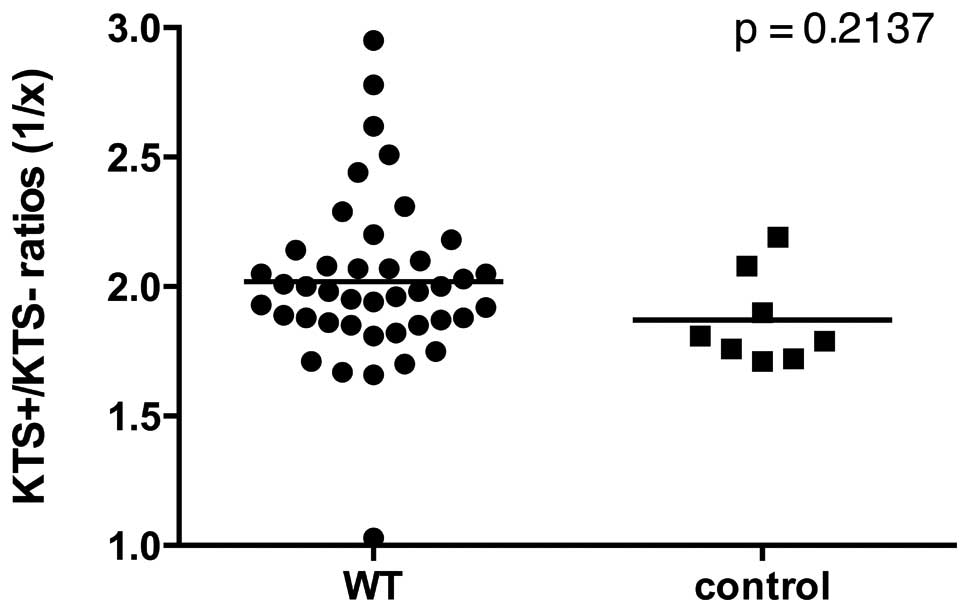
KTS+/KTS− splicing variants are not
differentially expressed in WT
Ratio for KTS+/KTS− transcript was found to be
stable in healthy tissues at a value of ~2:1 (7,21). We
determined the median KTS+/KTS− ratio to be 1.86:1 for controls and
1.97:1 for WTs, respectively (Fig.
5). Apart from four outliers with a ratio >2.5:1 and one
outlier with a ratio of 1:1, ratios presented to be quite constant
in both tumors and controls. Consequently, no statistically
relevant deviation between transcript ratios of WT and controls
were found (Fig 5). Similarly,
there was no statistical correlation between the KTS+/KTS− ratios
and the methylation status of the CpGs 2–4 at the CTCF binding
sites (p=0.914), but a trend toward a correlation of KTS+/KTS−
ratios and WT1 mRNA expression (p=0.061, data not shown).
Discussion
The outstanding role of WT1 in urinary tract
development is well known. Its function as a TSG was assumed due to
the findings that mutations of WT1 led to both anomalies in the
urinary tract and malignancies in different tissues, particularly
in kidneys. However, mutations of WT1 are detectable in
approximately 30% of sporadic WTs, only. Moreover, it is known that
WT1 mRNA is overexpressed in a large number of WTs. In our study,
16 (36.4%) tumors showed WT1 overexpression, further casting the
generally perceived doubt on an exclusive function of WT1 as a TSG.
In fact, previous studies indicated that specific WT1 splice
variants can act as an oncogene. The KTS+/KTS− splicing variants in
particular seem to be relevant in this manner (21).
However, mechanisms other than mutation of TSG are
known to be involved in tumorigenesis, particularly in childhood
tumors. We previously described that methylation of imprinted genes
is disturbed in a large number of WTs (22). While WT1 is not exclusively
expressed from a single allele in human kidney, it can be imprinted
in selected human tissues. Jinno et al reported a biallelic
or exclusively maternal expression in fetal brain and uterus
(15). Mitsuya et al
confirmed a paternal imprinting in human fibroblasts and
lymphocytes in some cases (23).
Malik et al and later Hancock et al reported an
epigenetic regulation of a WT1 antisense transcript (17,24).
An alternative transcript of WT1 (AWT1) has also been described
(24), which may undergo loss of
imprinting during WT tumorigenesis (25).
Considering these data, we investigated an impact of
DNA methylation on WT1 mRNA expression. As we have demonstrated the
influence of CTCF binding site methylation on both NNAT and
IGF2/H19 loci (12), we searched
for such a binding site in the surrounding of WT1. The detected
binding site is located downstream the WT1 gene and inside a CpG
island. Correlating the methylation with the WT1 expression, we
displayed a distinct overexpression for tumors with
hypermethylation at this site, whereas tumors with normal
methylation pattern had an mRNA expression equal to the control.
Renda et al confirmed that the affinity of the CTCF factor
may be inhibited by methylation of specific CpGs in the CTCF core
sequence in chicken β-globulin (26). In addition, Xu et al
described that even flanking CpGs in the surrounding of a CTCF core
sequence affects the accessibility of CTCF at the breast cancer 1
(BRCA1) promoter (27). According
to these findings, we hypothesized that the hypermethylation of the
CTCF core binding site has an effect on the transcriptional
activity due to a lower affinity of CTCF to its binding site. In
line with this, we observed that only the CpGs 2–4 that span the
CTCF binding site had higher levels of methylation in tumors
compared to controls (Fig. 2).
These findings are consistent with a theory proposed by Phillips
and Corces that describes a retaining of the CTCF factor during the
whole cell cycle to protect its binding sites against DNA
methylation, particularly those involved in DNA looping (13).
Although aberrant WT1 expression is a common
phenomenon in WTs, another mechanism may also be involved in
tumorigenesis, which is the different functionality of WT1
transcripts due to the KTS+/KTS− splice variants. Thus, we
investigated whether CTCF is not only involved in the regulation of
the expression, but also in the maintenance of the KTS+/KTS− ratio.
In our analysis, there was no statistical correlation between the
methylation of the CTCF binding site and the KTS+/KTS− ratio
(p=0.941), but a trend toward a correlation between WT1 mRNA
expression and the KTS+/KTS− ratio (p=0.061). This stands for an
independent regulation of the KTS+/KTS− ratio from the CTCF binding
site methylation. Even if there is a true correlation between the
KTS+/KTS− ratio and the WT1 mRNA expression, this cannot be
extracted from our data at this time and opens the field for
further investigations.
In conclusion, we have detected an aberrant
methylation pattern at a CTCF binding site downstream the WT1 gene,
which is associated with an elevated WT1 transcriptional activity.
However, regulation of the KTS+/KTS− transcripts is independent of
the CTCF binding site methylation. Thus, methylation of the CTCF
binding site may be partially responsible for the transcriptional
activation of the WT1 locus and hypermethylation of this site may
be one important oncogenic mechanism in WT.
References
|
1
|
Hohenstein P and Hastie ND: The many
facets of the Wilms’ tumour gene, WT1. Hum Mol Genet.
15:R196–R201. 2006.PubMed/NCBI
|
|
2
|
Rivera MN and Haber DA: Wilms’ tumour:
connecting tumorigenesis and organ development in the kidney. Nat
Rev Cancer. 5:699–712. 2005.
|
|
3
|
Pelletier J, Bruening W, Li FP, et al:
WT1 mutations contribute to abnormal genital system
development and hereditary Wilms’ tumour. Nature. 353:431–434.
1991. View
Article : Google Scholar
|
|
4
|
Brown KW, Wilmore HP, Watson JE, et al:
Low frequency of mutations in the WT1 coding region in Wilms’
tumor. Genes Chromosomes Cancer. 8:74–79. 1993.PubMed/NCBI
|
|
5
|
Wells J, Rivera MN, Kim WJ, Starbuck K and
Haber DA: The predominant WT1 isoform (+KTS) encodes a
DNA-binding protein targeting the planar cell polarity gene
Scribble in renal podocytes. Mol Cancer Res. 8:975–985.
2010.
|
|
6
|
Yang L, Han Y, Suarez Saiz F, Saurez Saiz
F and Minden MD: A tumor suppressor and oncogene: the WT1 story.
Leukemia. 21:868–876. 2007.PubMed/NCBI
|
|
7
|
Haber DA, Sohn RL, Buckler AJ and Housman
DE: Alternative splicing and genomic structure of the Wilms tumor
gene WT1. Proc Natl Acad Sci USA. 88:9618–9622. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Klamt B, Koziell A, Poulat F, et al:
Frasier syndrome is caused by defective alternative splicing of
WT1 leading to an altered ratio of WT1 +/−KTS splice
isoforms. Hum Mol Genet. 7:709–714. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bor YC, Swartz J, Morrison A, Rekosh D,
Ladomery M and Hammarskjöld ML: The Wilms’ tumor 1 (WT1) gene (+KTS
isoform) functions with a CTE to enhance translation from an
unspliced RNA with a retained intron. Genes Dev. 20:1597–1608.
2006.
|
|
10
|
Hammes A, Guo JK, Lutsch G, et al: Two
splice variants of the Wilms’ tumor 1 gene have distinct functions
during sex determination and nephron formation. Cell. 106:319–329.
2001.
|
|
11
|
Lewis A and Reik W: How imprinting centres
work. Cytogenet Genome Res. 113:81–89. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hubertus J, Zitzmann F, Trippel F, et al:
Selective methylation of CpGs at regulatory binding sites controls
NNAT expression in Wilms tumors. PLoS One. 8:e676052013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Phillips JE and Corces VG: CTCF: master
weaver of the genome. Cell. 137:1194–1211. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fiorentino FP and Giordano A: The tumor
suppressor role of CTCF. J Cell Physiol. 227:479–492. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jinno Y, Yun K, Nishiwaki K, et al: Mosaic
and polymorphic imprinting of the WT1 gene in humans. Nat Genet.
6:305–309. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mares J, Kríz V, Weinhäusel A, et al:
Methylation changes in promoter and enhancer regions of the WT1
gene in Wilms’ tumours. Cancer Lett. 166:165–171. 2001.PubMed/NCBI
|
|
17
|
Hancock AL, Brown KW, Moorwood K, et al: A
CTCF-binding silencer regulates the imprinted genes AWT1 and
WT1-AS and exhibits sequential epigenetic defects during
Wilms’ tumourigenesis. Hum Mol Genet. 16:343–354. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
de Kraker J, Graf N, van Tinteren H, et
al: Reduction of postoperative chemotherapy in children with stage
I intermediate-risk and anaplastic Wilms’ tumour (SIOP 93-01
trial): a randomised controlled trial. Lancet. 364:1229–1235.
2004.PubMed/NCBI
|
|
19
|
Pfaffl MW: A new mathematical model for
relative quantification in real-time RT-PCR. Nucleic Acids Res.
29:e452001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Méreau A, Anquetil V, Cibois M, et al:
Analysis of splicing patterns by pyrosequencing. Nucleic Acids Res.
37:e1262009.
|
|
21
|
Burwell EA, McCarty GP, Simpson LA,
Thompson KA and Loeb DM: Isoforms of Wilms’ tumor suppressor gene
(WT1) have distinct effects on mammary epithelial cells. Oncogene.
26:3423–3430. 2007.
|
|
22
|
Hubertus J, Lacher M, Rottenkolber M,
Müller-Höcker J, Berger M, et al: Altered expression of imprinted
genes in Wilms tumors. Oncol Rep. 25:817–823. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mitsuya K, Sui H, Meguro M, Kugoh H, Jinno
Y, et al: Paternal expression of WT1 in human fibroblasts and
lymphocytes. Hum Mol Genet. 6:2243–2246. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Malik K and Brown KW: Epigenetic gene
deregulation in cancer. Br J Cancer. 83:1583–1588. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Brown KW, Power F, Moore B, Charles AK and
Malik KT: Frequency and timing of loss of imprinting at 11p13 and
11p15 in Wilms’ tumor development. Mol Cancer Res. 6:1114–1123.
2008.PubMed/NCBI
|
|
26
|
Renda M, Baglivo I, Burgess-Beusse B,
Esposito S, Fattorusso R, et al: Critical DNA binding interactions
of the insulator protein CTCF: a small number of zinc fingers
mediate strong binding, and a single finger-DNA interaction
controls binding at imprinted loci. J Biol Chem. 282:33336–33345.
2007. View Article : Google Scholar
|
|
27
|
Xu X, Gammon MD, Zhang Y, Cho YH, Wetmur
JG, et al: Gene promoter methylation is associated with increased
mortality among women with breast cancer. Breast Cancer Res Treat.
121:685–692. 2010. View Article : Google Scholar : PubMed/NCBI
|