Introduction
Mitogen-inducible gene-6 (Mig-6) is an immediate
early response gene that can be induced by a variety of external
stimuli, including growth factors, hypoxia and stress (1). The Mig-6 locus (chromosome 1p36.12–33)
falls within the region 1p36.1–3 and exhibits a high frequency of
allelic loss in various human cancer types (2–4). Mig-6
downregulation has been reported in various cancer types, and
accumulating evidence suggests that Mig-6 is a tumor-suppressor
gene in both mice and humans (5–9). A
study by Amatschek et al (13) found that Mig-6 is downregulated in
breast cancer patients with short survival time. That study
indicated that Mig-6 may have a potential prognostic value in human
cancer. Mig-6 binds to epidermal growth factor receptor (EGFR)
family tyrosine kinases via its EGFR-binding domain thus leading to
inhibition of EGFR autophosphorylation and reduced MAPK activity
(10–13). Many experiments confirmed that Mig-6
exhibits a measurable effect on tumor cell proliferation and
invasion, but its role in apoptosis remains unclear.
Our previously reported study demonstrated that
Mig-6 is downregulated in non-small cell lung cancer (NSCLC) and
knockdown of Mig-6 expression in H1299 and BE1 cells promotes
EGF-induced lung cancer cell proliferation and migration (14). However, little is known about the
role of Mig-6 in NSCLC apoptosis, nor has the contribution of
upregulated Mig-6 on biological behaviors of A549 and H157 cells
previously been reported.
In the present study, statistical analysis was
performed on the potential correlation between Mig-6 expression and
poor survival outcome after lung cancer surgery. The increase of
the Mig-6 expression affecting proliferation, invasion of A549 and
H157 cells were examined. We also investigated the effects and
regulatory mechanisms underlying the impact of Mig-6 on apoptosis
in human NSCLC cells.
Materials and methods
Patients and specimens
The present study was conducted with the approval of
the Ethics Committee of China Medical University. Primary tumor
specimens were obtained from the First Affiliated Hospital of China
Medical University during the period of 2008 to 2013. Through the
surgery consent form, patients were informed that the resected
specimens were kept by our hospital and may be used for scientific
research, and that their privacy would be maintained. Follow-up
information was obtained by reviewing patient medical records. None
of the patients received radiotherapy or chemotherapy before
surgical resection, and all patients were treated with routine
chemotherapy after the operation. The histological diagnosis and
grade of differentiation of the tumors were defined by evaluation
of the hematoxylin and eosin-stained tissue sections, according to
the World Health Organization guidelines of classification. All 150
specimens were re-evaluated with respect to their histological
subtypes, differentiation status and tumor stages. For NSCLC
samples, squamous-cell carcinoma (SCC) and adenocarcinoma were
identified in 69 and 81 of the 150 cases, respectively. Lymph node
metastases were observed in 60 of the 150 patients. The p-TNM
staging system of the International Union Against Cancer (7th
edition) was used to classify specimens.
Cell culture and transfection
A549 and H1299 were obtained from the American Type
Culture Collection (Manassas, VA, USA), and H157 was purchased from
the Cell Bank, Chinese Academy of Sciences (Shanghai, China). The
BE1 were gifts from Dr Jie Zheng (College of Medicine, Beijing
University, China). The cells were cultured in RPMI-1640 containing
10% fetal calf serum (both from Invitrogen), 100 IU/ml penicillin
and 100 μg/ml streptomycin (both from Sigma). The cells were seeded
in 6-well plates 24 h prior to the experiment. Transfection with
siRNA for Mig-6 or non-targeting siRNA (GenePharma, Shanghai,
China), was conducted using DharmaFECT 1 (5 μl/well; Thermo Fisher
Scientific, Rockford, IL, USA) according to the manufacturer’s
recommendations. The sequences of siRNAs were: sense,
5′-GGAUAUCCAACUGUUGUAUTT-3′ and antisense,
5′-AUACAACAGUUGGAUAUCCTT-3′ (6).
For control scrambled siRNA: sense, 5′-UUCUCCGAACGUGUCACGUTT-3′ and
antisense, 5′-ACGUGACACGUUCGGAGAATT-3′. Cells were transfected with
control vector (pcDNA3) or Mig-6 overexpression vector
(pcDNA3-Mig-6) (both from Takara Biotechnology, Dalian, China)
using Attractene transfection reagent (Qiagen, Valencia, CA, USA)
according to the manufacturer’s protocol. The mRNA and protein
levels were assessed 48 h following transfection.
Immunohistochemistry
Surgically excised tumor specimens were fixed with
10% neutral formalin, embedded in paraffin and 4-μm thick sections
were prepared. Immunostaining was performed using the
avidin-biotin-peroxidase complex method (Ultra Sensitive TM,
Maixin, Fuzhou, China). The sections were deparaffinized in xylene,
rehydrated in graded alcohol series and boiled in 0.01 M citrate
buffer (pH 6.0) for 2 min in an autoclave. Endogenous peroxidase
activity was blocked using hydrogen peroxide (0.3%), which was
followed by incubation with normal goat serum to reduce
non-specific binding. Tissue sections were incubated with Mig-6
rabbit polyclonal antibody (1:100 dilution; Sigma). Rabbit
immunoglobulin (at the same concentration as the antigen specific
antibody; Maixin) was used as a negative control. Staining for all
primary antibodies was performed at room temperature for 2 h.
HRP-polymer conjugated anti-mouse/rabbit IgG (ready-to-use; Maixin)
was used as the secondary antibody. After washing, the sections
were incubated with horseradish peroxidase-conjugated
streptavidin-biotin, followed by 3,3′-diaminobenzidine
tetrahydrochloride to develop the peroxidase reaction.
Counterstaining of the sections was carried out with hematoxylin,
and they were then dehydrated in ethanol before mounting. Two
independent investigators examined all tumor slides randomly. Five
views were examined per slide, and 100 cells were observed per view
at ×400 magnification. Immunostaining of Mig-6 was scored following
a semi-quantitative scale by evaluating in representative tumor
areas. The intensity of Mig-6 staining was scored as 0 (no
staining); 1 (weak) and 2 (marked). Percentage scores were assigned
as 1, 1–25%; 2, 26–50%; and 3, 51–100%. The scores of each tumor
sample were multiplied to give a final score of 0–6 and the total
expression of Mig-6 was determined as either negative or low
expression (−), score <3 or positive expression or high
expression (+), score ≥3.
Western blot analysis
Total protein from cell lines were extracted in
lysis buffer (Thermo Fisher Scientific) and quantified using the
Bradford method. Sixty micrograms of protein were separated by
SDS-PAGE (12%). After transferring, the polyvinylidene fluoride
(PVDF) membranes (Millipore, Billerica, MA, USA) were incubated
overnight at 4°C with the following antibodies: Mig-6 (1:2,000;
Sigma), Bcl-2 and β-actin (1:500; Santa Cruz Biotechnology, Santa
Cruz, CA, USA), anti-phospho-ERK (1:2,000; Cell Signaling
Technology, Danvers, MA, USA). After incubation with
peroxidase-coupled anti-mouse or rabbit IgG (Santa Cruz
Biotechnology) at 37°C for 2 h, bound proteins were visualized
using ECL (Thermo Fisher Scientific) and detected using BioImaging
Systems (UVP Inc., Upland, CA, USA). The relative protein levels
were calculated based on β-actin as the loading control.
Quantitative real-time PCR (SYBR-Green
method)
Quantitative real-time PCR was performed using
SYBR-Green PCR master mix in a total volume of 20 μl on 7900HT Fast
Real-Time PCR System (both from Applied Biosystems) as follows:
95°C for 30 sec, 40 cycles of 95°C for 5 sec, 60°C for 30 sec. A
dissociation step was performed to generate a melting curve to
confirm the specificity of the amplification. β-actin was used as
the reference gene. The relative levels of gene expression were
represented as ΔCt = Ct gene - Ct reference, and the fold-change of
gene expression was calculated by the 2−ΔΔCt method.
Cell apoptosis experiments
Apoptosis was detected using an Annexin V-FITC/PI
double staining kit (KeyGen, Nanjing, China). Cells were harvested
and washed twice with cold phosphate-buffered saline (PBS) by
gentle shaking. Cells were then resuspended and added to binding
buffer (1X); cell density was adjusted to 2–5×105/ml. In
the dark, 5 μl Annexin V-FITC was added to the cell suspension
volume of 195 μl and incubated for 10 min at room temperature
before the addition of 190 μl binding buffer (1X) and 10 μl PI. Ten
thousand events per sample were acquired using a FACScan flow
cytometer and the percentage of cell apoptosis was analyzed using
CellQuest analysis software (both from Becton-Dickinson, San Jose,
CA, USA).
Cell proliferation test
Cell proliferation assay was performed using Cell
Counting Kit-8® solution (Dojindo, Gaithersburg, MD,
USA) according to the manufacturer’s protocol. Briefly, cells were
seeded at a concentration of 5×103 cells/100 μl/well in
96-well culture plates and treated with 10 μl/well of Cell Counting
Kit-8® solution during the last 4 h of the culture.
Optical density of the wells was measured at 450 nm using a
microplate reader.
Colony formation assay
For colony formation assays, cells were grown on 6
cm dishes at a density of 5,000 cells/dish, and transfected with PC
or Mig-6. Colonies were washed twice with PBS, fixed, and stained
with formaldehyde-crystal violet 13–15 days after transfection. All
experiments were independently repeated a minimum of three times
under identical conditions.
Matrigel invasion assay
Cell invasion assay was performed using a 24-well
Transwell chamber with a pore size of 8 μm (Costar, Cambridge, MA,
USA). The inserts were coated with 20 μl Matrigel (1:3 dilution; BD
Biosciences, San Jose, CA, USA). Forty-eight hours after the
transfection, cells were trypsinized and 3×105 cells in
100 μl of serum-free medium were transferred to the upper Matrigel
chamber and incubated for 16 h. After incubation, the non-invaded
cells on the upper membrane surface were removed with a cotton tip,
and the cells that passed through the filter were fixed with 4%
paraformaldehyde and stained with hematoxylin. The number of
invaded cells was counted in 10 randomly selected high power fields
under the microscope. This experiment was performed in
triplicate.
Statistical analysis
SPSS version 19.0 for Windows was used for all
analyses. The χ2 test was used to examine possible
correlations between Mig-6 expression and clinicopathological
factors. The Kaplan-Meier method was used to estimate the
probability of patient survival, and differences in the survival of
subgroup of patients were compared by using log-rank test. The Cox
regression mode laws were used for multivariate analysis. Student’s
t-test was used to compare differences between groups. p-values
were based on the two-sided statistical analysis, and p<0.05 was
considered to indicate a statistically significant difference.
Results
Correlation between Mig-6 expression and
survival
We first stratified these 150 lung cancer patients
as Mig-6-positive and -negative groups according to the Mig-6
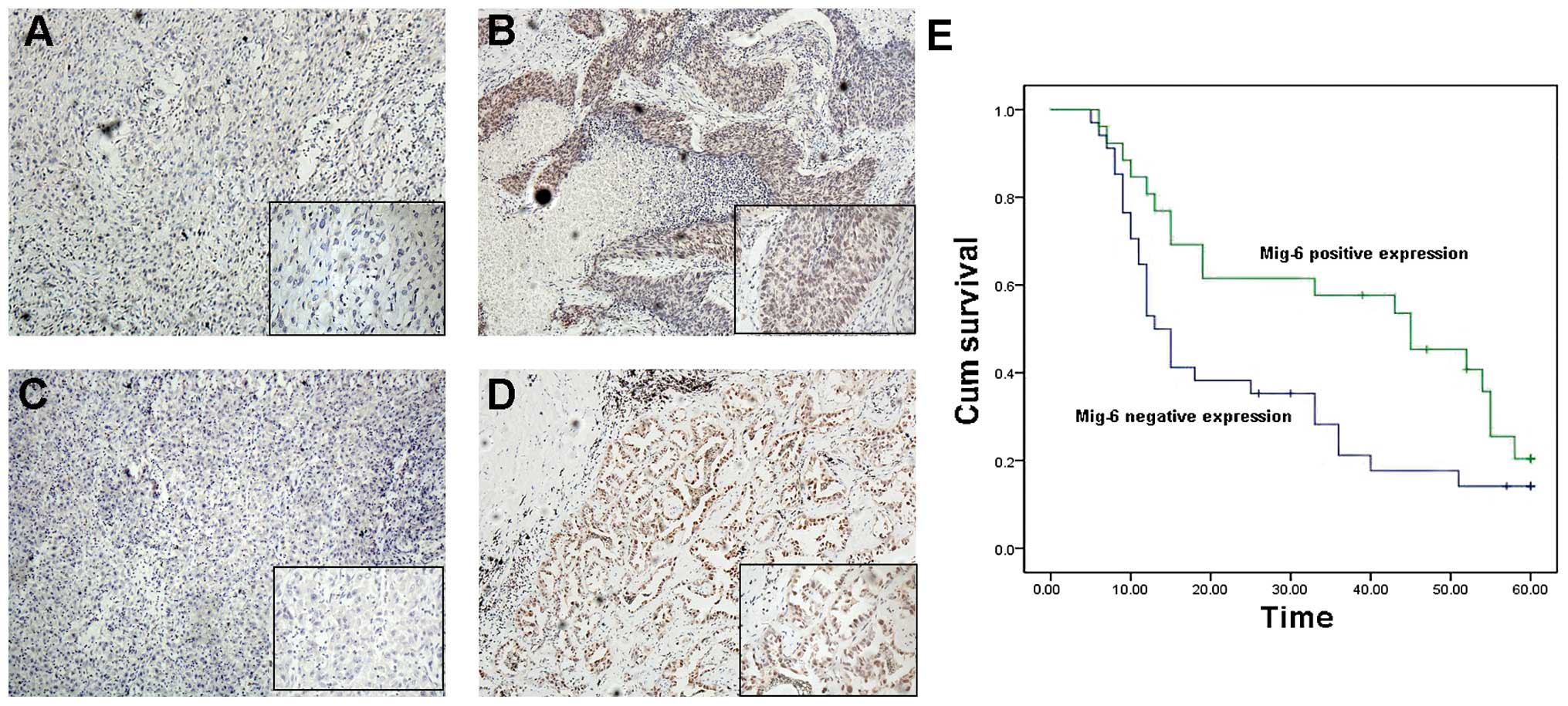
staining of tumor sections (Fig.
1A–D). As shown in Table I,
statistical differences were found between Mig-6 expression and the
characteristics of tumor size (T1+T2 vs. T3+T4, p=0.002),
differentiation (high vs. poor-moderate, p=0.002), TNM stage (I+II
vs. III+IV, p=0.014), as well as histological type (adenocarcinoma
vs. squamous cell carcinoma, p=0.005). We followed up on 60
patients and divided them into two groups according to Mig-6
expression. Patients with high expression of Mig-6 had a
statistically significantly longer survival (median survival,
45±11.132 months; 95% CI, 23.182–66.818 months) than those with low
expression of Mig-6 (median survival, 13±1.458 months; 95% CI,
10.143–15.857 months). Furthermore, a multivariate analysis using a
Cox regression model indicated that pTNM stage and loss of Mig-6
expression were independent, unfavorable prognostic factors [TNM
stage: Exp (B), 3.715, 95.0% CI, 2.227–6.198, p=0.000; Mig-6
expression: Exp (B), 0.329, 95.0% CI, 0.152–0.713, p=0.005)
(Table II)].
 | Table IRelationship between Mig-6 expression
in NSCLC and clinical pathological factors. |
Table I
Relationship between Mig-6 expression
in NSCLC and clinical pathological factors.
| | Mig-6 |
|---|
| |
|
|---|
| Characteristics | Patients | Positive n (%) | Negative n (%) | P-value |
|---|
| Age (years) | | | | 1.000 |
| <60 | 80 | 37 (46.25) | 43 (53.75) | |
| ≥60 | 70 | 33 (47.14) | 37 (52.86) | |
| Gender | | | | 0.324 |
| Male | 88 | 38 (43.18) | 50 (56.82) | |
| Female | 62 | 32 (51.61) | 30 (48.39) | |
| Histology | | | | 0.005 |
| ADC | 81 | 52 (64.20) | 29 (35.80) | |
| SCC | 69 | 28 (40.58) | 41 (59.42) | |
| Differentiation | | | | 0.002 |
| Well | 47 | 31 (65.96) | 16 (34.04) | |
| Moderate-Poor | 103 | 39 (37.86) | 64 (62.14) | |
| TNM stage | | | | 0.014 |
| I+II | 80 | 45 (56.25) | 35 (43.75) | |
| III+IV | 70 | 25 (35.71) | 45 (64.29) | |
| Tumor size | | | | 0.002 |
| T1+T2 | 65 | 40 (60.71) | 25 (39.29) | |
| T3+T4 | 85 | 30 (42.86) | 55 (57.14) | |
| Nodal status | | | | 0.323 |
| − | 90 | 39 (43.33) | 51 (56.67) | |
| + | 60 | 31 (51.67) | 29 (48.33) | |
| EGFR status | | | | 0.000 |
| Negative | 62 | 41 (66.67) | 21 (33.33) | |
| Positive | 88 | 29 (34.62) | 59 (65.38) | |
| Table IIMultivariate analysis for predictive
factors in patients with NSCLC (Cox regression model). |
Table II
Multivariate analysis for predictive
factors in patients with NSCLC (Cox regression model).
| Factors | Wald | Exp (B) | 95% CI for exp
(B) | P-value |
|---|
| Tissue
histology | 3.655 | 1.986 | 0.983–4.012 | 0.056 |
|
Differentiation | 0.209 | 0.839 | 0.395–1.781 | 0.648 |
| Gender | 0.009 | 1.030 | 0.554–1.915 | 0.925 |
| Age (years) | 2.644 | 1.028 | 0.994–1.064 | 0.104 |
| TNM stage | 25.251 | 3.715 | 2.227–6.198 | 0.000 |
| Mig-6
expression | 7.927 | 0.329 | 0.152–0.713 | 0.005 |
Mig-6 promotes apoptosis of NSCLC cell
lines by inhibiting the expression of Bcl-2
To determine whether Mig-6 influences the apoptosis
of NSCLC cells, we explored Mig-6 knockdown and overexpression.
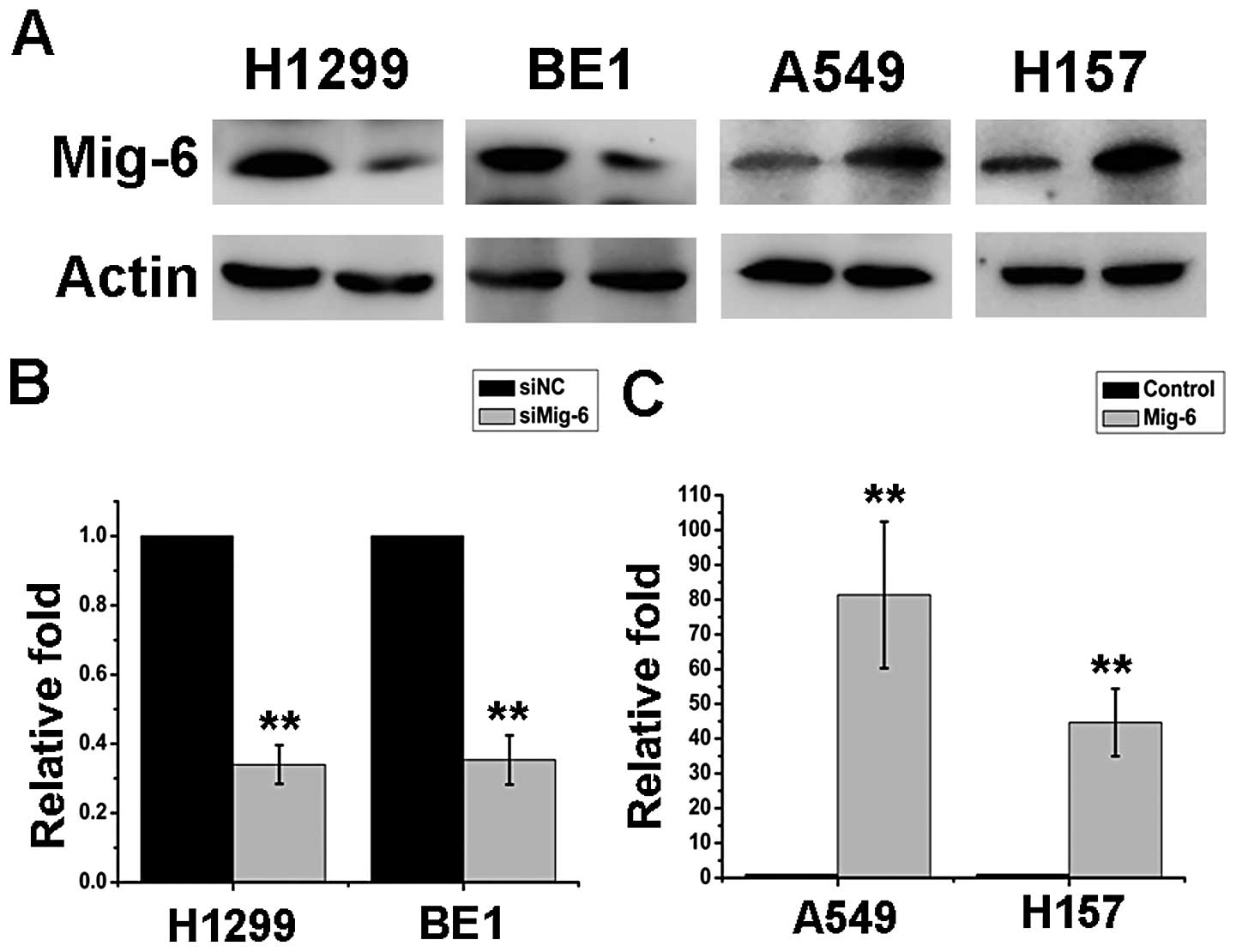
Interference efficiency was examined by mRNA and protein expression
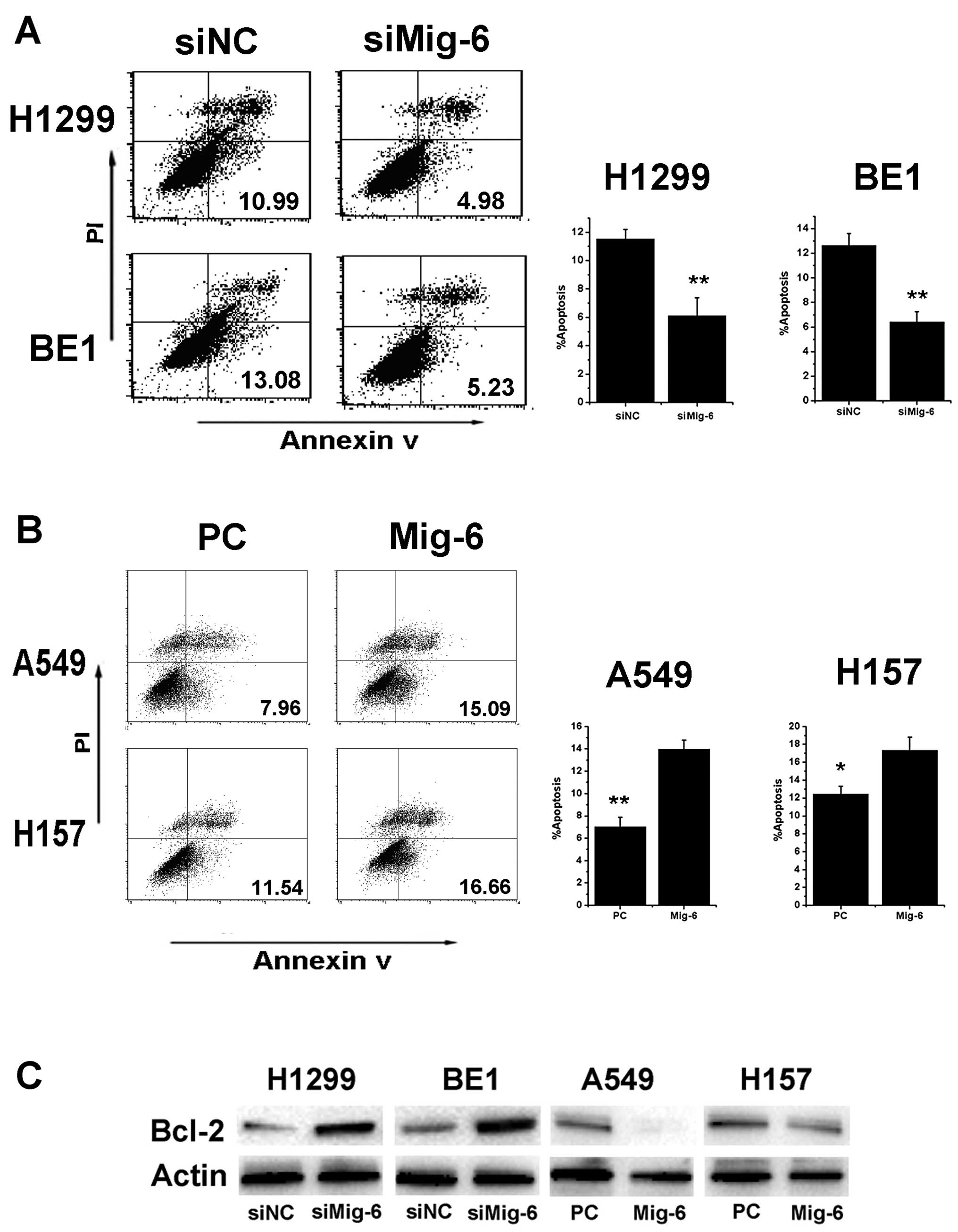
levels after 48 h of transfection treatment (Fig. 2A–C). Annexin V staining was
performed using flow cytometry to determine the effect of Mig-6 on
apoptosis in NSCLC cells. Mig-6 knockdown decreased apoptosis in
H1299 and BE1 cells (H1299 control vs. Mig-6 siRNA: 11.54±0.65 vs.
6.10±1.29%, p<0.01; BE1 control vs. Mig-6 siRNA: 12.63±0.96 vs.
6.42±0.83%, p<0.01) (Fig. 3A).
Mig-6 overexpression increased apoptosis in A549 and H157 cells
(A549 empty vector vs. Mig-6 plasmid: 7.05±0.82 vs. 13.98±0.79%,
p<0.01; H157 empty vector vs. Mig-6 plasmid: 12.46±0.87 vs.
17.35±1.44%, p<0.05) (Fig.
3B).
It has been established that apoptosis within cells
is chiefly involved in the expression of Bcl-2, Bax, or p53
(15–17). To determine a possible mechanism by
which Mig-6 promotes apoptosis of NSCLC cells, western blotting was
performed. As shown in Fig. 3C the
expression at both the protein and mRNA levels of anti-apoptotic
Bcl-2 was upregulated in the Mig-6 depletion group but
downregulated in the Mig-6 overexpression group. There were no
significant differences in Bax and p53 expression between each
group (data not shown). These results indicated that the function
of Mig-6 as a promotor of apoptosis in NSCLC cells is predominantly
implemented possibly by the pathways of Bcl-2 but not Bax and
p53.
Mig-6 downregulates Bcl-2 expression
through the ERK signaling pathway
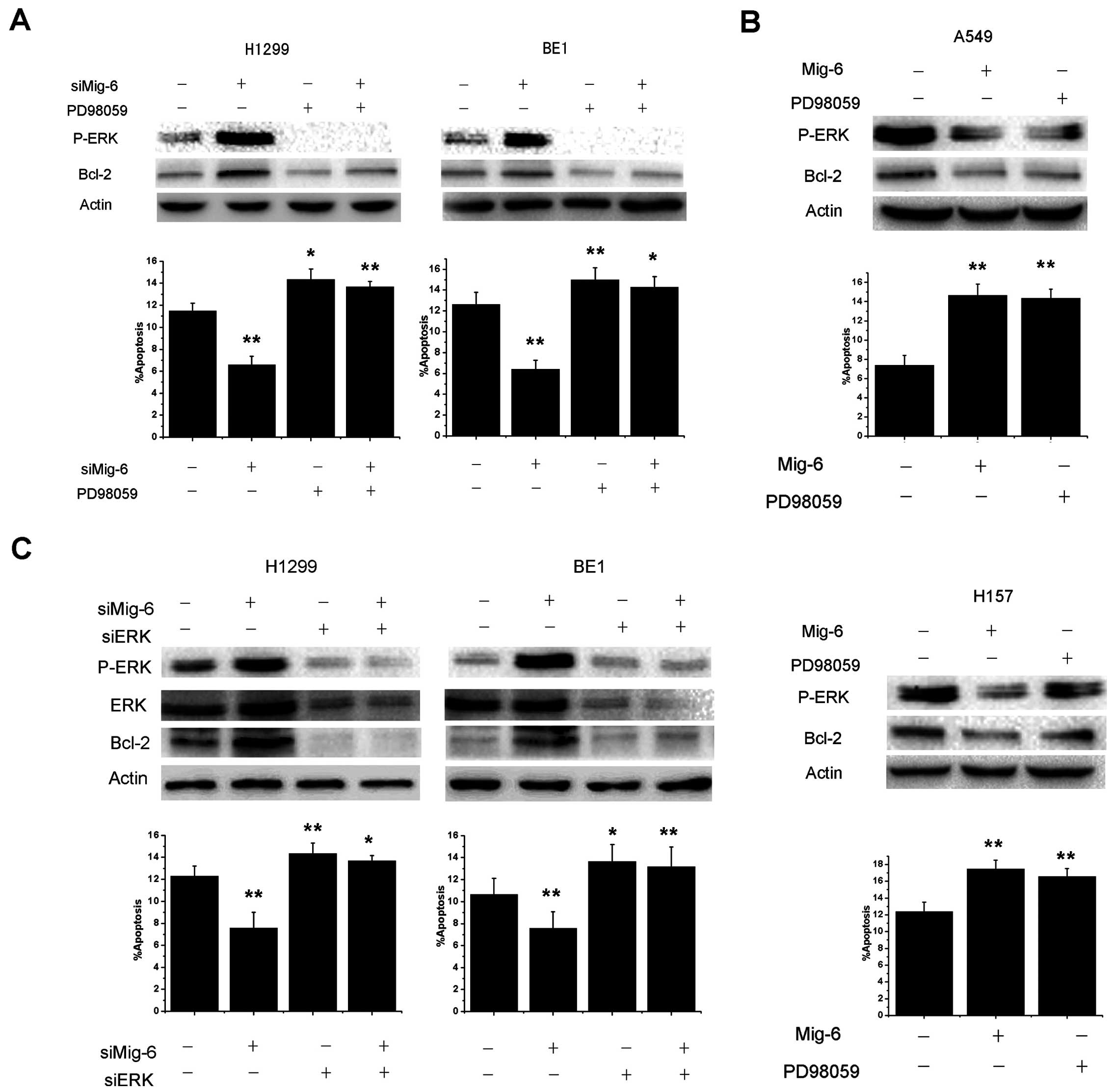
Our previous study confirmed that the suppression of
Mig-6 by a specific siRNA led to a marked increase in EGFR-ERK
signaling (14). Numerous reports
have shown that the ERK pathway plays an important role in the
apoptosis of NSCLC (18–20). We investigated whether ERK mediated
Mig-6 induction of Bcl-2 downregulation. Cells were treated with
PD98059, a selective inhibitor of MEK that disrupts the activation
of downstream ERK, for 1 h. We found that basal Bcl-2 production
and anti-apoptosis ability were inhibited by treatment of H1299 and
BE1 cells with 50 mol/l PD98059. Furthermore, PD98059 significantly
abolished Mig-6 siRNA-mediated Bcl-2 production and anti-apoptotic
effects (Fig. 4A). Consequently, we
overexpressed Mig-6 in A549 and H157 cells, we also blocked ERK
activities using PD98059. As shown in Fig. 4B, Mig-6 and PD98059 can inhibit the
expression of P-ERK and Bcl-2. Similar to Mig-6 overexpression,
inhibition of ERK by PD98059 can also promote apoptosis of both
cell lines. To further validate the role of ERK signaling in
Mig-6-induced apoptosis, we used siRNAs to deplete ERK1/2
expression in H1299 and BE1 cells. Then, we transfected Mig-6 siRNA
and examined Bcl-2 protein expression, as well as the apoptosis
ability of cells. As shown in Fig.
4C, the effect of Mig-6 siRNA-mediated Bcl-2 production and
anti-apoptotic effects were abolished in ERK1/2-depleted H1299 and
BE1 cell lines.
Effect of Mig-6 upregulation on the
proliferation and invasive potential of transfected cells
A549 and H157 cells were used in the present study
to investigate the effects of Mig-6 upregulation on proliferation
and invasion. These cells have low endogenous Mig-6 protein levels.
The Mig-6 expression levels were unchanged upon transient
transfection with the empty vector, whereas pcDNA3-Mig-6 plasmid
significantly upregulated both mRNA as well as protein expression
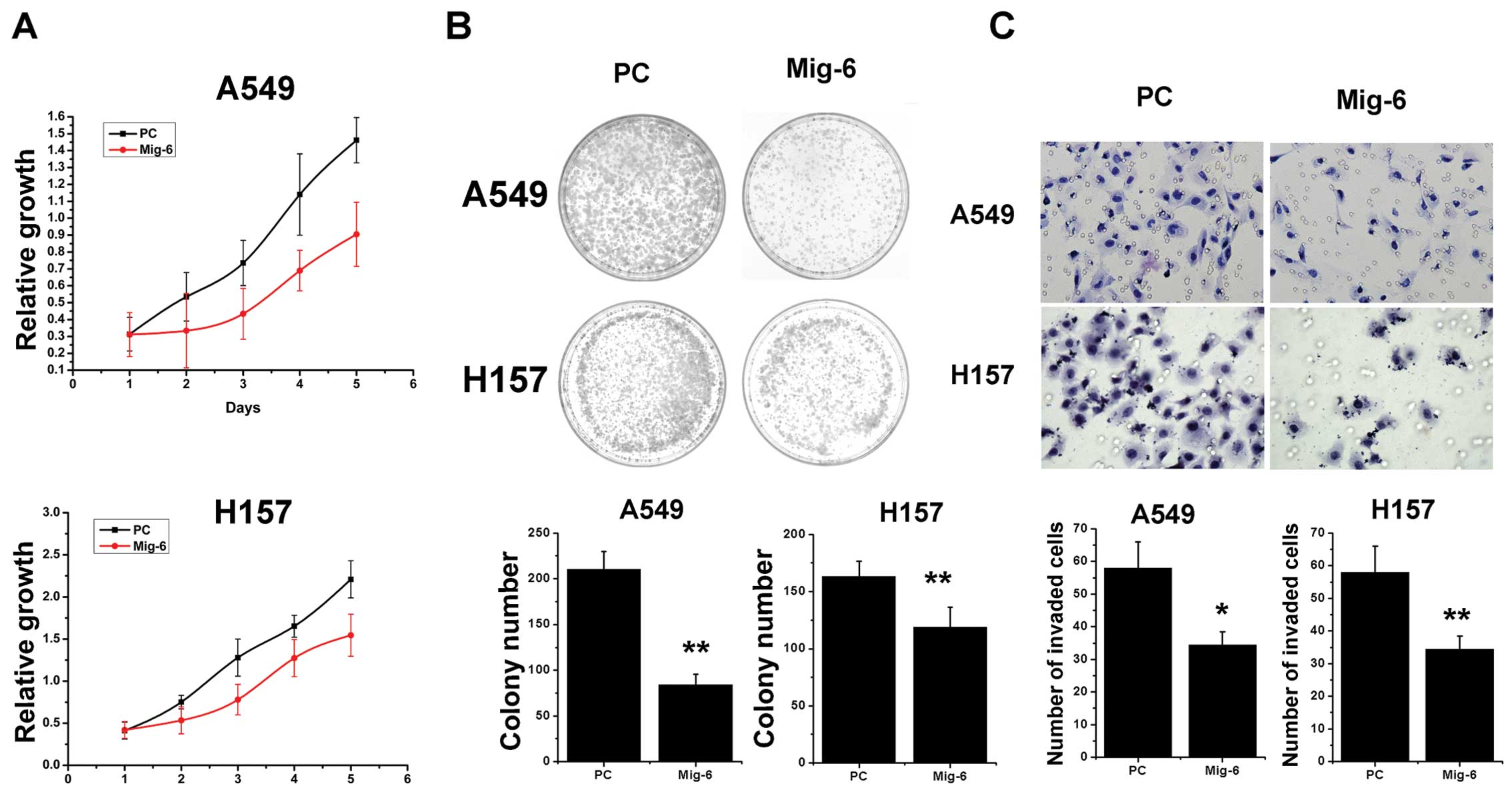
levels in the A549 and H157 cell lines (Fig. 2A). Next, we examined the cell growth
rate. With pcDNA3-Mig-6 plasmid transfection, A549 cells displayed
reduced growth rates when compared to the control (Fig. 5A). Similar to A549 cells, we found
that upregulation of Mig-6 also reduced the growth rate of H157
cells (Fig. 5A). We utilized an
independent method, colony formation assay, to validate the
antiproliferative effects of Mig-6 in lung cancer cells.
pcDNA3-Mig-6 plasmid led to a clear reduction of the colony
formation capacity of two tested lung cancer cell lines compared to
empty vector control (empty vector vs. Mig-6 plasmid: A549,
210.33±19.66 vs. 84.33±10.97, p<0.01; H157, 163.33±13.01 vs.
119±17.35, p<0.01) (Fig. 5B).
These overexpresssion studies demonstrated that Mig-6 inhibits
tumor cell proliferation. To further examine whether Mig-6
contributes to the invasive capabilities of NSCLC cells, we
conducted Matrigel invasion assays. The expression of Mig-6 was
enhanced in transfected cells in comparison to the control cells.
Our results demonstrated that the invasive capabilities of Mig-6
overexpressed A549 and H157 cancer cells were reduced compared to
the control cells (empty vector vs. Mig-6 plasmid: A549, 58±8 vs.
34.33±4.04, p<0.05; H157, 53.67±5.13 vs. 16.33±3.21, p<0.01)
(Fig. 5C). Collectively, these
results demonstrate that increased expression of Mig-6
significantly inhibited invasion of A549 and H157 cells.
Discussion
Mig-6 downregulation has been observed in various
types of cancer. Loss of Mig-6 in mice leads to impaired
differentiation of epidermal keratinocytes and these Mig-6 ablation
mice are highly susceptible to carcinogen-induced formation of
papillomas and melanomas (6).
Disruption of Mig-6 in mice also causes lung, gallbladder and bile
duct carcinogenesis (7). In
papillary thyroid cancer, tumor size was inversely correlated with
Mig-6 expression (21). In
immunohistochemical analysis using a tissue microarray of 111 liver
cancer patients, Reschke et al found that Mig-6 expression
was barely detectable in the majority of analyzed tumors (64%)
(22). We previously demonstrated
that Mig-6 expression was downregulated or absent in NSCLC
(14).
Despite the strong association between Mig-6
expression and cancer, reports on Mig-6 expression-based outcome in
tumor patients are limited. Using real-time polymerase chain
reaction (PCR), Ruan et al found that Mig-6 expression is
independently predictive of disease-free-survival in
BRAFV600E patients (23). Amatschek et al demonstrated
that the Mig-6 gene is downregulated relative to normal tissues in
patients with short survival using cDNA microarray analysis
(13). These data suggest that
Mig-6 expression may correlate closely with poor prognosis in some
types of human cancer.
However, the protein expression status of Mig-6
correlation with the outcome of the cancer patient has not been
elucidated. In the present study, we found Mig-6 downregulation
correlated with a poor prognosis in patients. We also found Mig-6
expression was lower in NSCLCs of poor differentiation and advanced
stage. Cox regression multivariate analysis showed tumor stage and
Mig-6 were the strongest predictors of survival. Our results
suggest a critical effect of Mig-6 silencing in tumorigenesis and
aggression of NSCLC.
The biological functions of Mig-6 low expression
have been investigated in several cell lines. Downregulation of
Mig-6 promotes proliferation and invasion in glioblastoma
multiforme (GBM) and PTC cell lines (21,24).
Mig-6 is also an endogenous inhibitor of EGF-induced cell migration
in human live cancer cell line HepG2 (22). Our previous study showed that Mig-6
specific siRNA promotes proliferation and invasion of H1299 and BE1
cells. However, how restored Mig-6 expression in lung cancer cells
affects proliferation and invasion of A549 and H157 cells has not
been fully clarified. In the present study, we further examined the
effects of exogenously transfected Mig-6 on proliferation, invasion
and apoptosis of A549 and H157 cells. We concluded that upregulated
Mig-6 decreased proliferated and invasive cells, and increased
apoptotic cells. Our data suggest that exogenously expressed Mig-6
may effectively inhibit progression of lung cancer.
The role of Mig-6 in apoptosis remains unclear. Our
findings that Mig-6 promotes apoptosis in NSCLC cell lines are in
concordance with previous reports in endometrial cancer (25), but appear inconsistent with the
findings reported in breast cancer cell lines (11). This observation led us to extend our
investigation into the underlying mechanisms of Mig-6-induced
apoptosis. Bcl-2 was one of the regulators of the cell intrinsic
apoptosis pathway, which regulates the integrity of the outer
mitochondria membrane (26). In
agreement with results from different cell models, our results
showed that Mig-6 specific siRNA prevented apoptosis, resulting in
an increase in the expression of anti-apoptotic Bcl-2. Next, we
investigated how Mig-6 specific siRNA increases expression of
Bcl-2. Our previous study showed that Mig-6 specific siRNA promoted
P-ERK expression. P-ERK, as a downstream factor of Mig-6, is
involved in the regulation of cell proliferation, differentiation,
apoptosis and cell cycle arrest, as well as the induction of drug
resistance (27,28). The Mig-6 siRNA-mediated Bcl-2
overexpression and anti-apoptotic effects can be reversed by ERK
inhibitor PD98059 or ERK siRNA. Furthermore, Mig-6 overexpression
or use of PD98059 can all reduce the expression of P-ERK, inhibit
the Bcl-2 production, which lead to the increase of the apoptosis
of cells. These results demonstrate that the effect of Mig-6 on
apoptosis involved in the expression of Bcl-2 may occur via the ERK
pathway in human NSCLC cells.
In conclusion, we have identified Mig-6 as a
potential biomarker for evaluation of tumor prognosis of lung
cancer. Our findings also suggest the potential important role of
Mig-6 in the control of lung cell apoptosis, an activity that may
be responsible, at least in part, for the development and/or
progression of lung cancer.
Meanwhile, the present study provides further
evidence for our previous results that Mig-6 plays a critical role
in inhibiting lung cancer cell proliferation and invasion. The
upregulation of Mig-6 may provide a helpful strategy for inhibitory
therapies of NSCLC.
Acknowledgements
The authors thank Dr Oreste Segatto (Regina Elena
Cancer Institute, Via Delle Messi d’Oro 156, Rome 00158, Italy) for
kindly providing the pcDNA3 vector- and pcDNA3-Mig-6 overexpression
vector. This study was supported by grants from the National
Natural Science Foundation of China (no. 30972967), and the
Specialized Research Fund for the Doctoral Program of Higher
Education (no. 20092104110018), and the Program for Liaoning
Excellent Talents in University. We thank International Science
Editing (Shannon Free Zone-West, Shannon, Co., Clare Ireland) for
the critical review of the manuscript.
References
|
1
|
Zhang YW and Vande Woude GF: Mig-6, signal
transduction, stress response and cancer. Cell Cycle. 6:507–513.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ogunbiyi OA, Goodfellow PJ, Gagliardi G,
et al: Prognostic value of chromosome 1p allelic loss in colon
cancer. Gastroenterology. 113:761–766. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Koshikawa K, Nomoto S, Yamashita K,
Ishigure K, Takeda S and Nakao A: Allelic imbalance at 1p36 in the
pathogenesis of human hepatocellular carcinoma.
Hepatogastroenterology. 51:186–191. 2004.PubMed/NCBI
|
|
4
|
Tseng RC, Chang JW, Hsien FJ, et al:
Genomewide loss of heterozygosity and its clinical associations in
non small cell lung cancer. Int J Cancer. 117:241–247. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jeong JW, Lee HS, Lee KY, et al:
Mig-6 modulates uterine steroid hormone responsiveness and
exhibits altered expression in endometrial disease. Proc Natl Acad
Sci USA. 106:8677–8682. 2009. View Article : Google Scholar
|
|
6
|
Ferby I, Reschke M, Kudlacek O, et al:
Mig6 is a negative regulator of EGF receptor-mediated skin
morphogenesis and tumor formation. Nat Med. 12:568–573. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang YW, Staal B, Su Y, et al: Evidence
that MIG-6 is a tumor-suppressor gene. Oncogene. 26:269–276.
2007.
|
|
8
|
Jin N, Gilbert JL, Broaddus RR, Demayo FJ
and Jeong JW: Generation of a Mig-6 conditional null allele.
Genesis. 45:716–721. 2007.
|
|
9
|
Anastasi S, Sala G, Huiping C, et al: Loss
of RALT/MIG-6 expression in ERBB2-amplified breast
carcinomas enhances ErbB-2 oncogenic potency and favors resistance
to Herceptin. Oncogene. 24:4540–4548. 2005.PubMed/NCBI
|
|
10
|
Anastasi S, Fiorentino L, Fiorini M, et
al: Feedback inhibition by RALT controls signal output by the ErbB
network. Oncogene. 22:4221–4234. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu D, Makkinje A and Kyriakis JM: Gene 33
is an endogenous inhibitor of epidermal growth factor (EGF)
receptor signaling and mediates dexamethasone-induced suppression
of EGF function. J Biol Chem. 280:2924–2933. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Anastasi S, Baietti MF, Frosi Y, Alema S
and Segatto O: The evolutionarily conserved EBR module of RALT/MIG6
mediates suppression of the EGFR catalytic activity. Oncogene.
26:7833–7846. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Amatschek S, Koenig U, Auer H, et al:
Tissue-wide expression profiling using cDNA subtraction and
microarrays to identify tumor-specific genes. Cancer Res.
64:844–856. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li Z, Dong Q, Wang Y, Qu L, Qiu X and Wang
E: Downregulation of Mig-6 in nonsmall-cell lung cancer is
associated with EGFR signaling. Mol Carcinog. 51:522–534. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu S, Xing W, Peng J, et al: Tumor
transfected with CCL21 enhanced reactivity and apoptosis resistance
of human monocyte-derived dendritic cells. Immunobiology.
213:417–426. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ling YH, Liebes L, Jiang JD, et al:
Mechanisms of proteasome inhibitor PS-341-induced
G2-M-phase arrest and apoptosis in human non-small cell
lung cancer cell lines. Clin Cancer Res. 9:1145–1154.
2003.PubMed/NCBI
|
|
17
|
Kim JW, Ferris RL and Whiteside TL:
Chemokine C receptor 7 expression and protection of circulating
CD8+ T lymphocytes from apoptosis. Clin Cancer Res.
11:7901–7910. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Faber AC, Li D, Song Y, et al:
Differential induction of apoptosis in HER2 and EGFR addicted
cancers following PI3K inhibition. Proc Natl Acad Sci USA.
106:19503–19508. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu Y, Yang Y, Ye YC, et al: Activation of
ERK-p53 and ERK-mediated phosphorylation of Bcl-2 are involved in
autophagic cell death induced by the c-Met inhibitor SU11274 in
human lung cancer A549 cells. J Pharmacol Sci. 118:423–432. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Okamoto K, Okamoto I, Okamoto W, et al:
Role of survivin in EGFR inhibitor-induced apoptosis in non-small
cell lung cancers positive for EGFR mutations. Cancer Res.
70:10402–10410. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lin CI, Du J, Shen WT, et al:
Mitogen-inducible gene-6 is a multifunctional adaptor protein with
tumor suppressor-like activity in papillary thyroid cancer. J Clin
Endocrinol Metab. 96:E554–E565. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Reschke M, Ferby I, Stepniak E, et al:
Mitogen-inducible gene-6 is a negative regulator of epidermal
growth factor receptor signaling in hepatocytes and human
hepatocellular carcinoma. Hepatology. 51:1383–1390. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ruan DT, Warren RS, Moalem J, et al:
Mitogen-inducible gene-6 expression correlates with survival and is
an independent predictor of recurrence in BRAFV600E
positive papillary thyroid cancers. Surgery. 144:908–914. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ying H, Zheng H, Scott K, et al: Mig-6
controls EGFR trafficking and suppresses gliomagenesis. Proc Natl
Acad Sci USA. 107:6912–6917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim TH, Franco HL, Jung SY, et al: The
synergistic effect of Mig-6 and Pten ablation on endometrial cancer
development and progression. Oncogene. 29:3770–3780. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ewings KE, Wiggins CM and Cook SJ: Bim and
the pro-survival Bcl-2 proteins: opposites attract, ERK repels.
Cell Cycle. 6:2236–2240. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen YR, Wang X, Templeton D, Davis RJ and
Tan TH: The role of c-Jun N-terminal kinase (JNK) in apoptosis
induced by ultraviolet C and γ radiation. Duration of JNK
activation may determine cell death and proliferation. J Biol Chem.
271:31929–31936. 1996.
|
|
28
|
Xia Z, Dickens M, Raingeaud J, Davis RJ
and Greenberg ME: Opposing effects of ERK and JNK-p38 MAP kinases
on apoptosis. Science. 270:1326–1331. 1995. View Article : Google Scholar : PubMed/NCBI
|