Introduction
Lung cancer is the most common cause of
cancer-related mortality in the world (1). Non-small cell lung cancer (NSCLC)
accounts for ~80–85% of lung cancer cases, and small cell lung
cancer (SCLC) accounts for the remaining 15–20%. More than half of
patients with NSCLC are diagnosed at an advanced stage (stage III
or IV), and chemotherapy is often the first choice of treatment for
these patients (2,3). However, the response to chemotherapy
as well as the associated prognosis remains unfavorable.
Pemetrexed (PEM) is a multi-targeted antifolate drug
that disrupts multiple enzymes involved in pyrimidine and purine
synthesis (4). It has been approved
for the treatment of NSCLC (5).
Combination chemotherapy with PEM and cisplatin has better
tolerability compared to cisplatin, and have been used as
first-line treatment or as single drugs for maintenance therapy in
advanced NSCLC patients (6–8). A phase III trial outlined that
patients with adenocarcinoma treated with a PEM-based regime had
prolonged overall survival than those with squamous cell carcinoma
(7). However, the majority of lung
adenocarcinoma patients treated with PEM exhibit either intrinsic
or acquired resistance. Previous research of PEM resistance has
primarily focused on enzymes in the folate metabolic pathway, and
some researchers have found that overexpression of thymidylate
synthase (TS) and dihydrofolate reductase (DHFR) is associated with
insusceptibility to PEM (9,10). Yet, one recent study found that
PEM-treated lung adenocarcinoma patients with EGFR mutations had a
better response rate and longer progression-free survival (11). However, whether EGFR expression is
associated with PEM-resistance in NSCLC has not yet been
reported.
The epidermal growth factor receptor (EGFR) and
ErbB3 (HER3) are members of the ErbB family of receptor tyrosine
kinases. They play a critical role in processes such as neoplastic
cell proliferation, anti-apoptosis, angiogenesis and metastasis.
Generally speaking, ErbB gene expression has a negative correlation
with clinical outcome (12). EGFR
overexpression and mutations are found in lung adenocarcinoma, and
its overexpression is recognized in many types of human cancers,
including breast, colorectal and gastric cancer (13–15).
High expression of EGFR is often associated with aggressive
phenotypes and resistance to chemotherapy, and it is used as a
multi-drug resistant marker in certain types of cancer (16,17).
ErbB3 is considered to stimulate intracellular signaling coupled
with other ErbB family members. Novel therapies or combinations
blocking ErbB3 may provide strategies to overcome acquired
resistance and to increase the effectiveness of therapy (18). EGFR and ErbB3 inhibited the cellular
response to sorafenib in hepotocellular cell lines (19). In addition, our preliminary
experiments revealed that PEM may be used beneficially in
combination with EGFR. This suggests that PEM has an antitumor
effect by acting on the EGFR and its family members.
To the best of our knowledge, this is the first
study to investigate the association of expression of the ErbB
genes and PEM resistance. In the present study, we established a
PEM-resistant lung adenocarcinoma cell line and provided a model
with which to explore relevant factors of acquired resistance to
PEM. By comparison with to the parental cell line, we aimed to
elucidate the correlation between EGFR and ErbB3 expression and PEM
resistance. This may provide novel predictive markers for the
clinical application of PEM.
Materials and methods
Preliminary experiments
The docking analysis was performed with the
Surflex-Dock model. The crystal structure of the EGFR-erlotinib
complex was collected from a protein data bank (PDB code: 1M17).
All of the hydrogen atoms were added to define the correct
configuration and tautomeric states. Then the model structure was
energy-minimized, and the Powell energy minimization algorithm was
used for energy minimization. After extracting the binding ligand
erlotinib, PEM was then docked into the binding pocket for
docking-scoring analysis (20).
Cell culture
The human lung adenocarcinoma cell line A549 was
obtained from the Cell Resource Center of the Shanghai Institutes
for Biological Sciences. A549 cells were cultured in RPMI-1640
medium supplemented with 10% fetal bovine serum (FBS) (both from
Gibco, Carlsbad, CA, USA), 100 U/ml of penicillin G, and 100 μg/ml
of streptomycin, and cells were cultivated in a incubator with 5%
CO2 at 37°C under humidified conditions.
Establishment of the PEM-resistant cell
line
The A549 cell line was exposed to a single high
concentration of PEM (the 50% inhibitory concentration of PEM for
A549 cells) over a period of 48 h repeatedly (10,12).
PEM was obtained from Eli Lilly and Company (Indianapolis, IN, USA;
A762406C). The treated cells were then washed with diluted
phosphate-buffered saline (PBS) and cultured in fresh growth medium
without PEM every day until all the dead cells were washed out.
After that, the surviving cells were cultivated as normal cells.
The treated A549 cells recovered and exhibited logarithmic growth
after 2 weeks. When cells were growing exponentially and
subcultured with trypsin, these cells were again exposed to PEM for
48 h. The degree of resistance of the treated cells was detected
discontinuously until it was in accordance with the requirement of
the experiment. The PEM-resistant cell line was established after 5
months, and it was named A549/PEM. Then the resistant cells were
cultured in an incubator with 5% CO2 at 37°C for 1 month
and passed several generations. The resistance was detected again,
and the A549/PEM cell line was proven to acquire stable resistance.
The resistant cells were used for subsequent experiments after
another month of culture in PEM-free medium.
Growth inhibition assay
Growth inhibition of the cells was detected by the
CCK-8 assay (Dojindo Molecular Technologies, Kumamoto, Japan).
Cells (2,000) were added into every well of a 96-well flat bottomed
microplate and cultured in 100 μl RPMI-1640 medium supplemented
with 10% FBS. The cells were divided into 7 group with different
concentrations of PEM. The cells were incubated at 37°C for 24 h in
a humidified incubator with 5% CO2. Subsequently, the
PEM concentrations were 0.001, 0.01, 0.1, 1.0, 10, 100 and 1,000
μg/ml, respectively. The well without PEM was set as the control
group. Those wells with 100 μl nutrient solution only were
considered as the blank control. After incubating for 48 h, the
drug-containing growth medium was replaced with 110 μl medium
containing CCK-8 reagent (10 μl CCK-8 and 100 μl RPMI). Following
lucifuge culturing for 2 h, the optical density (OD value) was
measured for each well (450 nm) by an automated spectrophotometer.
The resistance of the A549/PEM cell lines was calculated according
to the OD values. Each assay was performed in quintuplicate at
least 3 times.
The absorbance values at 450 nm in the experimental
wells relative to the initial value indicated cell growth or death,
respectively. The following formula was used to calculate the
surviving cell fraction: 1 − [(mean absorbance of experimental
cells − mean absorbance of blank control cells)/(mean absorbance of
control cells − mean absorbance of blank control cells)] × 100%
(10). The mean and standard
deviation (SD) were calculated, respectively. The lower the
IC50 value, the higher was the ability for inhibition of
cell proliferation (21).
Flow cytometric analysis
A cell cycle analysis kit was obtained from Beyotime
Biotechnology Co. Ltd. (Shanghai, China). A single-cell suspension
of A549 and A549/PEM cells was collected respectively, and washed
with ice-cold PBS 3 times, and then the cells were fixed with
ice-cold 70% ethanol for 30 min. The supernatant was discarded
after centrifugation, and the cells were again washed with cold PBS
twice. These cells were then treated with RNase for 30 min at 37°C.
Subsequently, the cells were dyed with propidium iodide (PI), for
analysis of the cell cycle by flow cytometry (FCM).
A549 and A549/PEM cells were cultured for 24 h with
media containing PEM at a final concentration of 0.5 μg/ml. A
single-cell suspension (1–5×106) was washed twice with
cold PBS, and the supernatant fluid was discarded by
centrifugation. Then cells were fixed with 70% ethanol at 4°C
overnight. Cells were resuspended with 100 μl 1X Annexin-binding
buffer, and then 5 μl Annexin V and 5 μl PI (Invitrogen Life
Technologies Corporation, Carlsbad, CA, USA) was added to each
column. Subsequently, cells were incubated for 15 min at 37°C
without light. After adding 400 μl 1X Annexin-binding buffer, cells
in the ice were detected by FCM within 30 min. The early apoptotic
cells (Annexin V-positive, PI-negative), late apoptotic cells
(double-positive) and living cells (double-negative) (12) were detected by FCM and subsequently
analyzed by CellQuest software (Becton-Dickinson, USA).
Construction and infection of short
hairpin RNAs
Silencing of gene expression was achieved using
short hairpin RNA (shRNA) technology. shRNAs targeting EGFR (sense,
5′-CAC CGA AGA CGA CAC CGC CTC ACC TCC ACC GTG CAA CTC ATC ACG CTT
CAA GAG AGC G-3′ and antisense, 5′-TGA GGG ATC CAA AAA ACT CAC CTC
CAC CGT GCA ACT CAT CAC GCT CTC TTG AAG CGG G-3′); and/or ErbB3
(sense, 5′-CCG GAA TAT TCG CCC AAC CTT TAA ACT CGA GTT TAA AGG TTG
GGC GAA TAT TTT TTT G-3′ and antisense, 5′-AAT TCA AAA AAA TAT TCG
CCC AAC CTT TAA ACT CGA GTT TAA AGG TTG GGC GAA TAT T-3′) were
cloned into PLKO.1-Puro plasmid (Addgene, Cambridge, MA, USA).
Lentiviral particles containing PLKO.1 (empty vector control,
sh-control), PLKO.1-anti-EGFR shRNA (shEGFR) and PLKO.1-anti-ErbB3
shRNA (shErbB3) were produced. For infection, A549/PEM cells were
grown in 75 mm2 flasks and transduced at 60% confluency
with 10 ml HEK-293T medium containing virions, in the presence of
polybrene (10 μg/ml). After 48 h, the expression of mRNA and
protein was determined by real-time PCR and western blotting,
respectively.
Relative quantitative real-time PCR
Total RNA was extracted from A549 and A549/PEM cells
using TRIzol reagent (Gibco). The concentration and purity of RNA
were determined by measuring the absorbance at 260 nm using a
NanoDrop® ND-1000 spectrophotometer (Thermo Scientific,
Wilmington, DE, USA). Reverse transcription was proceeded with
PrimeScript® reverse transcriptase (Takara Bio, Tokyo,
Japan) at 37°C for 15 min, and at 85°C for 5 sec, and then chilled
at 4°C immediately. The generated cDNA samples were stored at
−20°C.
Real-time PCR was performed using PCR amplification
equipment (Roche Diagnostics, Basel, Switzerland). The reaction mix
of 20 μl contained PCR forward primer 1.6 μl (5 μM), PCR reverse
primer 1.6 μl (5 μM), cDNA 2.0 μl, SYBR Premix Ex Taq II (Takara
Bio) 10.0 μl and ultrapure water 4.8 μl. PCR reactions were
performed under the following conditions: 95°C for 10 sec followed
by 1 cycle at 95°C for 5 sec, 60°C for 20 sec, and 72°C for 20 sec
followed by 40 cycles. Glyceraldehyde-3-phosphate dehydrogenase
(GAPDH) was used as the internal control, since its expression has
been demonstrated to remain stable during the protocol (10). The standard curves and the threshold
cycle (Ct) of target genes were obtained from the instrument’s
software. The relative expression of mRNA was represented as
2−ΔΔCt, and it was calculated as follows: ΔCt = Ct
(target gene) − Ct (GAPDH), ΔΔCt = ΔCt (treatment) − ΔCt (control),
and R = 2 − [ΔCt (treatment) − ΔCt (control)] (22).
All primers used in the present study were designed
by Sangon Biotech Co. Ltd. (Shanghai, China). The primer sequences
were as follows: EGFR forward primer, 5′-AGG CAC GAG GAA CAA GCT
CAC-3′ and reverse primer, 5′-ATG AGG ACA TAA CCA GCC ACC-3′; ErbB3
forward primer, 5′-TGC TGA GAA CCA ATA CCA GACA-3′ and reverse
primer, 5′-CTG TCA CTT CAC GAA TCC ACTG-3′; GAPDH forward primer,
5′-CTG CAC CAC CAA CTG CTT AG-3′ and reverse primer, 5′-TGA AGT CAG
AGG AGA CCA CC-3′.
Western blotting
Whole-cell proteins in the A549 and A549/PEM cell
lines were isolated. The lysates were centrifuged, and the
supernatant was collected and stored at −80°C according to the
manufacturer’s instructions. Total protein (10 μg) was loaded per
well, separated by 10–15% SDS-PAGE, and transferred to
polyvinylidene difluoride membranes at 60 V for 1 h at 4°C. The
membranes were blocked and incubated with primary antibodies
(Bioworld Co., USA; diluted 1:1,000 in TBS-A). The membranes were
rinsed thrice with 1% Tween-20-PBS for 30 min. The secondary
antibodies (Abcam Co., Cambridge, UK; diluted 1:1,200 in TBS-A)
were used with peroxidase-conjugated AffiniPure goat anti-mouse IgG
(1:8,000) and peroxidase-conjugated AffiniPur goat anti-rabbit IgG
(1:8,000) for 1 h at room temperature. The blotted membranes were
washed 3 times with 0.1% Tween-20-PBS for 15 min and 3 times with
PBS for 15 min. The immunoblots were detected using an
electrochemiluminescence kit and exposed to the Vilber Fusion FX5
automatic gel imaging analysis system (Vilber, Marne La Vallée,
France).
Statistical analysis
Differences between resistant cells and parental
cells were analyzed using Student’s t-test. All statistical
analysis was performed with SPSS 13.0 software. P<0.05 was
considered to indicate a statistically significant result.
Results
Molecular docking
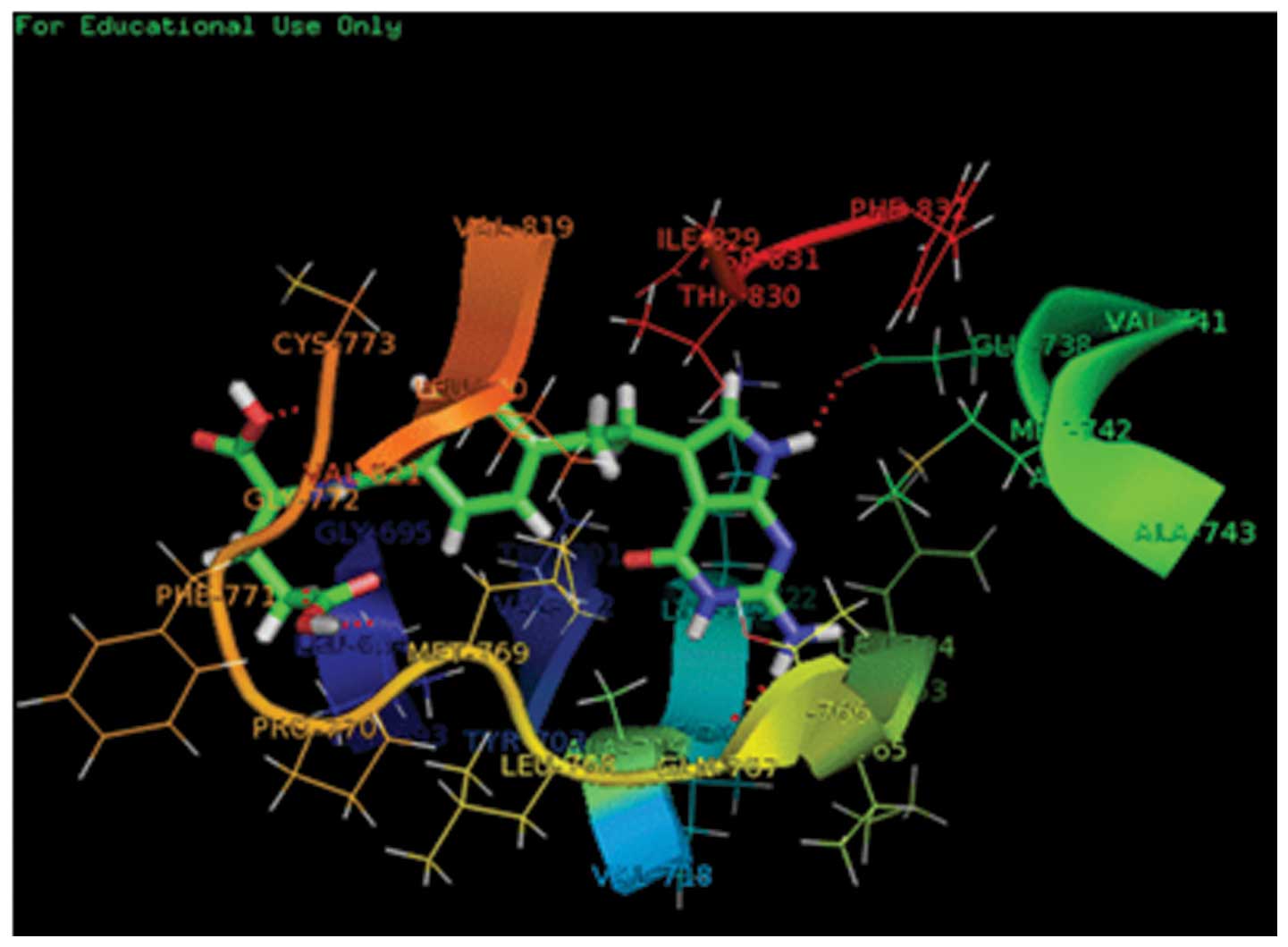
The docking studies indicated that PEM may bind to
the pocket of the EGFR (Fig. 1). In
the model, PEM was nicely bound to 1M17 and had hydrogen bonds, and
the score was 6.22 by the SYBYL 7.3 software. The length of the
hydrogen bonds formed between PEM and GLU738, GLY772, ALA719,
CYS773 and MET769 were 2.093, 2.424, 2.228, 2.035 and 2.048 Å,
respectively.
Establishment of the PEM-resistant cell
line
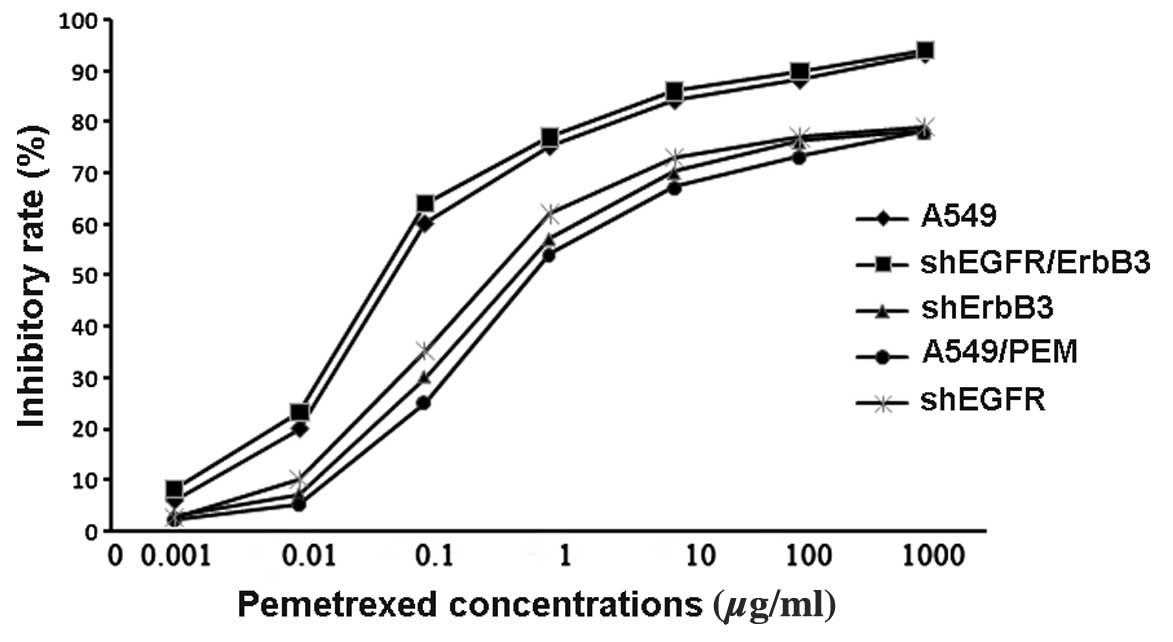
The PEM-resistant cell line A549/PEM was
successfully induced after 5 months. In the CCK-8 assay, the
IC50 values of PEM for A549 and A549/PEM cells were
0.22±0.04 and 4.37±0.26 μg/ml, respectively. The A549/PEM cell line
was significantly more resistant than the A549 cell line to PEM
(P=0.0024) (Fig. 3), and the
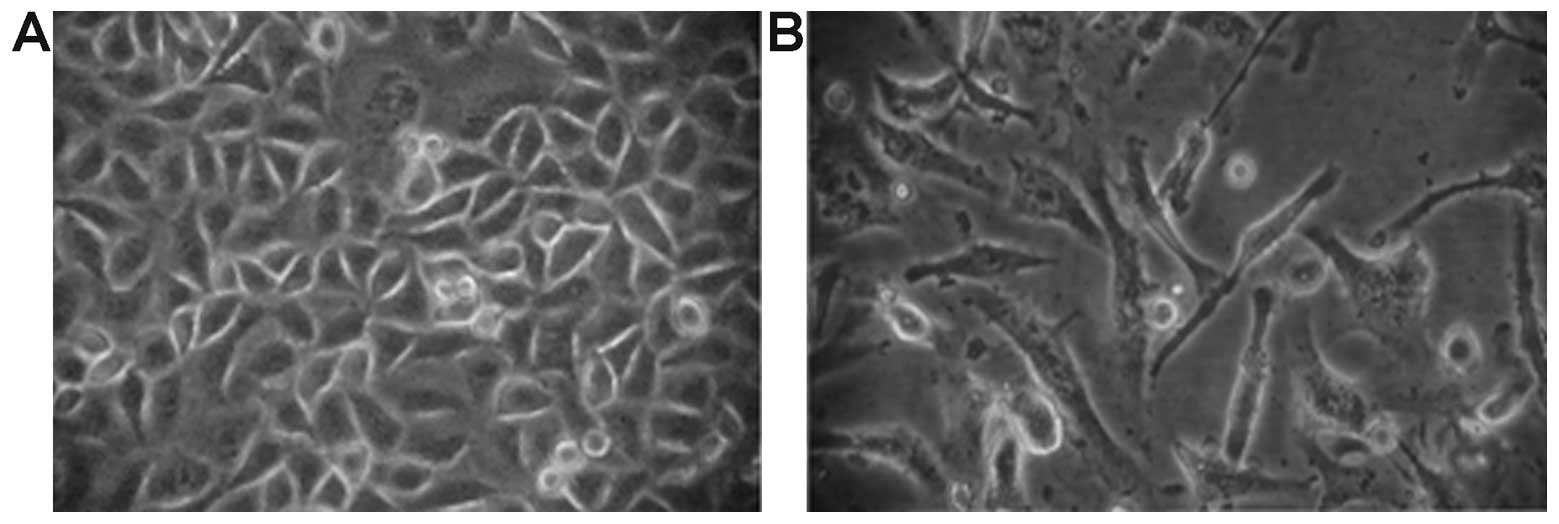
resistant index (RI) was 19.86. The morphologic observation showed
that most of the cells died after being treated with PEM. The
volume of the surviving cells was altered. Cells had an irregular
shape, and the cell membrane was vague (Fig. 2). A549/PEM cells showed a longer
doubling time than the parental cell line (33.5±1.71 vs. 27.1±1.15
h, P=0.02).
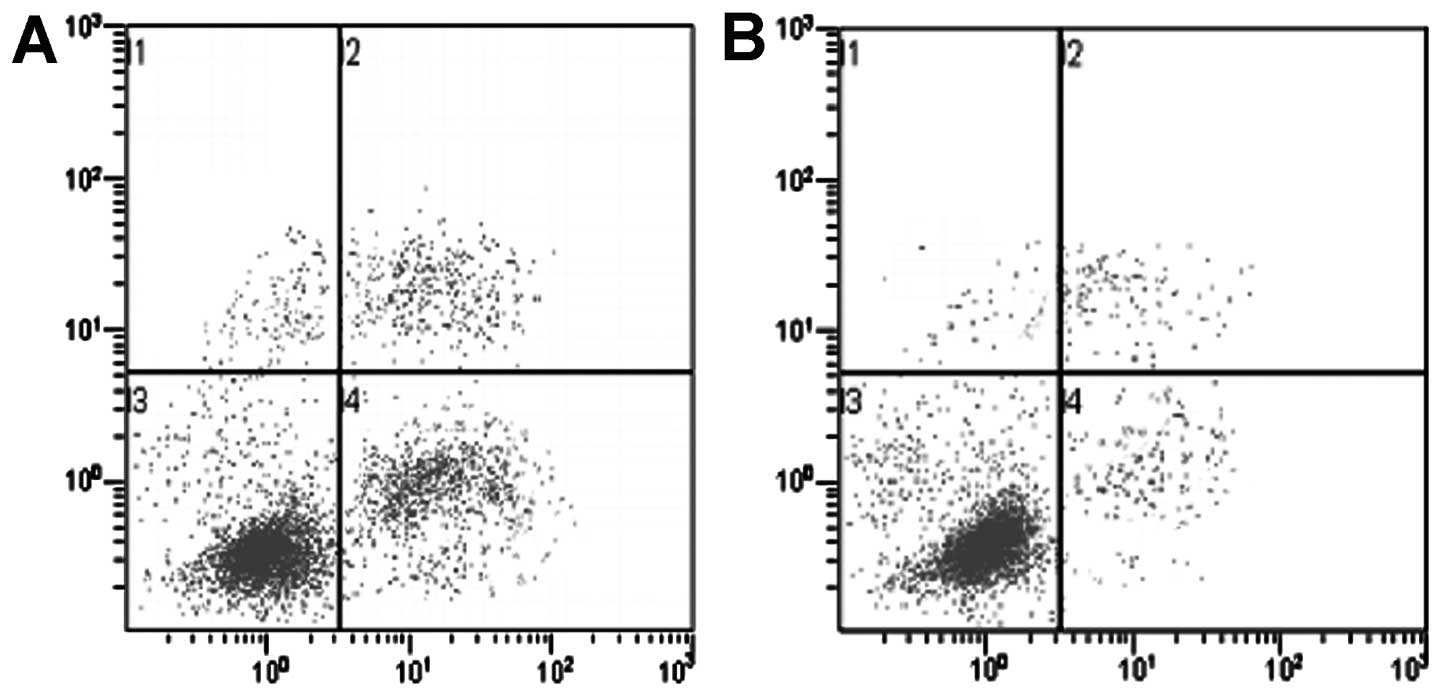
Flow cytometric analysis revealed significant
apoptosis after A549 cells were exposed to a high concentration
(5×10−5 mol/l) of PEM for 24 h, whereas A549/PEM cells
showed a much lower apoptosis rate (P<0.001) (Fig. 4). The percentage of apoptotic A549
cells increased from 2.6±0.1% (without exposure to PEM) to
23.52±3.2% (with exposure to PEM) (P<0.001). The percentage of
apoptotic A549/PEM cells increased from 1.7±0.1% (without exposure
to PEM) to 4.16±2.1% (with exposure to PEM) (P=0.072). In addition,
the percentages of cells in the G1/G0, S and G2/M phases were
analyzed. The number of cells in the G1/G0 phase increased, and
that in the S phase decreased in the A549/PEM cells (P<0.05)
(Table I).
 | Table IQuantitative assessment of the cell
cycle distribution of A549 and A549/PEM cells. |
Table I
Quantitative assessment of the cell
cycle distribution of A549 and A549/PEM cells.
| Cell cycle phases
|
|---|
|
|---|
| Cell lines | G1/G0 (%) | S (%) | G2/M (%) |
|---|
| A549 | 58.05±1.37 | 37.63±1.25 | 4.32±1.15 |
| A549/PEM | 66.09±1.63a | 30.03±0.91a | 3.88±1.01 |
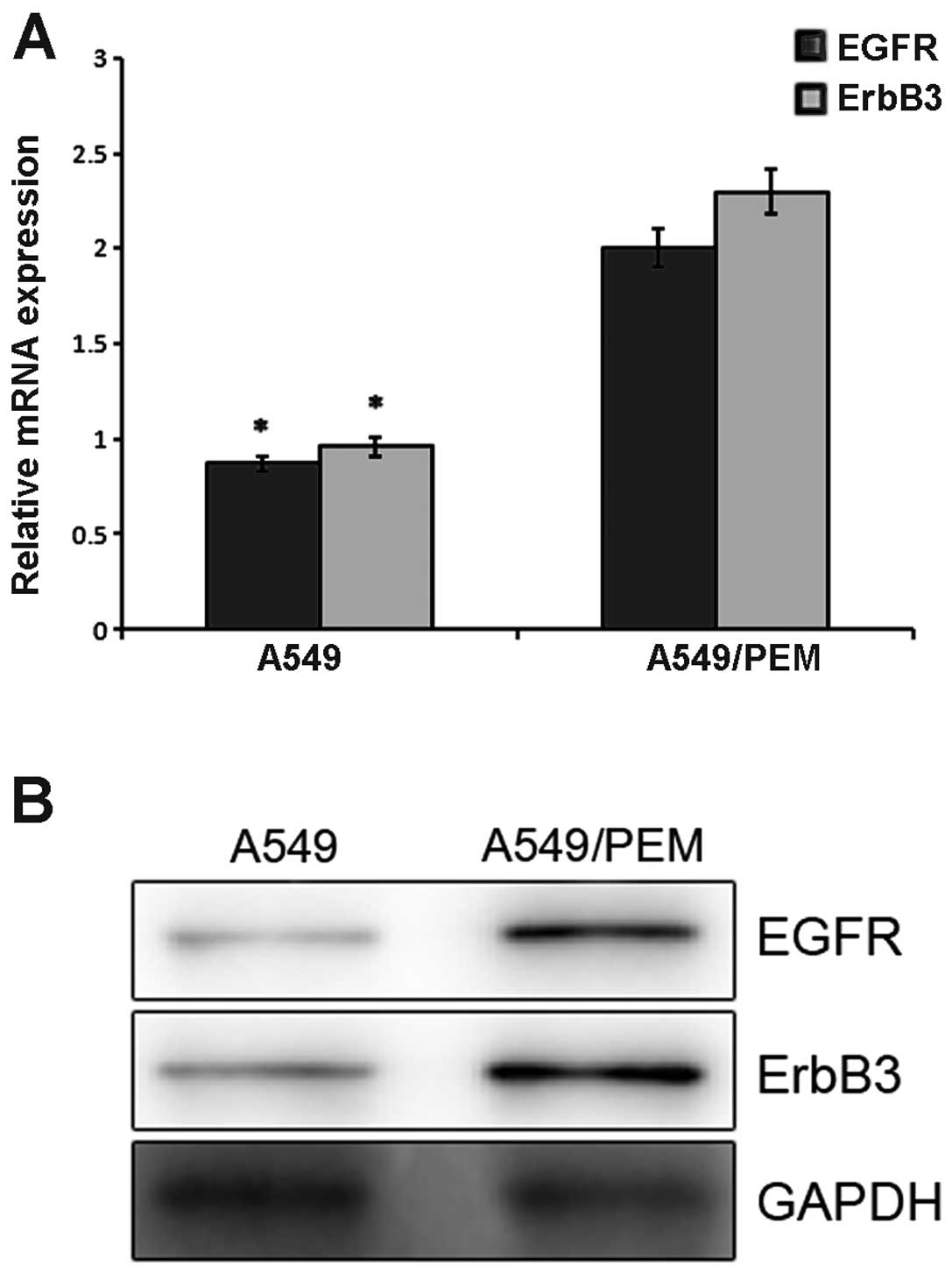
Expression of EGFR and ErbB3 in the A549
and A549/PEM cells
The mRNA expression of EGFR in the A549 and A549/PEM
cells was 0.87±0.08 and 2.01±0.12, respectively (P=0.0089)
(Fig. 5A). ErbB3 mRNA expression in
the two cell lines was 0.96±0.04 and 2.31±0.22, respectively
(P=0.034). Compared with the parental cell line, the EGFR and ErbB3
expression levels were significantly higher. Western blotting was
used to analyze their expression at the protein level. The gray
scale of the stained area was measured under identical conditions.
Higher average optical densities for EGFR and ErbB3 were observed
in the A549/PEM cells when compared with that in the parental cell
line (Fig. 5B). The difference was
statistically significant (P<0.05).
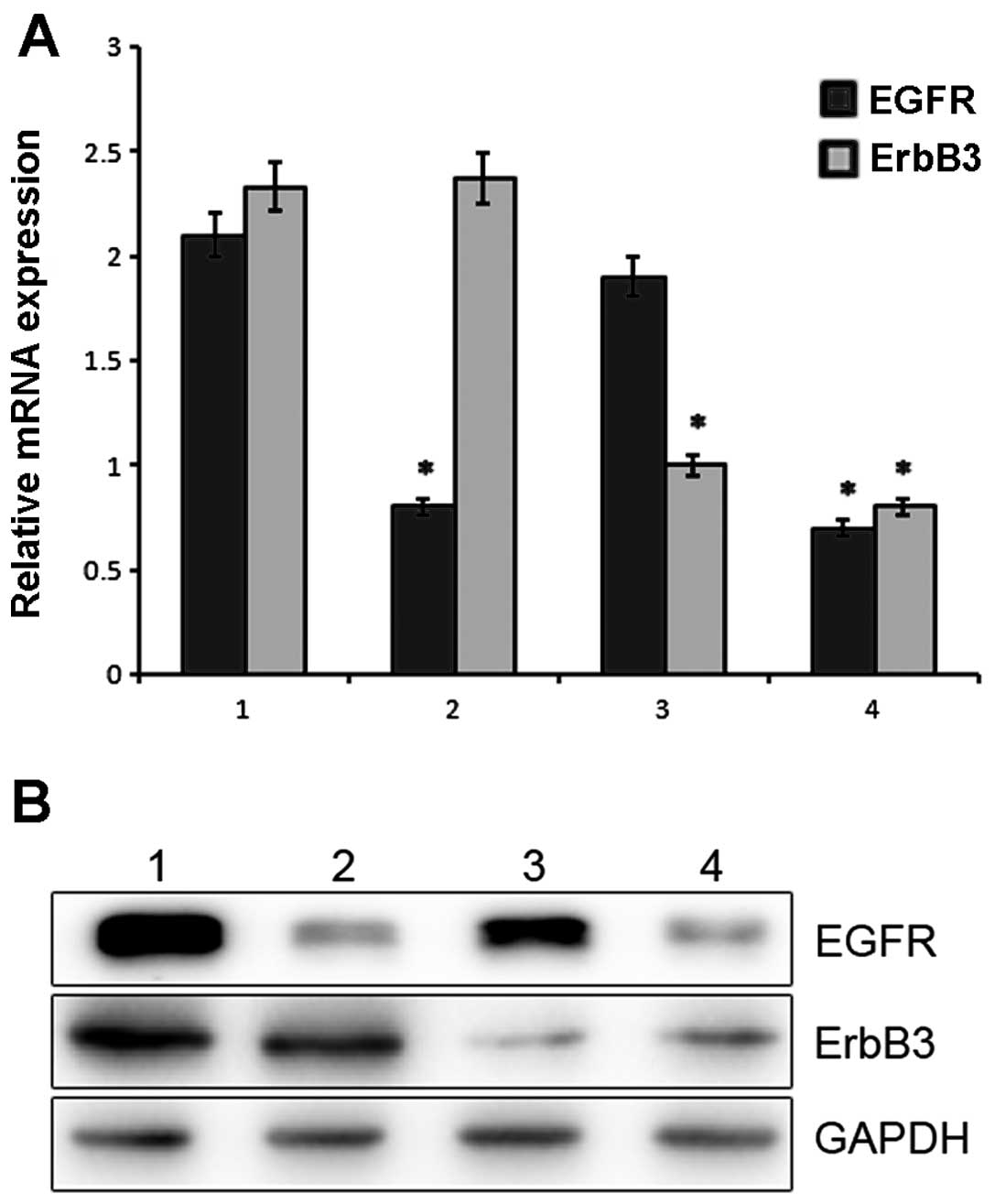
Expression levels of EGFR and ErbB3 in
the A549/PEM cells following EGFR, ErbB3 or EGFR/ErbB3
downregulation
Following lentiviral infection of the A549/PEM cell
line, the mRNA expression of EGFR and ErbB3 was examined by
real-time PCR. As shown in Fig. 6A,
a significant difference was noted between the silenced and control
cells (P<0.05). The relative level of EGFR mRNA in the
A549/PEM-shEGFR cells was significantly downregulated by 2.4-fold
when compared with that in the A549/PEM-sh-control cells. In the
A549/PEM cells infected with ErbB3 shRNA, we observed that the
level of ErbB3 mRNA was successfully downregulated by 2.5-fold
compared with that in the A549/PEM-sh-control cells.
The protein expression of EGFR and ErbB3 was
evaluated by western blotting. The average optical density for EGFR
protein expression in the shEGFR-infected cells was lower when
compared with the value in the A549/PEM-sh-control cells. The
observed differences were significant (P<0.05) (Fig. 6B). Consistent with the above mRNA
results, the average optical density for ErbB3 protein expression
in the A549/PEM-shErbB3, A549/PEM-shEGFR/ErbB3 and
A549/PEM-sh-control cells showed significant differences between
the cell groups (P<0.05). The results were in accordance with
the mRNA levels.
Cellular response to PEM following
downregulation of EGFR or ErbB3
A549/PEM cells that overexpress EGFR and ErbB3 are
more resistance to PEM than A549 cells that do not overexpress EGFR
and ErbB3. After inhibiting EGFR expression, the IC50
value (index of chemotherapy sensitivity to PEM) of the
EGFR-shRNA-infected cells was 4.52±0.47 μg/ml, and this value for
A549/PEM was 4.37±0.26 μg/ml (P=0.33). The IC50 value of
the ErbB3-shRNA infected cells that reduced the expression of ErbB3
was 4.46±0.31 μg/ml (P=0.42). This difference was not statistically
significant (Fig. 3). Following the
silencing of both EGFR and ErbB3, the IC50 value
detected by CCK-8 assay was 0.19±0.17 μg/ml (P=0.0015). These
results indicate that overexpression of EGFR or ErbB3 may increase
the cell resistance to PEM treatment.
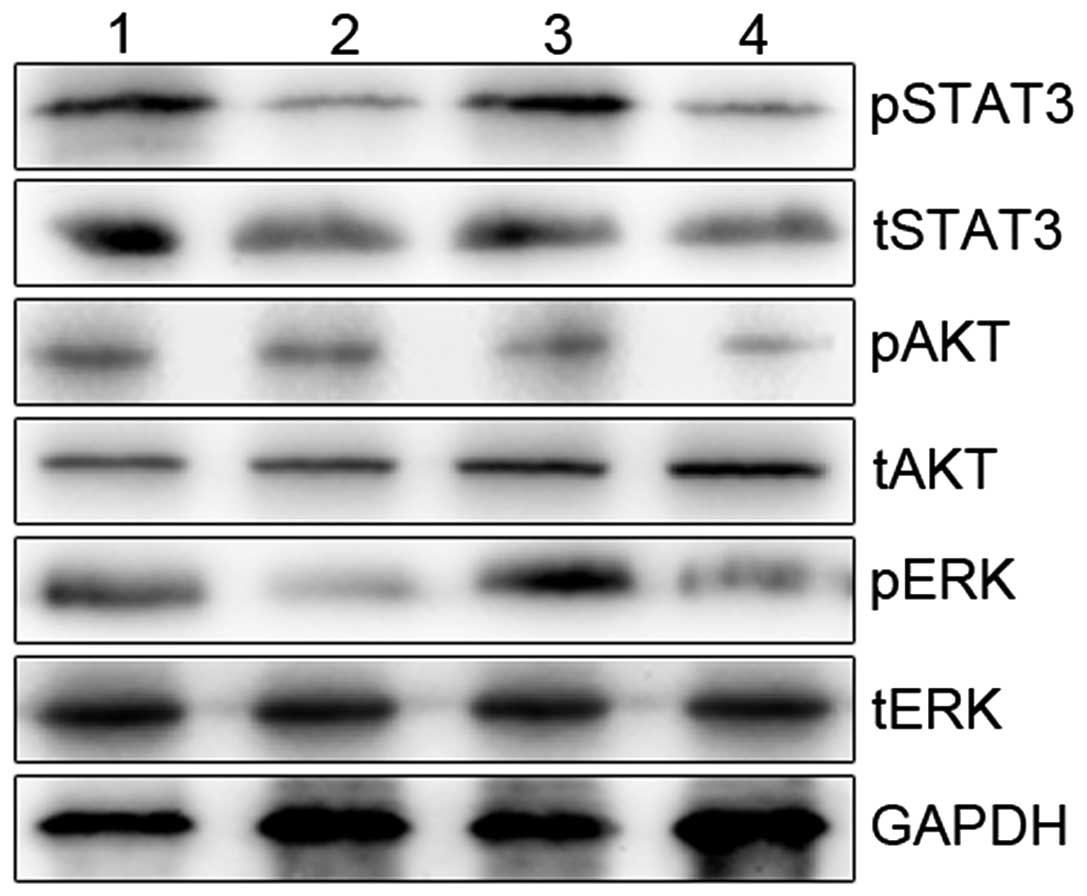
Effects of the downregulation of EGFR,
ErbB3 or EGFR/ErbB3 on EGFR/ErbB3-dependent pathways in the
A549/PEM cell lines
We next investigated the mechanistic basis for the
resistance to PEM of the A549/PEM cells (A549/PEM-sh-control,
A549/PEM-shEGFR, A5490PEM-shErbB3 and A549/PEM-shEGFR/ErbB3).
Phosphorylation of STAT3, AKT and ERK was inhibited by knockdown of
both EGFR and ErbB3 (P<0.05), whereas these levels were mildly
suppressed by silencing of ErbB3 alone (P>0.05) (Fig. 7). Downregulation of EGFR resulted in
marked inhibition of the phosphorylation of STAT3 and ERK. We
provide proof that dual silencing of EGFR and ErbB3 in A549/PEM
cells improved the sensitivity to PEM. We observed that the
phosphorylation of STAT3, AKT and ERK in the A549/PEM-shEGFR/ErbB3
cells was markedly abrogated.
Discussion
Drug resistance is a great obstacle to the
successful treatment of NSCLC. To data, it has been shown that lung
cancer multidrug resistance involves a variety of mechanisms,
including expression of drug transporters, activation of
detoxification system, structural change in targets or inactivation
of tumor-suppressor genes and activation of oncogenes (23). However, no single mechanism can
reasonably explain the primary or secondary chemotherapy resistance
phenomenon. For this reason, inducing drug-resistance in cell lines
in vitro is an important method with which to study the
mechanisms of chemotherapy resistance and investigate the functions
of potential resistance-induced genes or proteins (24). PEM-resistant A549 cell lines have
been reported in a previous study. The cell lines were exposed to
step-wise increasing concentrations of PEM (10). Cells show low resistance when the
resistance index (RI) is <5-fold, moderate resistance when RI is
5–15, and high resistance when RI is >15-fold (25). Here, we established a PEM-resistant
lung adenocarcinoma cell line successfully through high
concentration intermittence, named A549/PEM, with RI of 19.86. The
A549/PEM cell line showed a lower apoptosis rate than the A549
cells following treatment with PEM. Previous in vitro
experiments suggest that antifolate drugs can affect the cell
cycle, and cells are arrested in the G1 phase (26,27).
It is generally acknowledged that when the cycle of tumor cell
proliferation is short, the drugs targeting the DNA synthesis
process are more sensitive. Under contrary, conditions, the
sensitivity is reduced (28). The
percentage of A549/PEM cells in the S phase was decreased while the
percentage in the G1/G0 phase was increased. Thus, cells in the DNA
synthesis phase were decreased and cell proliferation was slowed
down. This is agreement with other research results, and it may be
one of the reasons for the resistance to PEM.
Although the relationship between expression of TS,
DHFR, GARFT genes and PEM resistance has been demonstrated
(29,30), we focused on ErbB genes since high
expression of EGFR and ErbB3 was observed in our established
PEM-resistant cell line. Further investigation confirmed that
downregulation of both EGFR and ErbB3 in A549/PEM cells by
lentiviral infection reversed the cell resistance to PEM. These
findings suggest that a high expression level of EGFR or ErbB3 is
one of the resistance factors for PEM. The chromosomes of lung
adenocarcinoma cells are damaged by PEM (31), and the function of damaged
chromosomes may be recovered by upregulation of EGFR or ErbB3, thus
reducing the effect of PEM. In addition, cell signal transduction
and regulation mechanisms are attributed to tumor resistance. The
major signal transduction pathways that ultimately result in
proliferative signals to the cell nucleus include
Ras/Raf/MEK/ERK/MAPK, PI3K/AKT and JAK/STAT pathways (32,33).
The altered expression of EGFR and ErbB3 and activity of these
signaling pathways may influence the resistance of lung
adenocarcinoma cells to PEM. Thus, further study was performed to
explore those pathways. Downregulation of EGFR and/or ErbB3 was
found to result in a decrease in the phosphorylation of STAT3, AKT
and ERK in the A549/PEM cell line. However, A549/PEM-shEGFR and
A549/PEM-shErbB3 cells did not exhibit reduced resistance to PEM
with the phosphorylation of AKT in common. Significant inhibition
of AKT phosphorylation almostly restored sensitivity to PEM after
silencing of both EGFR and ErbB3. The
phosphatidylinositol-3-kinase/protein base B (PI3K/AKT) is involved
in a variety of tumor biological markers as a classic survival
signaling pathway of anti-apoptosis. This pathway plays a major
role in tumor development and the potential tumor response to
chemotherapy. Studies found that sustained activation of the
PI3K/AKT pathway is related to acquired resistance to chemotherapy
(34). Together, these studies
outline that PI3K/AKT signaling may be a feasible pathway for
PEM-resistance.
Gene polymorphisms or mutations may also influence
the response to PEM. Although patients with EGFR mutations respond
to TKIs, the second-site point mutation of the EGFR is a major
cause of acquired resistance to TKIs (35,36).
Increasing EGFR gene copy may be a more valuable marker than EGFR
mutations in the prognosis of NSCLC with TKI treatment (37). Research also showed that gefitinib
treatment led to increased ErbB3 mRNA levels (38). Given that patients with
overexpression of ErbB3 in lung cancer are likely to benefit from
EGFR tyrosine kinase inhibitors (39), we may turn to EGFR tyrosine kinase
inhibitors based on the expression of EGFR and ErbB3 when tumors
treated with PEM progress. Prospective clinical studies are
required to confirm the relationship between gene amplification and
chemosensitivity or therapy transformation.
In summary, drug-resistance is the result of the
influence of multiple factors together. Upregulation of gene
expression is one of the important factors. Our results
demonstrated that overexpression of EGFR or ErbB3 may be a
predictive marker for patients treated with PEM. The possible
mechanism of PEM resistance in A549/PEM cells may be associated
with overexpression of EGFR and/or ErbB3 through the PI3K/AKT
signaling pathway. Our study showed that the expression of EGFR and
ErbB3 increased, while structure changes in the genes were
undetermined. It is possible that a secondary mutation of EGFR
hindered the effect of PEM. Therefore, further study of the
resistant mechanisms and the impact of ErbB genes in PEM resistance
is warranted.
Acknowledgements
The authors are grateful to Dr Hongming Zhai,
Department of Chemistry, Lanzhou University, Lanzhou, P.R. China,
for the assistance on the docking studies by SYBYL 7.3 software.
This study was supported by the Jieping Wu Foundation of China (no.
320.6753.1219), and the Youth Foundation of the Affiliated Hospital
of the Medical College, Qingdao University (AHMCQ201232). We also
thank the Affiliated Hospital of the Medical College, Qingdao
University Research Fund for financial support.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Herbst RS, Heymach JV and Lippman SM: Lung
cancer. N Engl J Med. 359:1367–1380. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schiller JH, Harrington D, Belani CP, et
al: Comparison of four chemotherapy regimens for advanced
non-small-cell lung cancer. N Engl J Med. 346:92–98. 2002.
View Article : Google Scholar
|
|
4
|
Assaraf YG: Molecular basis of antifolate
resistance. Cancer Metastasis Rev. 26:153–181. 2007. View Article : Google Scholar
|
|
5
|
Furrukh M, Al-Moundhri M, Zahid KF, Kumar
S and Burney I: Customised, individualised treatment of metastatic
non-small-cell lung carcinoma (NSCLC). Sultan Qaboos Univ Med J.
13:202–217. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Longo-Sorbello GS, Chen B, Budak-Alpdogan
T and Bertino JR: Role of pemetrexed in non-small cell lung cancer.
Cancer Invest. 25:59–66. 2007. View Article : Google Scholar
|
|
7
|
Scagliotti GV, Parikh P, von Pawel J, et
al: Phase III study comparing cisplatin plus gemcitabine with
cisplatin plus pemetrexed in chemotherapy-naive patients with
advanced-stage non-small-cell lung cancer. J Clin Oncol.
26:3543–3551. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Esteban E, Casillas M and Cassinello A:
Pemetrexed in first-line treatment of non-small cell lung cancer.
Cancer Treat Rev. 35:364–373. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ceppi P, Volante M, Saviozzi S, et al:
Squamous cell carcinoma of the lung compared with other histotypes
shows higher messenger RNA and protein levels for thymidylate
synthase. Cancer. 107:1589–1596. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang D, Ochi N, Takigawa N, et al:
Establishment of pemetrexed-resistant non-small cell lung cancer
cell lines. Cancer Lett. 309:228–235. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu SG, Yang CH, Yu CJ, et al: Good
response to pemetrexed in patients of lung adenocarcinoma with
epidermal growth factor receptor (EGFR) mutations. Lung
Cancer. 72:333–339. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Benavente S, Huang S, Armstrong EA, et al:
Establishment and characterization of a model of acquired
resistance to epidermal growth factor receptor targeting agents in
human cancer cells. Clin Cancer Res. 15:1585–1592. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Giltnane JM, Moeder CB, Camp RL and Rimm
DL: Quantitative multiplexed analysis of ErbB family coexpression
for primary breast cancer prognosis in a large retrospective
cohort. Cancer. 115:2400–2409. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sharma SV and Settleman J: ErbBs in lung
cancer. Exp Cell Res. 315:557–571. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hayashi M, Inokuchi M, Takagi Y, et al:
High expression of HER3 is associated with a decreased survival in
gastric cancer. Clin Cancer Res. 14:7843–7849. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mimeault M, Hauke R and Batra SK: Recent
advances on the molecular mechanisms involved in the drug
resistance of cancer cells and novel targeting therapies. Clin
Pharmacol Ther. 83:673–691. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schmidt M and Lichtner RB: EGF receptor
targeting in therapy-resistant human tumors. Drug Resist Updat.
5:11–18. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Desbois-Mouthon C: The HER3/ErbB3
receptor: a promising target in cancer drug therapy. Gastroenterol
Clin Biol. 34:255–259. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Blivet-Van Eggelpoël MJ, Chettouh H,
Fartoux L, et al: Epidermal growth factor receptor and HER-3
restrict cell response to sorafenib in hepatocellular carcinoma
cells. J Hepatol. 57:108–115. 2012.PubMed/NCBI
|
|
20
|
Lu S, Zheng W, Ji L, et al: Synthesis,
characterization, screening and docking analysis of
4-anilinoquinazoline derivatives as tyrosine kinase inhibitors. Eur
J Med Chem. 61:84–94. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhou Y, Ling XL, Li SW, Li XQ and Yan B:
Establishment of a human hepatoma multidrug resistant cell line in
vitro. World J Gastroenterol. 16:2291–2297. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ikari A, Sato T, Watanabe R, Yamazaki Y
and Sugatani J: Increase in claudin-2 expression by an
EGFR/MEK/ERK/c-Fos pathway in lung adenocarcinoma A549 cells.
Biochim Biophys Acta. 1823:1110–1118. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kaszubiak A, Holm PS and Lage H:
Overcoming the classical multidrug resistance phenotype by
adenoviral delivery of anti-MDR1 short hairpin RNAs and ribozymes.
Int J Oncol. 31:419–430. 2007.PubMed/NCBI
|
|
24
|
Amati P and Lago C: Sensitivity to
amphotericin B of in vitro established cell lines. Nature.
247:466–469. 1974. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Snow K and Judd W: Characterisation of
adriamycin- and amsacrine-resistant human leukaemic T cell lines.
Br J Cancer. 63:17–28. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lu X, Errington J, Curtin NJ, Lunec J and
Newell DR: The impact of p53 status on cellular sensitivity
to antifolate drugs. Clin Cancer Res. 7:2114–2123. 2001.
|
|
27
|
Hsu HF, Huang KH, Lu KJ, et al:
Typhonium blumei extract inhibits proliferation of human
lung adenocarcinoma A549 cells via induction of cell cycle arrest
and apoptosis. J Ethnopharmacol. 135:492–500. 2011. View Article : Google Scholar
|
|
28
|
Heinemann L, Simpson GR, Annels NE, et al:
The effect of cell cycle synchronization on tumor sensitivity to
reovirus oncolysis. Mol Ther. 18:2085–2093. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hanauske AR, Eismann U, Oberschmidt O, et
al: In vitro chemosensitivity of freshly explanted tumor cells to
pemetrexed is correlated with target gene expression. Invest New
Drugs. 25:417–423. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ozasa H, Oguri T, Uemura T, et al:
Significance of thymidylate synthase for resistance to pemetrexed
in lung cancer. Cancer Sci. 101:161–166. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu MF, Hsiao YM, Huang CF, et al: Genetic
determinants of pemetrexed responsiveness and nonresponsiveness in
non-small cell lung cancer cells. J Thorac Oncol. 5:1143–1151.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sebastian S, Settleman J, Reshkin SJ,
Azzariti A, Bellizzi A and Paradiso A: The complexity of targeting
EGFR signalling in cancer: from expression to turnover. Biochim
Biophys Acta. 1766:120–139. 2006.PubMed/NCBI
|
|
33
|
Durmuş Tekir S, Yalçin Arga K and Ulgen
KO: Drug targets for tumorigenesis: insights from structural
analysis of EGFR signaling network. J Biomed Inform. 42:228–236.
2009.PubMed/NCBI
|
|
34
|
Fresno Vara JA, Casado E, de Castro J,
Cejas P, Belda-Iniesta C and González-Barón M: PI3K/Akt signalling
pathway and cancer. Cancer Treat Rev. 30:193–204. 2004.PubMed/NCBI
|
|
35
|
Kosaka T, Yatabe Y, Endoh H, et al:
Analysis of epidermal growth factor receptor gene mutation in
patients with non-small cell lung cancer and acquired resistance to
gefitinib. Clin Cancer Res. 12:5764–5769. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yamamoto H, Toyooka S and Mitsudomi T:
Impact of EGFR mutation analysis in non-small cell lung
cancer. Lung Cancer. 63:315–321. 2009.
|
|
37
|
Cappuzzo F, Hirsch FR, Rossi E, et al:
Epidermal growth factor receptor gene and protein and gefitinib
sensitivity in non-small-cell lung cancer. J Natl Cancer Inst.
97:643–655. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Grøvdal LM, Kim J, Holst MR, Knudsen SL,
Grandal MV and van Deurs B: EGF receptor inhibitors increase ErbB3
mRNA and protein levels in breast cancer cells. Cell Signal.
24:296–301. 2012.PubMed/NCBI
|
|
39
|
Kawano O, Sasaki H, Endo K, et al: ErbB3
mRNA expression correlated with specific clinicopathologic features
of Japanese lung cancers. J Surg Res. 146:43–48. 2008. View Article : Google Scholar : PubMed/NCBI
|