Introduction
Gastric cancer is the fourth most common cancer
worldwide with 989,600 new cases diagnosed in 2008, and it is the
second leading cause of cancer-related death (1,2).
Gastric cancer is either asymptomatic or causes non-specific
symptoms at an early stage, and thus the majority of patients
present with advanced stage disease at the time of initial
diagnosis, leading to the poor prognosis of gastric cancer. Gastric
cancer treatment involves surgery, chemotherapy, radiation therapy
and their combinations. However, current drugs are confronted with
low efficacy due to the high rate of patients at the advanced stage
of gastric cancer. Thus, the development of novel effective drugs
for gastric cancer therapy is urgently needed.
Thioridazine, an antagonist of the dopamine receptor
D2 family proteins, was initially developed as an antipsychotic
drug, and recently its anticancer function was revealed. Studies
have revealed the antiproliferative and apoptosis induction
capacities of thioridazine in neuroblastoma, glioma, leukemia,
breast cancer, cervical cancer and endometrial cancer (3–6). A
gene signature-based approach identified thioridazine as an
inhibitor of PI3K/Akt signaling in ovarian cancer cells. It
downregulated cell cycle regulators cyclin D1 and CDK4, and
upregulated p21, p16 and p-CDC25A, leading to the G0/G1 phase
arrest of the cells (7). Research
on cervical and endometrial cancer cells disclosing the involvement
of the PI3K/Akt/mTOR pathway in thioridazine-induced apoptosis
further supported this finding (3).
Thioridazine was also found to exhibit an anti-angiogenic effect
through the FAK/mTOR pathway (8),
which may act in inhibiting tumor growth in vivo. In
addition, Byun et al (8)
found that thioridazine inhibited cancer cell growth while sparing
various types of normal cells (6).
Chemoresistance is one of the limitations to gastric
cancer therapy and this issue must be remedied. Research has
revealed the capacity of thioridazine to reverse the
chemoresistance of cancer cells. Thioridazine was found to overcome
the resistance of P388 murine leukaemia cells to doxorubicin
(9). Thioridazine was also able to
reverse the chemoresistance of human KB carcinoma cells by
increasing the accumulation of chemotherapeutic drugs doxorubicin,
vinblastine and dactinomycin in the cells (10). Thioridazine in combination with
verapamil exhibited enhanced ability to reverse the multidrug
resistance of cancer cells (11).
Moreover, thioridazine was found to inhibit the expression of
P-glycoprotein and induce the apoptosis of ABCB1-overexpressing
drug-resistant mouse lymphoma (12,13).
In 2012, through small molecule library screening,
Sachlos et al (14)
discovered that thioridazine exhibited anti-neoplastic pluripotent
stem cell ability, without interfering with normal pluripotent stem
cells. Furthermore, it reduced the function of leukemia stem cells
while sparing the normal human repopulation cells, demonstrating
thioridazine’s specificity in targeting CSCs. As CSCs play a vital
role in drug resistance, cancer relapse and metastasis,
thioridazine may be an effective drug for cancer therapy by
targeting CSCs. Since thioridazine has been safely used in the
clinic for many years as an antipsychotic, it can be rapidly
evaluated in the clinic as an anticancer drug. However, the
antitumor effects of thioridazine on gastric cancer have never been
disclosed, and to the best of our knowledge, the in vivo
tumor inhibition capacity of thioridazine has only been reported in
murine cancer cells (15). In the
present study, we examined the cytotoxic effects of thioridazine on
gastric cancer cell lines NCI-N87 and AGS. Thioridazine reduced the
cell viability of gastric cancer cells and inhibited the colony
formation of NCI-N87 and AGS cells. Thioridazine induced gastric
cancer cell apoptosis dependent on caspases and through the
mitochondrial pathway. Moreover, thioridazine pretreatment
inhibited the growth of NCI-N87 cell-derived tumors. The present
study demonstrated the anticancer effects of thioridazine in
gastric cancer cells, and its ability to inhibit human gastric
cancer cell tumor growth in vivo, suggesting thioridazine as
a candidate for gastric cancer therapy.
Materials and methods
Cell lines and cell culture
Gastric cancer cell lines NCI-N87 and AGS were
obtained from the Cell Bank of the Shanghai Institutes for
Biological Sciences, Chinese Academy of Sciences (Shanghai, China).
NCI-N87 cells were maintained in DMEM supplemented with 10%
heat-inactivated fetal bovine serum (FBS), and AGS cells were
cultured in F-12K with 10% FBS. Both types of cells were incubated
at 37°C in an atmosphere with 5% CO2.
Analysis of cell viability
The NCI-N87 and AGS cells were plated in 96-well
plates at a density of 104 cells/well, and were treated
with thioridazine (Sigma) at indicated concentrations for 48 h.
After incubation with 0.5 mg/ml
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT)
(Beyotime Institute of Biotechnology, Haimen, China) for another 4
h, the supernatant was discarded, and 100 μl DMSO (dimethyl
sulfoxide) was added to dissolve the produced formazan crystals.
The absorbance was measured on a microplate reader (Thermo Fisher
Scientific) at a dual wavelength of 595 nm and 630 nm. The
difference in the outcome of OD595 and OD630
was considered as the measure of cell viability.
Observation of cytotoxicity
To directly observe the cytotoxic effects of
thioridazine, NCI-N87 and AGS cells were seeded in 24-well plates
and incubated with thioridazine at the indicated concentrations.
Forty-eight hours post-treatment, the cells were directly observed
using microscopy or stained with 2% crystal violet which was
dissolved in 20% methanol solution.
Colony formation assay
The NCI-N87 cells were seeded into 6-well plates at
3,000 cells/well and treated with thioridazine at indicated
concentrations after the cells recovered. The formed colonies were
stained with 2% crystal violet 10 days later, washed and
photographed. The AGS cells were seeded at 1,000 cells/well in
6-well plates, treated similarly to NCI-N87 cells, and were stained
with crystal violet 6 days later.
Western blot assay
Western blot analysis was performed according to
standard procedures. In brief, cells were trypsinized following
treatment for the indicated times, lysed using RIPA lysis buffer
and boiled in SDS-PAGE sample loading buffer (both from Beyotime
Institute of Biotechnology). Total protein (40 μg) was
electrophoretically separated on 10 or 12% SDS-PAGE gel and
transferred onto a polyvinylidene difluoride (PVDF) membrane. The
PVDF membrane was blocked with milk, incubated with the primary
antibodies and then with the secondary antibodies. Primary
antibodies for caspase-9, caspase-8, caspase-3 and PARP were
purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and
GAPDH was from CoWin Bioscience Co. (Beijing, China). All the
secondary antibodies were obtained from Santa Cruz Biotechnology.
Subsequently, the protein on the membrane was revealed using ECL
reagents (Pierce Biotechnology, Rockford, IL, USA).
Hoechst 33258 staining
The AGS cells were treated with thioridazine at
indicated concentrations, and fixed with 4% paraformaldehyde (PFA)
for 15 min 48 h post-treatment. The cells were then stained with
Hoechst 33258 (Molecular Probes, Eugene, OR, USA) at 1 μg/ml for 1
min, washed with PBS and photographed under a fluorescence
microscope.
Cell cycle analysis
The NCI-N87 and AGS cells were trypsinized following
treatment with thioridazine for 48 h, and then resuspended in PBS
and fixed with 70% ethanol overnight at −20°C. The fixed cells were
centrifugated, washed with PBS and incubated with 20 μg/ml RNAase
in a water bath at 37°C for 30 min. After been stained with 50
μg/ml PI for another 30 min, they were subjected to a
fluorescence-activated cell sorting (FACS) instrument for
analysis.
Detection of apoptosis
The NCI-87 and AGS cells were treated with
thioridazine for 48 h. Cells were trypsinized into single cells,
washed with PBS and binding buffer, and then resuspended in 500 μl
binding buffer. Annexin V-FITC (5 μl) and 10 μl propidium iodide
(PI, 20 μg/ml) were subsequently added to the cells for incubation.
Fifteen minutes later, the cells were washed with binding buffer
and subjected to FACS for analysis.
Analysis of mitochondrial membrane
potential alteration
Alteration in mitochondrial membrane potential is
detected by JC-1 staining assay. Unimpaired cells retain high
mitochondrial membrane potential, and JC-1 accumulates in the
matrix of the mitochondria and propagates as aggregates, which emit
red fluorescence in the presence of excitation. However, the
mitochondrial membrane potential decreases as cells are impaired,
and then JC-1 diffuses into the cytoplasm and forms monomers, which
emit green fluorescence after excitation. The experiment was
performed as follows.
The cells were seeded in 6-well plates at a density
of 3×106 cells/well and treated with thioridazine for 48
h. After been trypsinized, cells were resuspended in 500 μl DMEM,
and then mixed with 500 μl JC-1 working solution and incubated for
20 min at 37°C in the dark. Afterwards, the cells were washed twice
with JC-1 staining buffer, resuspended in 300 μl JC-1 staining
buffer and assessed by FACS.
Animal experiments
The animal experiments were performed according to
the U.S. Public Health Service Policy on Humane Care and the Use of
Laboratory Animals, and were also approved by the Institutional
Animal Care and Use Committee. Four-week-old female BALB/c nude
mice were purchased from Shanghai Laboratory Animal Center (SLAC;
Shanghai, China). To test the ability of thioridazine to inhibit
tumor growth, the NCI-N87 cells were incubated with DMSO or 5 μM
thioridazine for 24 h. The treated cells were trypsinized and
counted after been stained with trypan blue. Viable DMSO-pretreated
(1×106) and thioridazine-pretreated NCI-N87 cells were
subcutaneously injected into the left and right rear of mice,
respectively. After visible tumors emerged, they were measured
every three days. The volume was calculated as length × width ×
width/2.
Statistical analysis
Comparison of groups was analyzed by the Student’s
t-test. A p-value of less than 0.05 was considered to indicate a
statistically significant difference.
Results
Thioridazine exhibits cytotoxicity in
gastric cancer cells
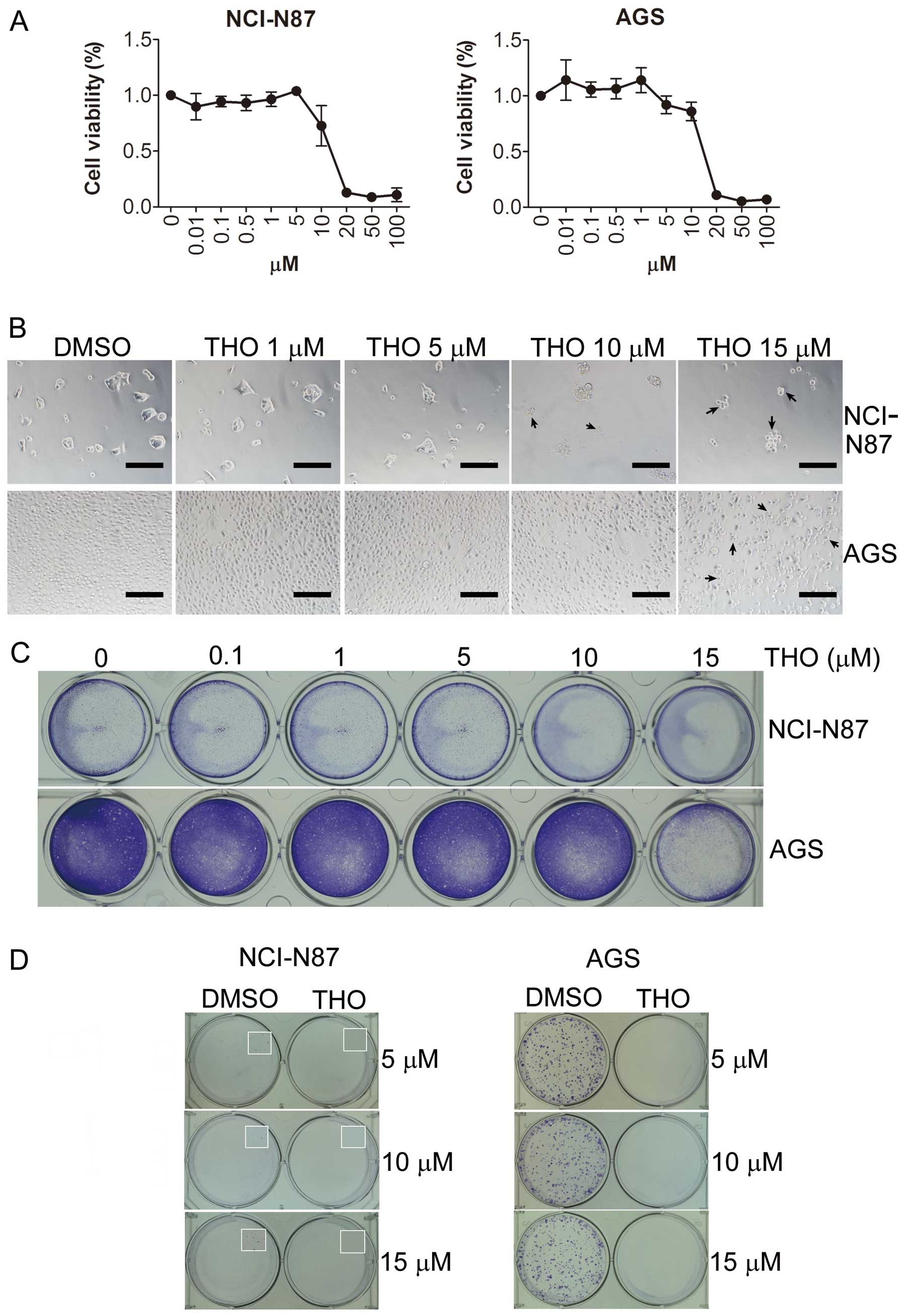
To initially assess the anticancer effect of
thioridazine on gastric cancer cells, MTT assay was performed on
treated cells. Thioridazine reduced the cell viability of NCI-N87
and AGS cells in a concentration-dependent manner (Fig. 1A). Moreover, severe morphological
alterations were observed in the majority of the cells treated with
15 μM thioridazine (Fig. 1B).
Results of the crystal violet staining clearly disclosed the
cytotoxic effect of thioridazine on NCI-N87 and AGS cells (Fig. 1C). Furthermore, 5 μM thioridazine,
which showed a slight effect on the cell viability of NCI-N87 and
AGS cells, markedly inhibited their colony formation ability
(Fig. 1D). Thioridazine at a
concentration of 2 μM also exhibited a slight inhibitory effect on
the colony formation of NCI-N87 and AGS cells.
Thioridazine induces gastric cancer cell
apoptosis
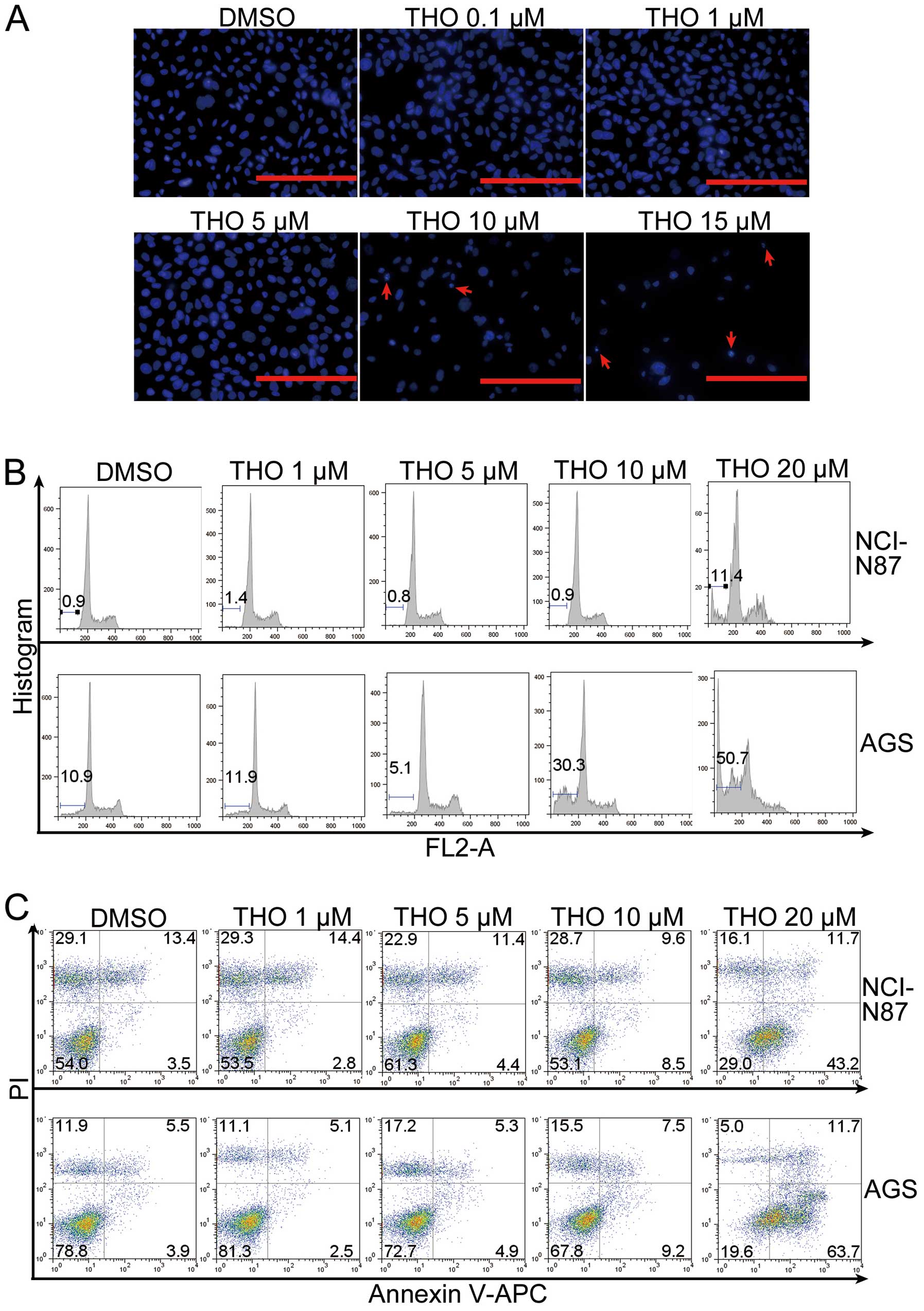
Thioridazine was previously found to induce cervical
and endometrial cancer apoptosis. To detect whether thioridazine
induces gastric cancer cell apoptosis, Hoechst 333258 staining was
carried out. Severe nuclear fragmentation was observed in the AGS
cells following treatment with thioridazine at 10 and 15 μM, but
not in the cells treated with DMSO or thioridazine at a low dosage
(Fig. 2A). As the appearance of
nuclear fragmentation indicates the occurrence of apoptosis, the
cell cycle distribution was analyzed in the treated and untreated
cells. The proportion of sub-G1 phase cells in the NCI-N87 and AGS
cells increased as the dosage of thioridazine was increased
(Fig. 2B). Further detection by
Annexin V/PI double staining assay revealed that thioridazine
treatment resulted in an increase in the percentage of Annexin
V-positive cells in both the NCI-N87 and AGS cells (Fig. 2C). The results indicate the
occurrence of apoptosis in the thioridazine-treated gastric cancer
cells.
Thioridazine induces gastric cancer cell
apoptosis dependent on the caspases
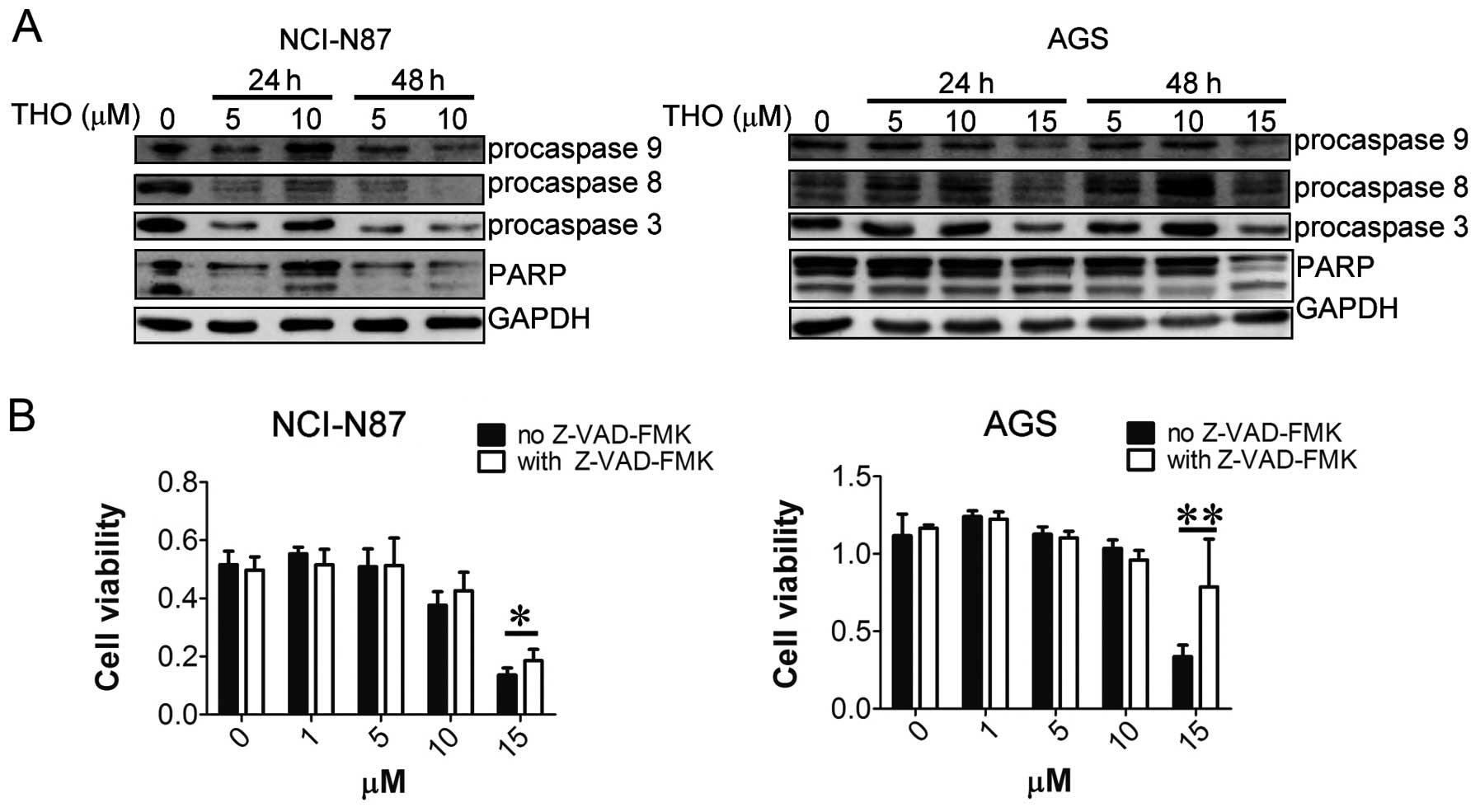
To further clarify the mechanism of
thioridazine-induced apoptosis, expression of a number of
apoptosis-related proteins was analyzed. PARP cleavage was observed
in the presence of thioridazine, which further supported the
occurrence of apoptosis. Precursors of caspase-9, caspase-8 and
caspase-3 were decreased in the NCI-N87 cells following the
treatment of thioridazine for 48 h. Moreover, downregulation of
precursors of caspase-9, caspase-8 and caspase-3 was observed in
the AGS cells treated with 15 μM thioridazine for 24 or 48 h
(Fig. 3A). These results indicate
the involvement of caspases in thioridazine-induced apoptosis in a
time- and dose-dependent manner. Furthermore, caspase inhibitor
Z-VAD-FMK was able to reverse the cytotoxicity of thioridazine both
in the NCI-N87 and AGS cells (Fig.
3B), suggesting that thioridazine induced gastric cancer cell
apoptosis in a caspase-dependent manner.
Thioridazine induces cell apoptosis via
the mitochondrial pathway
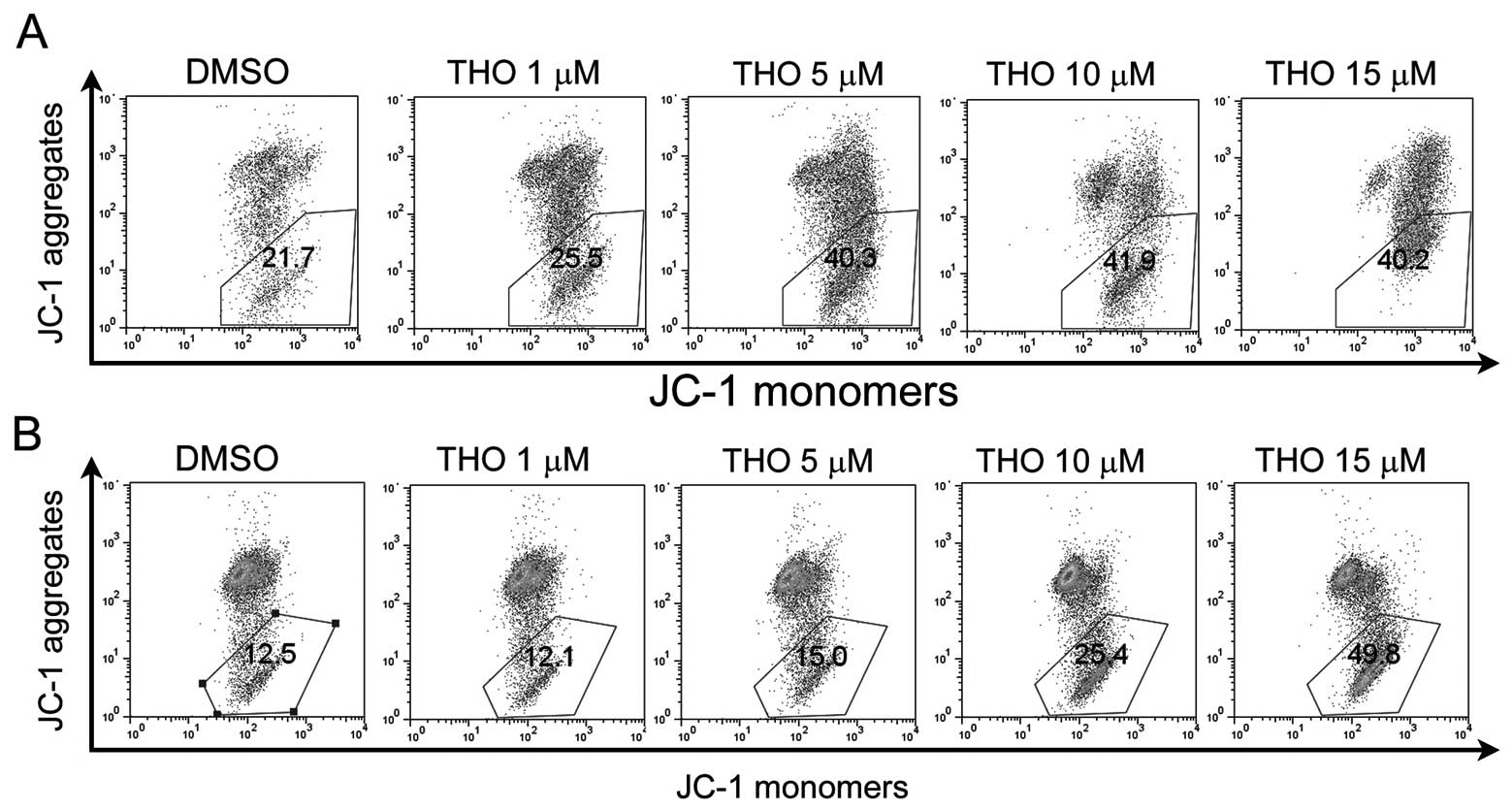
As caspase-9 is involved in the mitochondrial
apoptosis pathway, a decreased level of caspase-9 precursor implied
the breakdown of mitochondria in the thioridazine-induced
apoptosis. JC-1 staining assay was then carried out to examine the
alteration of mitochondria. The percentage of cells with loss of
mitochondrial membrane potential increased from 21.7 to 40.2% in
the NCI-N87 cells following treatment with 15 μM thioridazine
(Fig. 4A), and this proportion
increased from 12.5 to 49.8% in AGS cells (Fig. 4B). The loss of mitochondrial
membrane potential, along with decreased caspase-9 (Fig. 3A), suggest that thioridazine induced
gastric cancer cell death via the mitochondrial apoptosis
pathway.
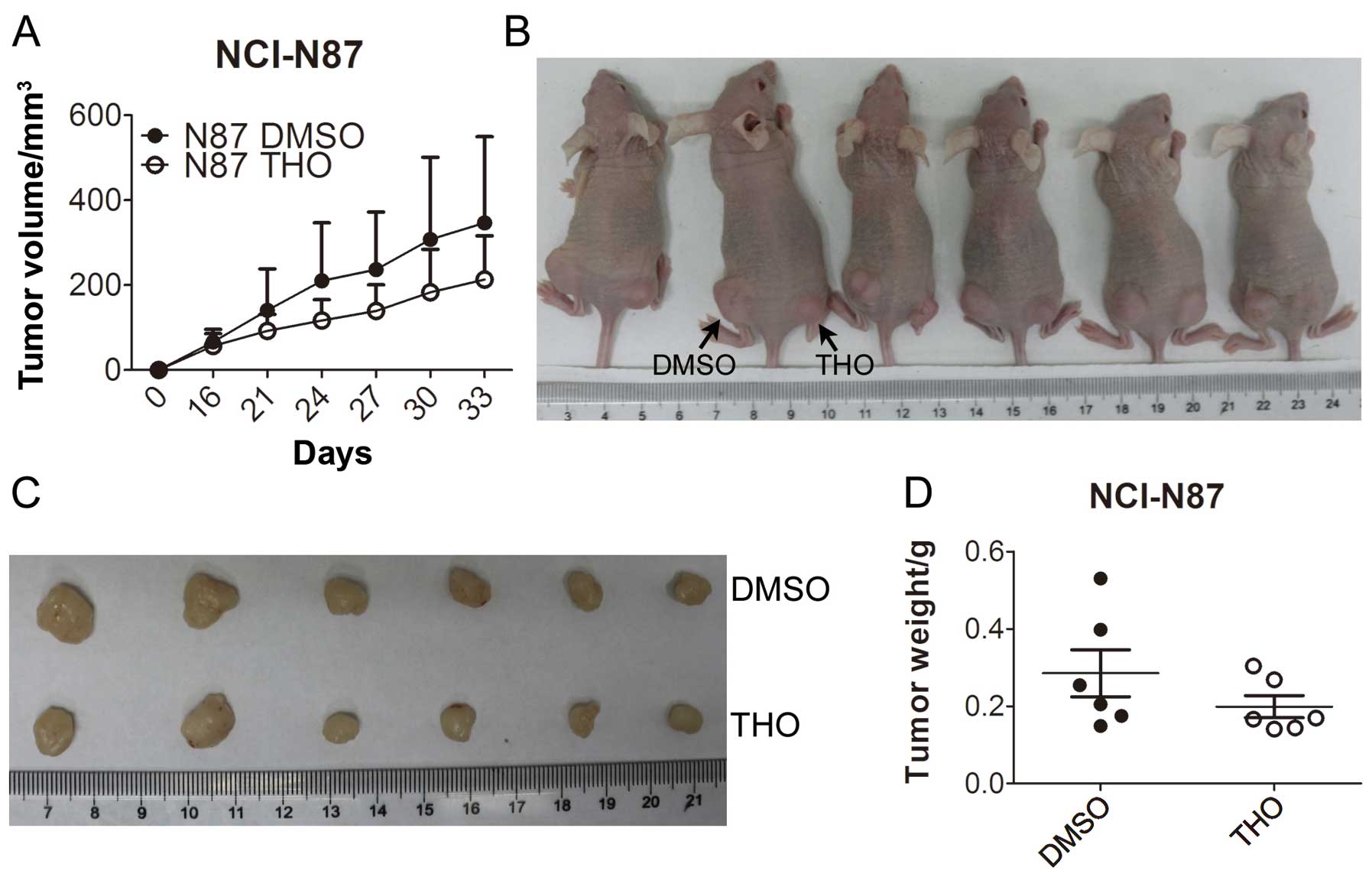
Thioridazine inhibits tumor growth in
vivo
To test the in vivo activity of thioridazine
on gastric cancer cells, NCI-N87 cells were pretreated with DMSO or
5 μM thioridazine for 24 h, and the cells were then injected
subcutaneously into the nude mice at the left or right rear to
compare their tumor formation ability. Thioridazine (5 μM) elicited
little change in the cell viability of NCI-N87 cells (Fig. 1A); however, pretreatment with 5 μM
thioridazine inhibited the growth of NCI-N87 cell-derived tumors
(Fig. 5A), and this was consistent
with thioridazine’s ability to inhibit colony formation (Fig. 1D). Obviously smaller tumors were
observed at the right rear region of the mice which were derived
from the thioridazine-pretreated cells, and the resected tumors
exhibited a similar result (Fig. 5B and
C). Moreover, the weight of the tumors derived from the
thioridazine-pretreated cells was lower than the tumors derived
from the cells pretreated with DMSO (Fig. 5D).
Discussion
Thioridazine has been reported to have
antiproliferative capacity in cancer cells, and is capable of
inducing cancer cell apoptosis in various types of cancers
(3–6). However, the cytotoxic effect of
thioridazine in gastric cancer cells has never been reported. The
present study revealed that thioridazine reduced the cell viability
of gastric cancer cells, inhibited gastric cancer cell colony
formation, and induced gastric cancer cell apoptosis via the
mitochondrial pathway. These results indicate that thioridazine
possesses anti-gastric cancer ability. Further experiments
disclosed that thioridazine pretreatment inhibited the in
vivo growth of tumors derived from NCI-N87 cells. To the best
of our knowledge, to date, there is no research concerning the
in vivo inhibition of human tumor growth by thioridazine,
but only research disclosing its ability to inhibit murine tumor
growth (15). The present study
further demonstrated the in vivo tumor inhibitory ability of
thioridazine and disclosed its cytotoxic effect on gastric cancer
cells, suggesting thioridazine as a candidate drug for gastric
cancer therapy.
Previous studies have disclosed the ability of
thioridazine to reversie chemoresistance, and thioridazine was able
to prevent the exclusion of small molecules from cancer cells
(10,12,16).
This may explain the anti-CSC ability of thioridazine that was
discovered by small molecule library screening (17). Thioridazine was found to have an
influence on cyclin and cyclin-dependent kinase (CDK) (3,7), which
are correlated with the cell cycle and cell growth. Moreover,
thioridazine inhibited the PI3K/Akt pathway (3,7), which
plays a critical role in CSCs (18). Moreover, DRD2, which was antagonized
by thioridazine, specifically regulated Wnt and Akt signaling
(17). Thus, thioridazine may
elicit its anti-CSC capacity by preventing the exclusion of small
molecules out of CSCs and by inhibiting the Wnt and PI3K/Akt
pathways in CSCs. However, this needs to be confimred by further
experiments. Thioridazine was shown to have anti-gastric cancer
ability both in vitro and in vivo in the present
study, yet whether it has an effect on gastric CSCs was not
elucidated. Studies are needed to isolate gastric CSCs (19) and to assess the effects of
thioridazine on gastric CSCs.
Thioridazine was previously found to reverse the
chemoresistance of cancer cells, and a significant therapeutic
outcome in combination with verapamil was noted (11). Thus, thioridazine may be considered
as an adjuvant in combination with chemotherapeutic drugs for
cancer therapy. Moreover, it may also be utilized to treat cancer
cells together with viruses. Thioridazine is an antagonist of the
dopamine receptor D2 family proteins, and analogues of thioridazine
have also exhibited antitumor effects (15), implying the involvement of the
dopamine receptor signaling pathway in cancer. Dopamine receptor
family proteins have been reported to be correlated with cancer
therapy (20,21). Thioridazine may elicit tumor growth
inhibition in a dopamine receptor-dependent and -independent
manner. Negative relationship of the expression of DRD2 family
protein and the prognosis of leukemia was revealed, and other
antagonists of the DRD2 family were found to exhibit an effect
against CSCs as well (17). These
results suggest that blocking of DRD2 family proteins may inhibit
the growth of cancer cells, even CSCs. Development of specific
small molecules that target or knock down DRD2 family proteins may
result in a favorable outcome. Overexpressing shRNA targeting DRD2
family proteins by oncolytic viruses, particularly specific tumor
targeting vectors (22,23), may exhibit a great antitumor effect.
However, selective agonist of DRD2 family proteins induced lung
cancer cell apoptosis (24,25), and this was contrary with the effect
of the function of DRD2 family proteins in leukemia (17). Thus, the strategy of targeting DRD2
needs to be considered in relation to different types of
cancers.
To overcome gastric cancer, it is vital to combine
prevention as well as effective therapy. Habits including the
eating of fresh food, avoiding the consumption of cured food and
smoking, detecting Helicobacter pylori infection and
screening will reduce the incidence of gastric cancer (2). However, in regards to patients
diagnosed with gastric cancer or even advanced gastric cancer,
treatment using effective drugs is vital. In the present study,
thioridazine exhibited favorable anti-gastric cancer effects in
vitro and in vivo, suggesting its use as a promising
candidate drug for gastric cancer therapy. Furthermore, its
combination with other therapeutic drugs may exhibit favorable
outcome in gastric cancer therapy.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (nos. 81172026, 81272402,
81301816 and 81172029), the Foundation of Shanghai Outstanding
Academic Leaders (no. 11XD1403800), the National High Technology
Research and Development Program (863 Program) (no. 2012AA022606),
the Post-doctoral Research Foundation of China (no. 2012M511107),
the Foundation for Interdisciplinary Research of Shanghai Jiao Tong
University (no. YG2011ZD07), the Shanghai Science and Technology
Commission Inter-governmental International Cooperation Project
(12410705900), the Shanghai Science and Technology Commission
Medical-Guiding Project (12401905800), the Program for Changjiang
Scholars and the Post-doctoral Research Program of Shanghai (no.
12R21415300).
References
|
1
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar
|
|
2
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
3
|
Kang S, Dong SM, Kim BR, Park MS, Trink B,
Byun HJ and Rho SB: Thioridazine induces apoptosis by targeting the
PI3K/Akt/mTOR pathway in cervical and endometrial cancer cells.
Apoptosis. 17:989–997. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Strobl JS, Kirkwood KL, Lantz TK, Lewine
MA, Peterson VA and Worley JF III: Inhibition of human breast
cancer cell proliferation in tissue culture by the neuroleptic
agents pimozide and thioridazine. Cancer Res. 50:5399–5405.
1990.PubMed/NCBI
|
|
5
|
Gil-Ad I, Shtaif B, Levkovitz Y, Dayag M,
Zeldich E and Weizman A: Characterization of phenothiazine-induced
apoptosis in neuroblastoma and glioma cell lines: clinical
relevance and possible application for brain-derived tumors. J Mol
Neurosci. 22:189–198. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhelev Z, Ohba H, Bakalova R, Hadjimitova
V, Ishikawa M, Shinohara Y and Baba Y: Phenothiazines suppress
proliferation and induce apoptosis in cultured leukemic cells
without any influence on the viability of normal lymphocytes.
Phenothiazines and leukemia. Cancer Chemother Pharmacol.
53:267–275. 2004. View Article : Google Scholar
|
|
7
|
Rho SB, Kim BR and Kang S: A gene
signature-based approach identifies thioridazine as an inhibitor of
phosphatidylinositol-3′-kinase (PI3K)/AKT pathway in ovarian cancer
cells. Gynecol Oncol. 120:121–127. 2011.PubMed/NCBI
|
|
8
|
Byun HJ, Lee JH, Kim BR, Kang S, Dong SM,
Park MS, Lee SH, Park SH and Rho SB: Anti-angiogenic effects of
thioridazine involving the FAK-mTOR pathway. Microvasc Res.
84:227–234. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ramu A, Spanier R, Rahamimoff H and Fuks
Z: Restoration of doxorubicin responsiveness in
doxorubicin-resistant P388 murine leukaemia cells. Br J Cancer.
50:501–507. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Akiyama S, Shiraishi N, Kuratomi Y,
Nakagawa M and Kuwano M: Circumvention of multiple-drug resistance
in human cancer cells by thioridazine, trifluoperazine, and
chlorpromazine. J Natl Cancer Inst. 76:839–844. 1986.PubMed/NCBI
|
|
11
|
Castaing M, Loiseau A and Cornish-Bowden
A: Synergy between verapamil and other multidrug-resistance
modulators in model membranes. J Biosci. 32:737–746. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kamiwatari M, Nagata Y, Kikuchi H,
Yoshimura A, Sumizawa T, Shudo N, Sakoda R, Seto K and Akiyama S:
Correlation between reversing of multidrug resistance and
inhibiting of [3H]azidopine photolabeling of
P-glycoprotein by newly synthesized dihydropyridine analogues in a
human cell line. Cancer Res. 49:3190–3195. 1989.
|
|
13
|
Spengler G, Molnar J, Viveiros M and
Amaral L: Thioridazine induces apoptosis of multidrug-resistant
mouse lymphoma cells transfected with the human ABCB1 and inhibits
the expression of P-glycoprotein. Anticancer Res. 31:4201–4205.
2011.PubMed/NCBI
|
|
14
|
Sachlos E, Risueno RM, Laronde S, et al:
Identification of drugs including a dopamine receptor antagonist
that selectively target cancer stem cells. Cell. 149:1284–1297.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gil-Ad I, Shtaif B, Levkovitz Y,
Nordenberg J, Taler M, Korov I and Weizman A: Phenothiazines induce
apoptosis in a B16 mouse melanoma cell line and attenuate in
vivo melanoma tumor growth. Oncol Rep. 15:107–112.
2006.PubMed/NCBI
|
|
16
|
Efferth T and Volm M: Reversal of
doxorubicin-resistance in sarcoma 180 tumor cells by inhibition of
different resistance mechanisms. Cancer Lett. 70:197–202. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sutton LP and Rushlow WJ: The dopamine D2
receptor regulates Akt and GSK-3 via Dvl-3. Int J
Neuropsychopharmacol. 15:965–979. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dubrovska A, Kim S, Salamone RJ, Walker
JR, Maira SM, Garcia-Echeverria C, Schultz PG and Reddy VA: The
role of PTEN/Akt/PI3K signaling in the maintenance and viability of
prostate cancer stem-like cell populations. Proc Natl Acad Sci USA.
106:268–273. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Takaishi S, Okumura T, Tu S, Wang SS,
Shibata W, Vigneshwaran R, Gordon SA, Shimada Y and Wang TC:
Identification of gastric cancer stem cells using the cell surface
marker CD44. Stem Cells. 27:1006–1020. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gabalec F, Beranek M, Netuka D, Masopust
V, Nahlovsky J, Cesak T, Marek J and Cap J: Dopamine 2 receptor
expression in various pathological types of clinically
non-functioning pituitary adenomas. Pituitary. 15:222–226. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gatto F and Hofland LJ: The role of
somatostatin and dopamine D2 receptors in endocrine tumors. Endocr
Relat Cancer. 18:R233–R251. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xu HN, Huang WD, Cai Y, et al:
HCCS1-armed, quadruple-regulated oncolytic adenovirus specific for
liver cancer as a cancer targeting gene-viro-therapy strategy. Mol
Cancer. 10:1332011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ding M, Cao X, Xu HN, et al: Prostate
cancer-specific and potent antitumor effect of a
DD3-controlled oncolytic virus harboring the PTEN
gene. PLoS One. 7:e351532012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sheikhpour M, Ahangari G, Sadeghizadeh M
and Deezagi A: A novel report of apoptosis in human lung carcinoma
cells using selective agonist of D2-like dopamine receptors: a new
approach for the treatment of human non-small cell lung cancer. Int
J Immunopathol Pharmacol. 26:393–402. 2013.PubMed/NCBI
|
|
25
|
Senogles SE: D2 dopamine receptor-mediated
antiproliferation in a small cell lung cancer cell line, NCI-H69.
Anticancer Drugs. 18:801–807. 2007. View Article : Google Scholar : PubMed/NCBI
|