Introduction
Lung cancer is generally diagnosed too late to be
operable, and consequently, chemotherapy provides the main
treatment option for most lung cancer patients (1–3).
Furthermore, drug resistance of lung cancer to chemotherapeutic
drugs is one of the important causes of the failure of chemotherapy
(4,5). β-elemene (β-ELE) is a new anticancer
drug extracted from Curcuma zedoaria Roscoe, know as
zedoary, that includes α, β, α and δ forms. β-ELE accounts for the
main antitumor effect (6,7). β-ELE injection has been widely used to
treat a variety of malignancies including lung cancer (8), liver cancer (9), malignant tumors of the digestive tract
(10) and bladder cancer (11). Recently studies have shown that
β-ELE reverses the drug resistance of tumor cells (7,12,13).
To explore the mechanisms of action of β-ELE, we examined the
effects of β-ELE on the cisplatin (DDP)-resistant human lung
adenocarcinoma cell line A549/DDP. Our results define a pathway of
β-ELE function involving the regulation of mitochondrial membrane
potential and apoptosis signaling proteins leading to the reversal
of drug resistance.
Materials and methods
Reagents and equipment
The cisplatin-sensitive human lung adenocarcinoma
cell line, A549, and its cisplatin-resistant derivative, A549/DDP,
were purchased from the China Military Medical Science Academy of
the PLA (Beijing, China). Cisplatin (DDP) (Yunnan Biological
Pharmaceutical Co., Ltd.; batch number: 090202); β-elemene
injection (Dalian Jingang Pharmaceutical Co., Ltd., batch number:
081152); mouse monoclonal anti-human antibodies against cytochrome
c, caspase-3, Bcl-2, Bad and β-actin; and horseradish
peroxidase-labeled rabbit anti-mouse IgG were obtained from Santa
Cruz Biotechnology (Santa Cruz, CA, USA). 2′,7′-Dichlorofluorescein
diacetate (DCFH-DA) fluorescence probe was purchased from
Invitrogen (Carlsbad, CA, USA). Propidium iodide (PI), ECL
chemiluminescence reagent kits, Hoechst 33342 staining reagent, MTT
cell proliferation assay kits, dimethyl sulfoxide (DMSO), RPMI-1640
culture medium, cyclosporine A and Ac-DEVD-CHO were from Sigma (St.
Louis, MO, USA). The Annexin V-FLUOS staining kit was purchased
from Roche Diagnostics (Indianapolis, IN, USA). JC-1 mitochondrial
membrane potential kits were purchased from Nanjing KeyGen
Development Co., Ltd. (Nanjing, China). The GSH/GSSG detection kits
were purchased from Jiangsu Pik Wan Biotechnology Research
Institute (Jiangsu, China). Equipment used included FACSCalibur
flow cytometer (Becton-Dickinson), Tcs SP2 laser scanning confocal
microscope (Leica), spectrophotometer (Eppendorf), electrophoresis
and transfer film equipment (Bio-Rad), Olympus IX71 fluorescence
microscope (Olympus), AE31/CCIS inverted microscope (Moltic Co.,
Ltd.) and 5804R low speed centrifuge (Eppendorf). β-ELE and DDP
were diluted with both RPMI-1640 and 10% fetal bovine serum (FBS)
medium to various working concentrations when used.
Cell culture
Human lung adenocarcinoma A549 and A549/DDP cells
(final concentration of 2 μg/ml DDP to maintain drug resistance)
were cultured in RPMI-1640 medium supplemented with 10% FBS, 100
U/ml penicillin and 100 mg/l streptomycin in an atmosphere of 5%
CO2 at 37°C. The A549/DDP cells were cultured for one
week in the medium without DDP prior to experimentation.
Exponentially growing cells were used in all experiments.
Drug sensitivity assay
The sensitivity of cells to drugs was determined
using the MTT assay. Briefly, cells were plated in triplicate in
96-well plates at a density of 5×103 cells/well for the
drug sensitivity assays. Cells were treated with 0.25, 0.5, 1, 2,
4, 8, 16 or 32 μg/ml of DDP for 24 h, 20 μl MTT dye (5 mg/ml) was
added at 37°C for 4 h, and then the culture medium was removed and
150 μl of DMSO per well was added with oscillation for 10 min.
Spectrometric absorbance at 570 nm was measured by using a
microplate reader (reference wavelength 630 nm). The experiment was
repeated 3 times to generate a growth curve. The proliferation rate
(%) was determined by calculating the value of the experimental
group/the value of the control group × 100%.
MTT cytotoxicity assay
A549/DDP cells were plated in triplicate in 96-well
plates at a density of 5×103 cells/well. After cells
adhered to the plates, a final concentration of 10, 20, 40 or 80
μg/ml β-ELE was added to the experimental groups, and the same
amount of drug dissolution medium was added to the control group.
MTT assay was used to determine β-ELE cytotoxicity as previously
described. The cell proliferation inhibition rate was calculated as
1 - the proliferation rate (%). The 50% inhibitory concentration
(IC50) was calculated by linear regression, and the fold
of drug resistance was calculated as IC50 of resistant
cells/IC50 of sensitive cells.
For assessing the β-ELE-mediated reversal of
A549/DDP cell drug resistance, the control group was treated with
varying final concentrations of DDP (0.25–32 μg/ml), and the
experimental group was additionally treated with 20 μg/ml β-ELE.
After 24 h, MTT solution was added, and the absorbance was measured
as described above. The fold of reversal was calculated as the
IC50 value in the absence of β-ELE to that in the
presence of β-ELE.
Apoptosis assay
A549/DDP cells were cultured in 2 μg/ml DDP to
maintain drug resistance, and β-ELE was added at 20 or 40 μg/ml to
the experimental group. After 24 h, the cells were collected by
centrifugation for Hoechst and Annexin V-FITC staining. For Hoechst
staining, nuclei were stained with DNA fluorescent dye and observed
under a fluorescence microscope. For Annexin V-FITC staining, the
cell pellet was re-suspended in 100 μl binding buffer containing 10
mM HEPES/NaOH, 140 mM NaCl, 5 mM CaCl2 (pH 7.4),
supplemented with 5 μl Annexin V-FITC and 5 μl PI. After the
incubation period (30 min at 37°C in the dark), an additional 400
μl of binding buffer was added and Annexin V-FITC/PI staining was
analyzed within 1 h by flow cytometry. The fluorescence intensity
(green FL1-H and red FL2-H) was measured on the FACSCalibur flow
cytometer. CellQuest Pro software was used for acquisition and
analysis of data.
Assessment of mitochondrial membrane
potential (ΔΨm) by JC-1 assay and laser confocal fluorescence
microscopy
A549/DDP cells were plated in triplicate in 96-well
plates at a density of 1×105 cells/well. The control
group was plated in 2 μg/ml DDP, and an additional 40 μg/ml β-ELE
was added to the experimental group. Cells were cultured for 0, 6,
12 or 24 h in serum-free culture medium, and then 10 mg/l JC-1 dye
was added and cells were incubated at 37°C for 15 min. Cells were
centrifuged, and the excess waste dye was aspirated. Cells were
then photographed under a laser confocal microscope, and JC-1
monomer (green fluorescence) was detected at an excitation
wavelength of 488 nm (emission wavelength 530 nm), while JC-1
polymer (red fluorescence) was detected at an excitation wavelength
of 535 nm (emission wavelength 590 nm). Ten fields were randomly
selected for calculation of the average fluorescence intensity
(Leica, LCS Universal Imaging software). The red fluorescence/green
fluorescence optical density ratio indicated the mitochondrial ΔΨm
levels, while a decrease in the optical density ratio represented
mitochondrial ΔΨm decrease.
Assessment of intracellular reactive
oxygen species (ROS) and the level of glutathione (GSH)
In order to assess intracellular ROS levels,
A549/DDP cells were plated in triplicate in 96-well plates at a
density of 5×103 cells/well with 2 μg/ml DDP and 0, 20
or 40 μg/ml β-ELE for 24 h. Cells were collected, incubated with 5
μM DCF-DA probe at 37°C for 20 min in serum-free medium, and washed
3 times, and the fluorescence intensity at a 488-nm excitation
wavelength and 525-nm emission wavelength was detected by flow
cytometry. DCFH-DA itself has no fluorescence and can freely pass
through the cell membrane. After entering the cell,
2′,7′-dichlorofluorescein (DCFH) is oxidized by the superoxide
anion and hydrogen peroxide to fluorescent
2′,7′-dichlorofluorescein (DCF). The level of DCF reflects the
level of intracellular ROS expression.
At the same time, we measured GSH according to the
manufacturer’s instructions. We measured total glutathione (GSSG +
GSH) content, and then subtracted the amount of GSH in the sample
to calculate the GSSG content. The ratio of GSH/(GSSG + GSH) was
used as a measure of GSH. The principle of this assay is as
follows: GSH reacts with 5′-dinitrobenzene acid (DTNB) to form GSSG
and stable 5-mercapto-2-nitrobenzene acid (TNB); GSSG is reduced by
GSSG reductase and NADPH, releasing additional TNB (yellow color),
which can be detected by spectrophotometry (maximum absorbance
wavelength 412 nm). The amount of TNB is proportional to the GSH
released in the samples. All experiments were repeated 3 times.
Western blot analysis of cytochrome c,
caspase-3 and Bcl-2 expression
A549/DDP cells were plated in triplicate in 96-well
plates at a density of 5×103 cells/well with 2 μg/ml DDP
plus 0, 20 or 40 μg/ml β-ELE. Cells were then collected in lysis
buffer and incubated on ice for 15 min. The lysates were
centrifuged at 4°C for 10 min, and 50 μg of protein was
eletrophoresed by 12% SDS-PAGE and transferred to polyvinyl
difluoride ethylene membranes. The membranes were then blocked with
5% skim milk for 2 h, washed in TBST, and incubated with mouse
anti-human cytochrome c (1:800 dilution), caspase-3 (1:1,000
dilution), Bcl-2 (1:1,000 dilution), Bad (1:1,000 dilution),
procaspase-3 (1:1,000 dilution) or β-actin (1:2,000 dilution).
HRP-labeled secondary antibodies (1:2,000 dilution) were added for
2 h at room temperature, followed by ECL chemiluminescence. For
assessment of the effects of cyclosporine and Ac-DEVD-CHO, cells
were divided into 4 groups (each with 2 μg/ml DDP to maintain drug
resistance): control group; 40 μg/ml β-ELE treatment group; 40
μg/ml β-ELE + Ac-DEVD-CHO (50 μmol/l) treatment group; and 40 μg/ml
β-ELE + cyclosporine A (2 μmol/l) treatment group. Caspase-3
expression and cleavage were assessed by western blot analysis as
described above.
Statistical analysis
Statistical analysis was performed using SPSS 13.5
and Origin 8.5 software. Statistical data represent mean ± SD and
were determined using single factor analysis of variance.
Comparisons between two groups were performed using a Student’s
t-test or χ2 test. P<0.05 was considered to indicate
a statistically significant result.
Results
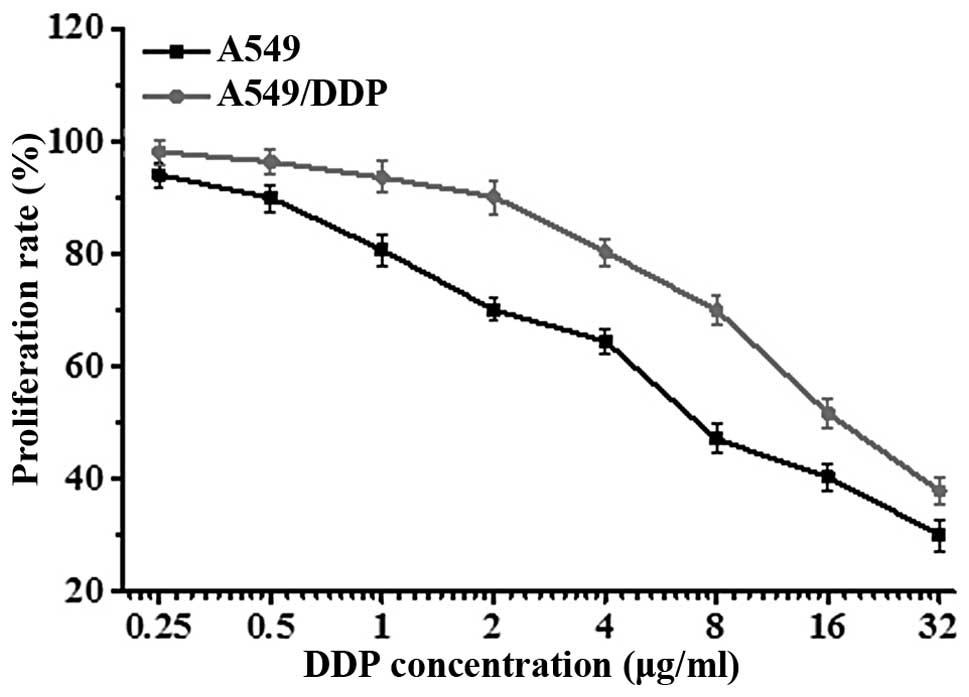
Determination of A549 and A549/DDP cell
drug sensitivity
To verify the differential sensitivity of A549 and
its derivative cell line, A549/DDP, to DDP (cisplatin), cells were
exposed to a gradient of DDP concentrations for 24 h, and cell
viability was assessed by MTT assay. Results showed that the
concentration of DDP required to inhibit the proliferation of A549
cells (IC50=5.73±2.11 μg/ml) was lower than the
concentration needed to inhibit the proliferation of A549/DDP cells
(IC50=15.34±1.05 μg/ml) (Fig. 1). The difference in IC50
was statistically significant (t=2.3571, P<0.01), confirming
that the A549/DDP cells were DDP-resistant.
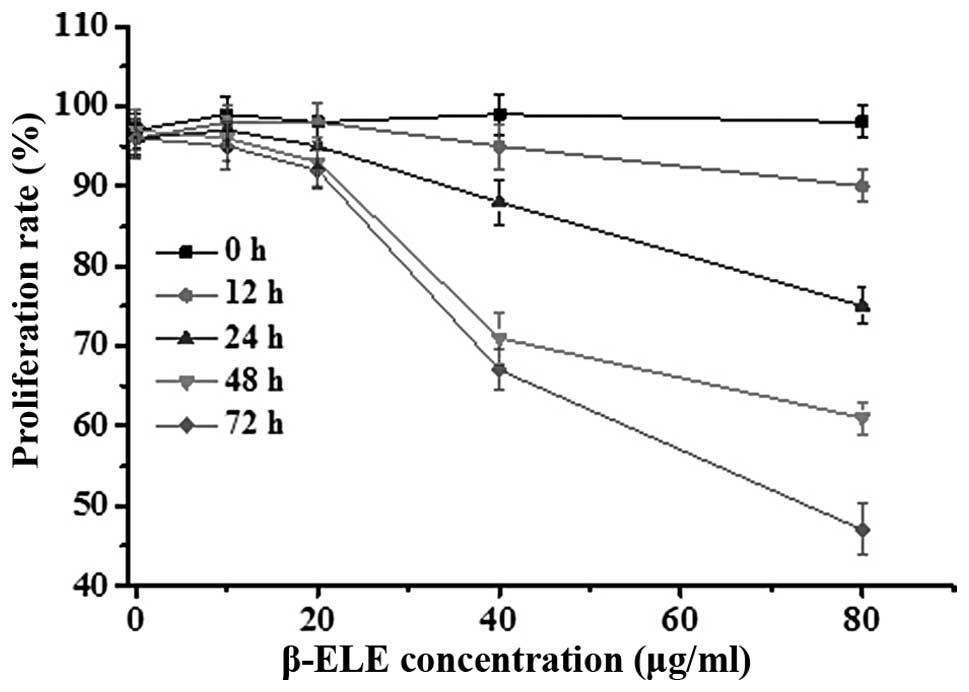
Effects of β-ELE on A549/DDP cell
toxicity
To assess the effects of β-ELE on A549/DDP cells, we
performed MTT assays over a range of doses and times. Results
showed that β-ELE inhibited A549/DDP cell growth in dose-dependent
manner (Fig. 2; 20 vs. 40 μg/ml
β-ELE: χ2=2.6249, P<0.05 at 24 h;
χ2=2.1449, P<0.05 at 48 h). This effect was also
partially time-dependent, depending on β-ELE dose (24 vs. 48 h for
20 μg/ml β-ELE: χ2=27.4632, P>0.05; for 40 μg/ml
β-ELE: χ2=2.4136, P<0.05). Based on these results, we
selected 20 μg/ml ELE treatment for 24 h as the optimum
concentration and time that ELE reverses the drug resistance of
A549/DDP cells.
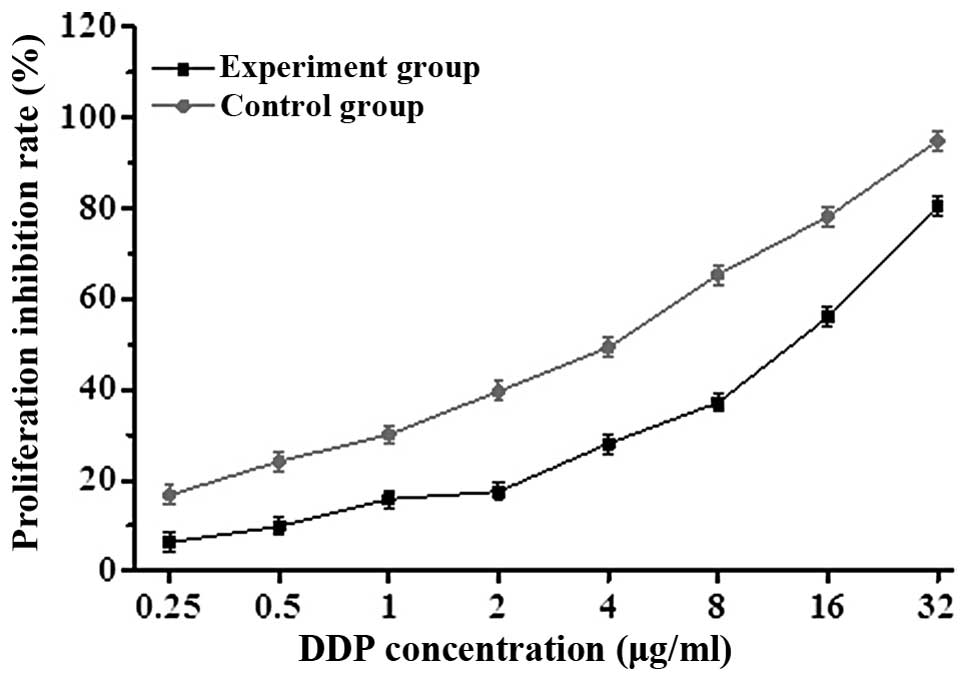
β-ELE reverses drug resistance of
A549/DDP cells
To determine whether β-ELE can reverse the drug
resistance of A549/DDP cells, we exposed cells for 24 h to a range
of doses of DDP in the absence or presence of 20 μg/ml β-ELE. The
β-ELE-treated cells showed increased sensitivity to DDP at all
concentrations (Fig. 3; P<0.05).
Furthermore, the IC50 value of the experimental group
(4.15±0.89 μg/ml) was significantly lower than the IC50
value of the control group (15.46±1.23 μg/ml) (t=1.4321,
P<0.01), with the drug resistance ratio reversed 3.73±0.38-fold
(Table I and Fig. 3). The results indicate that β-ELE
enhances the sensitivity of A549/DDP cells to DDP.
 | Table IEffect of β-ELE in reversing the drug
resistance of A549/DDP cells (n=3, mean ± SD). |
Table I
Effect of β-ELE in reversing the drug
resistance of A549/DDP cells (n=3, mean ± SD).
| Cell proliferation
inhibition rate (%) |
|---|
|
|
|---|
| DDP (μg/ml) | Control group | Experimental
group |
|---|
| 0.25 | 6.35±1.03 | 16.79±1.85a |
| 0.5 | 9.88±0.99 | 24.20±0.13a |
| 1 | 15.89±0.46 | 30.14±0.47a |
| 2 | 17.55±1.35 | 39.64±0.09a |
| 4 | 28.11±0.65 | 49.34±0.05a |
| 8 | 37.21±1.45 | 65.37±1.05a |
| 16 | 55.96±2.03 | 78.21±0.79a |
| 32 | 80.44±0.77 | 94.85±0.91a |
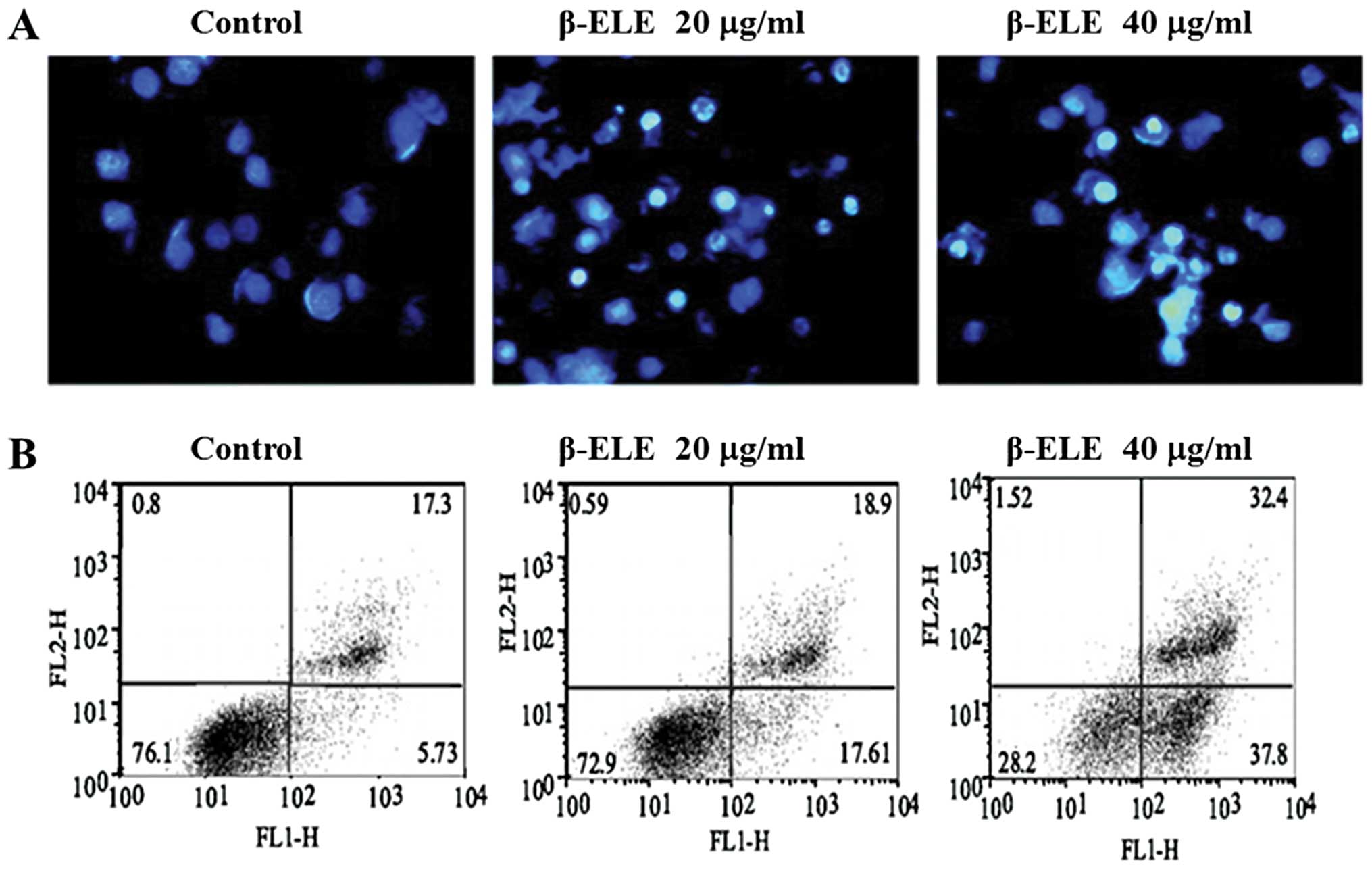
β-ELE increases levels of A549/DDP cell
apoptosis
To determine whether the enhanced sensitivity to DDP
conferred by β-ELE is related to increased levels of apoptosis, we
performed Hoechst 33342 fluorescent staining following β-ELE
treatment. Results showed that upon treatment with 20 and 40 μg/ml
β-ELE for 24 h, A549/DDP cell nuclei became progressively smaller
with more dense granular chromatin staining, which suggests typical
morphological changes of apoptosis (Fig. 4A). We verified these findings by
flow cytometry following Annexin V-FITC/PI staining. Our results
demonstrated a dose-dependent increase in the apoptosis rate for
cells treated with 20 and 40 μg/ml β-ELE as compared to the
control, which was apparent for cells in early apoptosis
(17.61±0.10 and 37.80±0.12% vs. 5.73±0.09%, respectively) and cells
in middle-late apoptosis (18.9±0.11 and 32.4±0.13 vs. 17.3±0.11%,
respectively). The differences between these values were
statistically significant (P<0.05) (Fig 4B). Collectively, these results
suggest that β-ELE increases the levels of apoptosis in A549/DDP
cells.
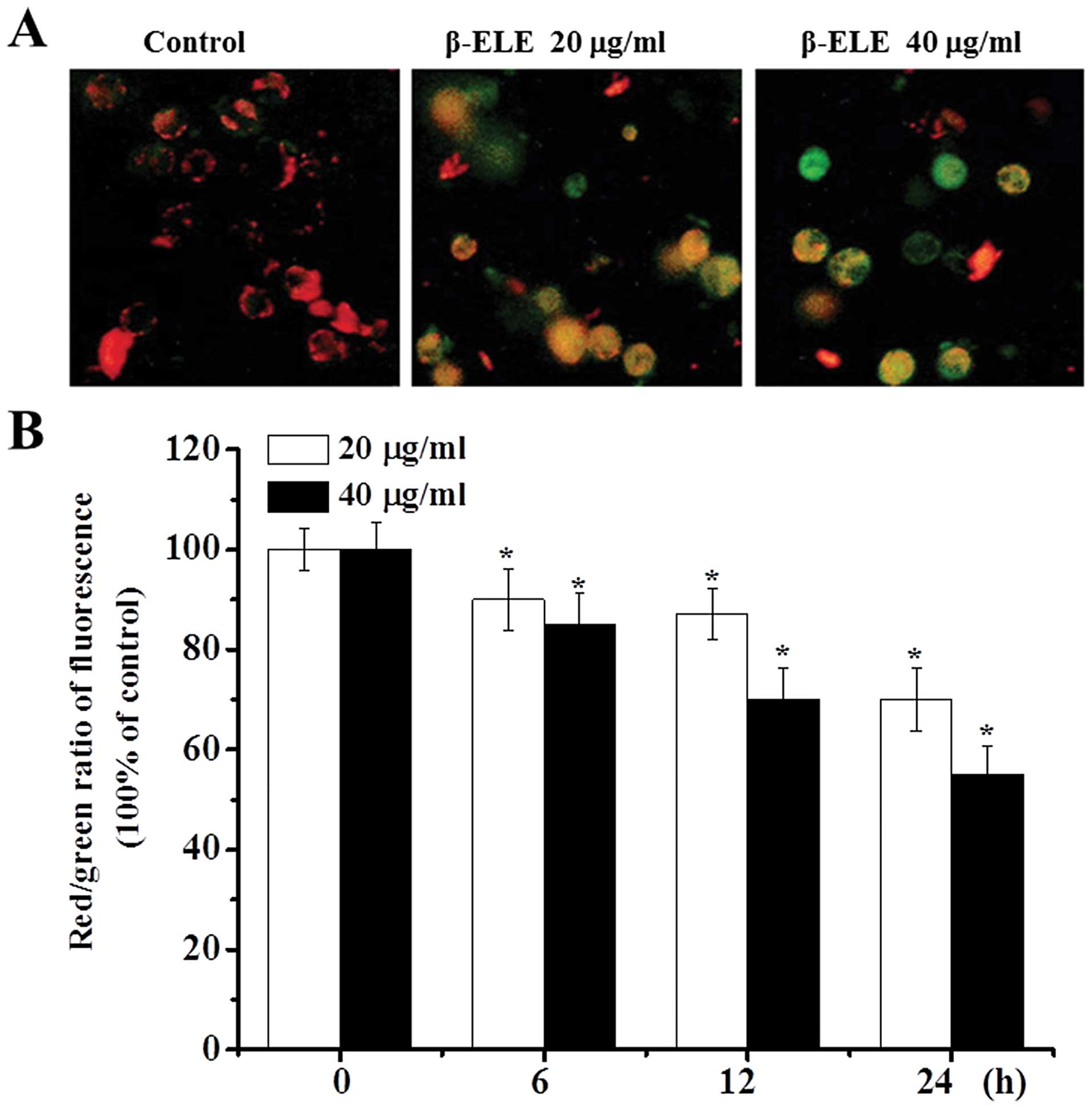
β-ELE decreases the mitochondrial
membrane potential of A549/DDP cells
During apoptosis, the mitochondrial membrane
potential decreases (14,15). As an additional verification of the
effects of β-ELE, we assessed the mitochondrial membrane potential
of A549/DDP cells before and after a 6-, 12- and 24-h drug
treatment. Most of the control cells were stained red by JC-1
assay, indicating an intact cell membrane, with clearly visible
nuclei. In contrast, cells treated with 20 or 40 μg/ml β-ELE showed
increasing amounts of green fluorescence, cell rupture and cell
content outflow, suggesting a decline in the mitochondrial ΔΨm. The
effect was more obvious for the 40 μg/ml β-ELE group, which showed
clear pyknosis (Fig. 5A). Analysis
of the red/green fluorescent light density ratio showed that the
decrease was time-dependent and was statistically significant for
the 20 and 40 μg/ml ELE groups (6 h: χ2=2.2447,
P<0.05, χ2=2.0256, P<0.05; 12 h:
χ2=2.0143, P<0.05, χ2=1.3121, P<0.01;
24 h: χ2=1.3084, P<0.01, χ2=1.0034,
P<0.01) (Fig. 5B).
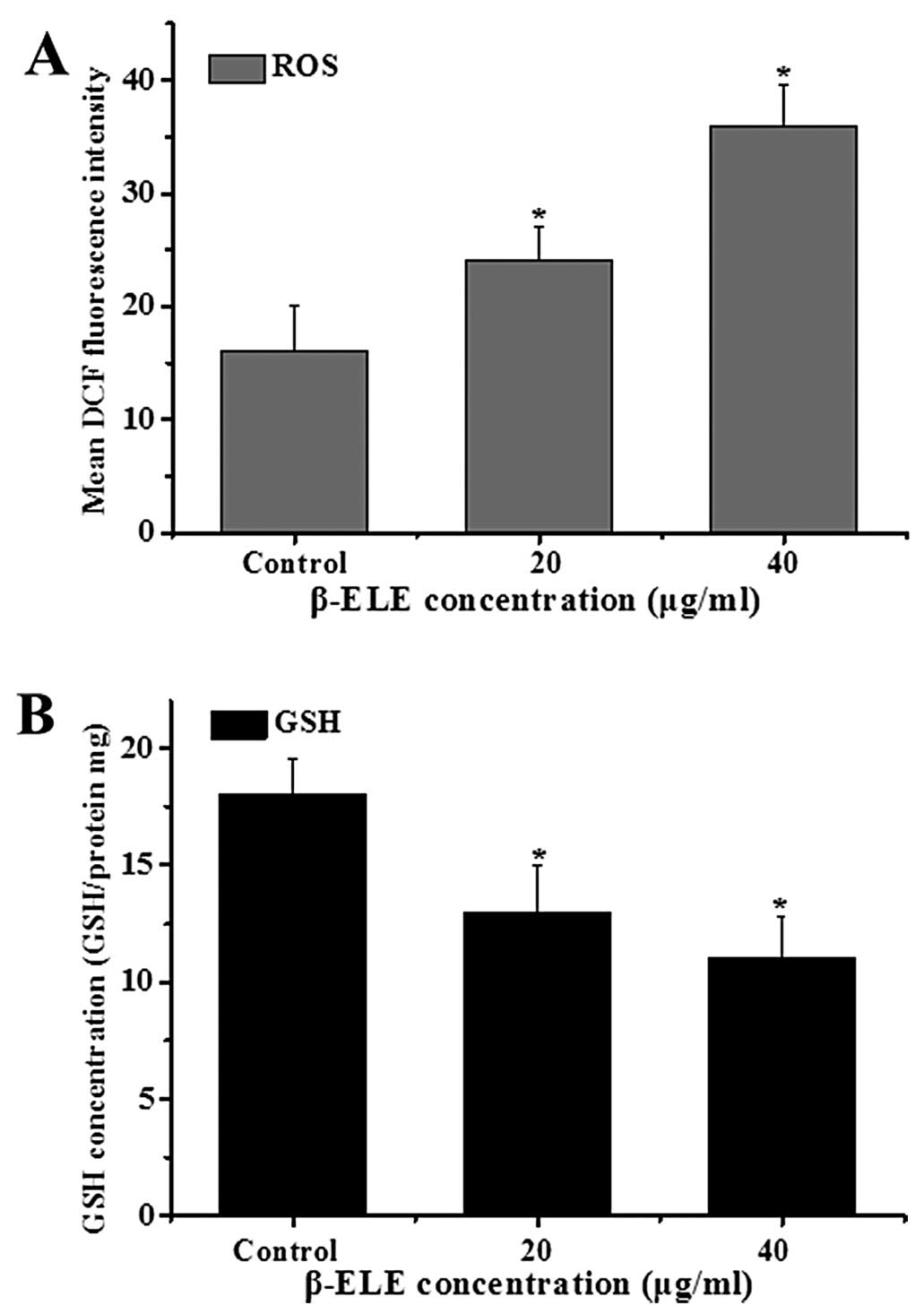
β-ELE increases levels of reactive oxygen
species (ROS) generation and glutathione (GSH) release in A549/DDP
cells
The generation of ROS and decline in intracellular
GSH levels are also associated with apotosis (16,17).
To examine the ROS levels, we performed a DCF assay following
treatment with β-ELE for 24 h. Results showed a statistical
increase in DCF fluorescence intensity (χ2=3.2443,
P<0.05; χ2=2.1254, P<0.05, respectively) in the
cells treated with 20 or 40 μg/ml β-ELE. These results suggest that
the content of ROS was increased by β-ELE treatment (Fig. 6A). Further assessment of GSH levels
showed that the GSH/(GSSG + GSH) ratio decreased, suggesting a
dose-dependent decrease in GSH content (χ2=2.8437,
P<0.05; χ2=2.1244, P<0.05) (Fig. 6B). These results suggest that β-ELE
activates a pathway of apoptosis that involves both the generation
of ROS and decline in intracellular GSH.
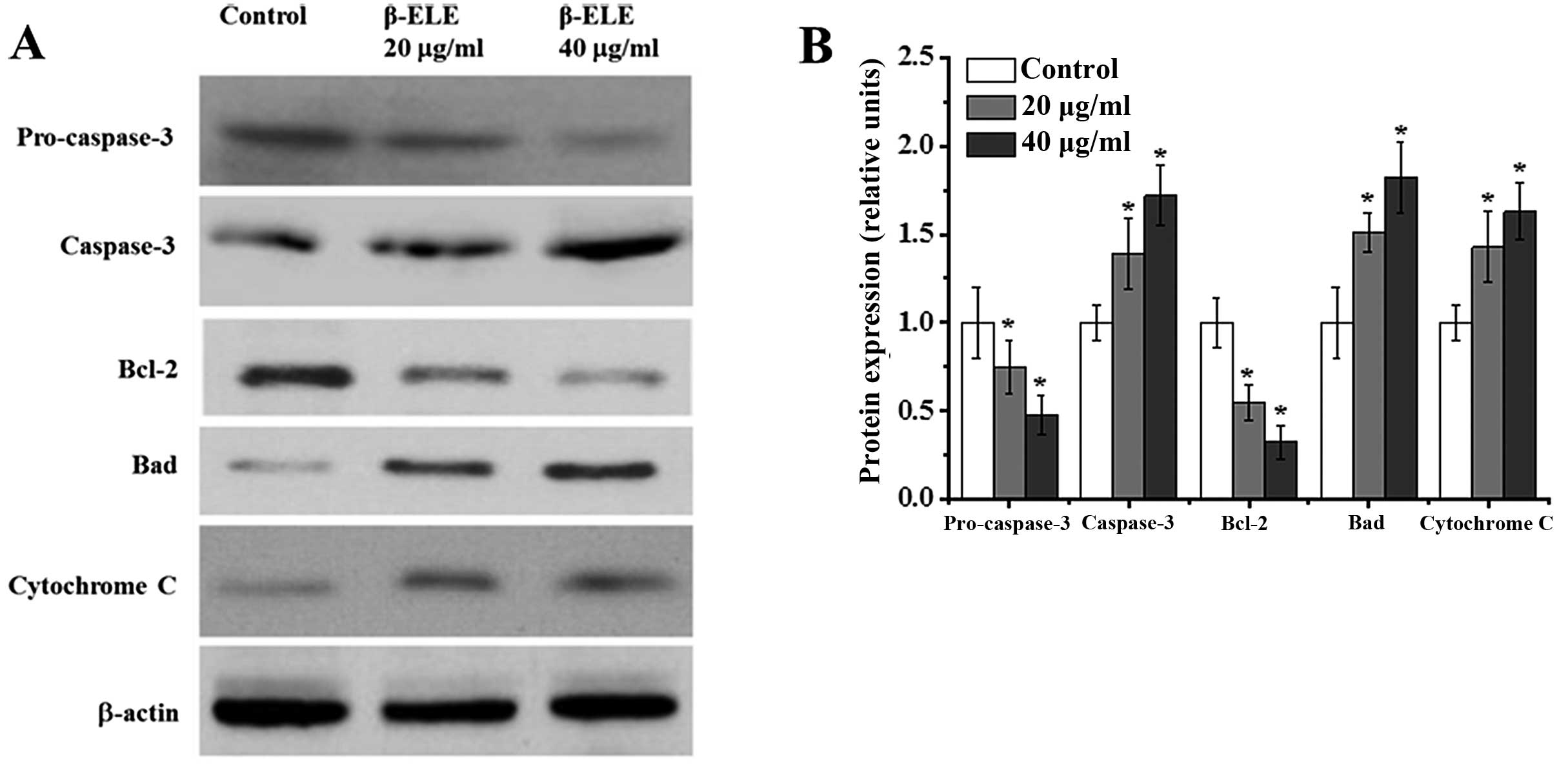
β-ELE activates apoptosis in A549/DDP
cells through a pathway involving caspase-3 activation, modulation
of Bcl-2 family protein expression and cytochrome c release
Apoptosis may be accompanied by the cleavage of
procaspase-3 to active caspase-3, the modulation of Bcl-2 family
protein expression and the release of cytochrome c (18–20).
To assess the effects of β-ELE on these signaling pathways, we
performed western blotting of A549/DDP cells following a 24-h
treatment. Compared with the control group, cells treated with 20
and 40 μg/ml β-ELE had 0.75- and 0.48-fold less procaspase-3
(χ2=3.8782, P<0.05; χ2=3.9644, P<0.05,
respectively), while a 1.39- and 1.72-fold increase was noted in
cleaved caspase-3 (χ2=2.1134, P<0.05;
χ2=2.3516, P<0.05, respectively) (Fig. 7). This indicates a shift from
inactive to active caspase-3. Cells treated with 20 and 40 μg/ml
β-ELE also had 0.55–0.33-fold less of the anti-apoptotic Bcl-2
(χ2=3.9442, P<0.05; χ2=4.0142, P<0.05)
and 1.51–1.82-fold more of the pro-apoptotic Bad protein
(χ2=1.9641, P<0.05; χ2=1.8746, P<0.05).
In addition, a 1.43- and 1.63-fold increase in cytoplasmic
cytochrome c levels (χ2=1.9642, P<0.05;
χ2=1.7692, P<0.05) was also consistent with increased
apoptosis. These results suggest that β-ELE damages the
mitochondrial membrane, leading to the release of mitochondrial
cytochrome c to the cytoplasm, which also leads to the
activation of caspase-3 and modulation of Bcl-2 family proteins to
activate an apoptotic pathway that reverses drug resistance.
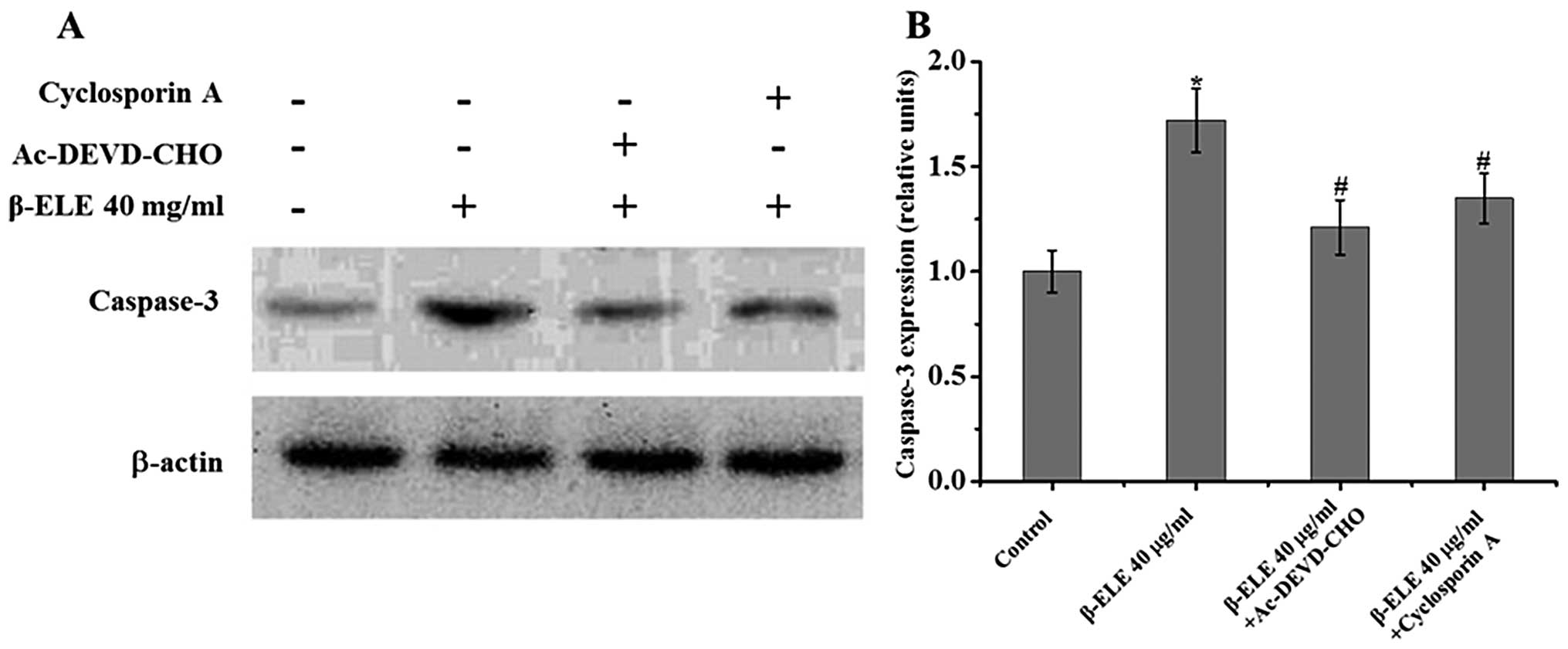
Cyclosporine A and Ac-DEVD-CHO inhibit
β-ELE-induced apoptosis in A549/DDP cells
To further understand the role of the mitochondrial
membrane and apoptotic pathways in the reversal of multidrug
resistance, we treated A549/DDP cells with 40 μg/ml β-ELE for 24 h
in combination with the mitochondrial membrane permeability
transition pore blocking agent cyclosporine A and the caspase-3
inhibitor Ac-DEVD-CHO. Results showed that the addition of either
cyclosporine A or Ac-DEVD-CHO activated a 1.72±0.13% increase in
the β-ELE-treated group compared with the control group;
0.68±0.14-fold for β-ELE cells + cyclosporine A vs. β-ELE group;
1.35±0.15-fold for β-ELE cells + Ac-DEVD-CHO vs. β-ELE group. The
decrease in β-ELE activation by cyclosporine A and Ac-DEVD-CHO was
statistically significant (P<0.05) (Fig. 8). These results indicate that the
mitochondrial membrane integrity and caspase-3 activation play an
important role in β-ELE-induced A549/DDP cell apoptosis.
Discussion
Apoptosis is a form of programmed cell death that
occurs naturally to maintain homeostasis, but can be circumvented
by cancer cells (21). Researchers
originally thought that apoptosis occurs primarily in response to
nuclear changes (22); however, it
is now acknowledged that the mitochondrion is the apoptosis control
center (23,24). We used the JC-1 fluorescent probe
detection method to assess changes in the mitochondrial membrane
potential following β-ELE treatment.
Our results indicated that β-ELE caused pyknosis,
cell rupture and outflow of contents. Further analysis revealed
that the effects were time- and dose-dependent and were accompanied
by a decrease in the mitochondrial membrane potential. Decline in
the membrane ΔΨm is a hallmark of apoptosis initiation and is
indicative of membrane damage. Therefore, these results indicate
that β-ELE reverses drug resistance in A549/DDP cells by inducing
damage to the mitochondrial membrane and decreasing membrane
potential.
Mitochondria are the ‘energy processing plants’ of
biology. Failure or inhibition of the respiratory electron
transport chain leads to increased mitochondrial ROS, an activated
oxygen free radical that attacks the mitochondrial membrane and
decreases mitochondrial ΔΨm, resulting in increased permeability
(25). We demonstrated that the
decreased mitochondrial membrane potential in A549/DDP cells was
accompanied by increased ROS, suggesting a pathway of apoptosis
induced by β-ELE. We also observed a decrease in intracellular GST
levels. GST is the scavenger of oxygen free radicals that removes
intracellular ROS that accumulates during cellular injury (26). Therefore, our results indicate that
β-ELE reverses A549/DDP cell drug resistance through increased ROS
contents in cells which leads to a reduced intracellular GSH
content. These intracellular mediators, in turn, cause further
aggravation of the damage to the mitochondrial membrane, resulting
in further progression towards apoptosis. This cycle is consistent
with the results of Yang et al (27) who demonstrated that application of
fucoidan to the hepatocellular carcinoma cell line SMMC-7721
activated an increase in ROS, a decreased in GSH, mitochondrial
membrane depolarization, and the induction of apoptosis.
A decline in mitochondrial membrane potential (ΔΨm)
and increase in permeability is also associated with cytochrome
c. Cytochrome c is released and activation of caspase
cascade ensues. Caspase-3 is irreversibly activated through
cleavage of procaspase-3, resulting in the activation of an
apoptotic program. Cytochrome c released to the cytoplasmic
is mediated by Bcl-2 family proteins on the outer mitochondrial
membrane. The Bcl-2 family of proteins includes both anti-apoptotic
(Bcl-2, Bcl-xL) and pro-apoptotic (Bax, Bad) proteins. Cytochrome
c is located in the mitochondrial intermembrane space, so it
cannot normally be detected in the cytoplasm. When the structure of
the mitochondrial membrane is damaged, particularly the
mitochondrial permeability transition pore (PTP) between the outer
and inner membranes, mitochondrial membrane permeability increases,
cytochrome c is released into the cytoplasm across the
membrane, which leads to depolarization, change in mitochondrial
membrane permeability and caspase-3 activation; thus, cytochrome
c is one of the key factors in apoptosis signal
transduction. However, the release of cytochrome c is
modulated by effects of the Bcl-2 family proteins on PTP on the
mitochondrial membrane. A decrease in anti-apoptotic Bcl-2 family
proteins (such as Bcl-2, Bcl-xL) and an increase in pro-apoptotic
proteins (e.g., Bax, Bad) further promote the release of cytochrome
c into the cytoplasm, triggering the caspase cascade and
inducing increased apoptosis (28–35).
We demonstrated that β-ELE promotes a dose-dependent increase in
cytoplasmic cytochrome c, transition of caspase-3 to its
active form, a decrease in the expression of anti-apoptotic Bcl-2,
and an increase in pro-apoptotic Bad. Therefore, our results
suggest a pathway of apoptosis activated by β-ELE that involves
characteristic mitochondrial changes. Further study demonstrated
that caspase-3 expression was reduced by the addition of either the
mitochondrial PTP inhibitor cyclosporine A or caspase-3 inhibitor
Ac-DEVD-CHO. These results verify the relationship between
mitochondrial permeability and caspase-3 activation in this
pathway. In summary, our results demonstrated that β-ELE reverses
A549/DDP cell drug resistance via cytochrome c release,
caspase activation and modulation of the expression of Bcl-2 family
proteins. These results provide a mechanism for β-ELE that may
explain its ability to overcome drug resistance and could be useful
in the treatment of drug-resistance cancers.
Acknowledgements
The present study was supported by the Fujian
Provincial Natural Science Foundation, China (no. 2010D014).
References
|
1
|
Cicenas S, Zaliene A and Atkocius V:
Treatment outcome of locally advanced stage IIIA/B lung cancer.
Medicina (Kaunas). 45:452–459. 2009.(In Lithuanian).
|
|
2
|
Szkorupa M, Klein J, Bohanes T, et al:
Neoadjuvant chemotherapy and surgical treatment in advanced stages
of non-small cell lung cancer. Rozhl Chir. 90:433–439. 2011.(In
Czech).
|
|
3
|
Spásová I: The position of neoadjuvant
chemotherapy in the treatment of non-small-cell lung carcinoma.
Vnitr Lek. 53:715–723. 2007.(In Czech).
|
|
4
|
Lee Y, Kim HY, Lee SH, et al: Clinical
significance of heterogeneity in response to retreatment with
epidermal growth factor receptor tyrosine kinase inhibitors in
patients with lung cancer acquiring secondary resistance to the
drug. Clin Lung Cancer. 15:145–151. 2014. View Article : Google Scholar
|
|
5
|
Goldberg SB, Oxnard GR, Digumarthy S, et
al: Chemotherapy with erlotinib or chemotherapy alone in advanced
non-small cell lung cancer with acquired resistance to EGFR
tyrosine kinase inhibitors. Oncologist. 18:1214–1220. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ju JF, Yu WP, Fu CS and Ma LM: Modern
research and clinical application of β-elemene. Qilu Pharm Affairs.
27:546–548. 2008.
|
|
7
|
Guo HQ, Zhang GN, Wang YJ, et al:
β-elemene, a compound derived from Rhizoma zedoariae, reverses
multidrug resistance mediated by the ABCB1 transporter. Oncol Rep.
31:858–866. 2014.
|
|
8
|
Hao SH, Zhang J and Zhang T: A study of
elemene emulsion treated advanced non small cell lung cancer on
platinum based chemotherapy. Clin Focus. 27:1529–1531. 2012.(In
Chinese).
|
|
9
|
Zhao C, Zhang YC, Sun YY, et al: A study
of elemene injection combined with interventional chemotherapy
treated primary liver cancer. Chin J Diffic Compl Cas. 11:882–883.
2012.(In Chinese).
|
|
10
|
Cheng HD, Yang Z, Zhang MJ, et al:
Clinical observation of elemene combined with paclitaxel/tegafur in
the treatment of advanced esophageal carcinoma. J Anhui Med Pharm.
16:1679–1681. 2012.(In Chinese).
|
|
11
|
Ren W and Du SK: Different effect of
elemene and BCG on preventing the recurrence of postoperative
superficial bladder. J Shaanxi Med. 41:1151–1152. 2012.(In
Chinese).
|
|
12
|
Li QQ, Lee RX, Liang H, Zhong Y and Reed
E: Enhancement of cisplatin-induced apoptosis by β-elemene in
resistant human ovarian cancer cells. Med Oncol. 30:424–444.
2013.
|
|
13
|
Zhang Y, Mu XD, Li EZ, et al: The role of
E3 ubiquitin ligase cbl proteins in reversing multi-drug resistance
of β-elemene human gastric adenocarcinoma cells. Int J Mol Sci.
14:10075–10089. 2013.
|
|
14
|
Miedlich SU, Zalutskaya A, Zhu ED, et al:
Phosphate-induced apoptosis of hypertrophic chondrocytes is
associated with a decrease in mitochondrial membrane potential and
is dependent upon Erk1/2 phosphorylation. J Biol Chem.
285:18270–18275. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hong J, Samudio I, Chintharlapalli S, et
al: 1,1-bis (3′-indolyl)-1- (p-substituted phenyl) methanes
decrease mitochondrial membrane potential and induce apoptosis in
endometrial and other cancer cell lines. Mol Carcinog. 47:492–507.
2008.
|
|
16
|
Zuo L and Motherwell MS: The impact of
reactive oxygen species and genetic mitochondrial mutations in
Parkinson’s disease. Gene. 532:18–23. 2013.
|
|
17
|
Circu ML and Aw TY: Glutathione and
modulation of cell apoptosis. Biochim Biophys Acta. 1823:1767–1777.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Park HJ, Jeon YK, You DH, et al: Daidzein
causes cytochrome c-mediated apoptosis via the Bcl-2 family in
human hepatic cancer cells. Food Chem Toxicol. 60:542–549. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Brentnall M, Rodriguez-Menocal L, De
Guevara RL, et al: Caspase-9, caspase-3 and caspase-7 have distinct
roles during intrinsic apoptosis. BMC Cell Biol. 9:32–40. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gyulkhandanyan AV, Mutlu A, Freedman J, et
al: Markers of platelet apoptosis: methodology and applications. J
Thromb Thrombolysis. 33:397–411. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pucci B, Kasten M and Giordano A: Cell
cycle and apoptosis. Neoplasia. 2:291–299. 2000. View Article : Google Scholar
|
|
22
|
Robertson JD, Orrenius S and Zhivotovsky
B: Review: nuclear events in apoptosis. J Struct Biol. 129:346–358.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tait JF: Imaging of apoptosis. J Nucl Med.
49:1573–1576. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Radović N, Cucić S and Altarac S:
Molecular aspects of apoptosis. Acta Med Croatica. 62:249–256.
2008.(In Croatian).
|
|
25
|
Prosperini A, Juan-García A, Font G, et
al: Beauvericin-induced cytotoxicity via ROS production and
mitochondrial damage in Caco-2 cells. Toxicol Lett. 222:204–211.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yan F, Yang WK, Li XY, et al: A
trifunctional enzyme with glutathione S-transferase, glutathione
peroxidase and superoxide dismutase activity. Biochim Biophys Acta.
1780:869–872. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang L, Wang P, Wang H, et al: Fucoidan
derived from Undaria pinnatifida induces apoptosis in human
hepatocellular carcinoma SMMC-7721 cells via the ROS-mediated
mitochondrial pathway. Mar Drugs. 11:1961–1976. 2013.
|
|
28
|
Liu XT, Wang YR and Zhang M:
Mitochondria-mediated apoptosis: a review of recent studies. J
Environ Health. 30:182–184. 2013.
|
|
29
|
Zhao D, Huo LF, Liu H, et al:
Mitochondria, cytochrome c, caspase and cell apoptosis. J Med Pest
Control. 28:1337–1340. 2012.
|
|
30
|
Ng KB, Bustamam A, Sukari MA, et al:
Induction of selective cytotoxicity and apoptosis in human
T4-lymphoblastoid cell line (CEMss) by boesenbergin a isolated from
Boesenbergia rotunda rhizomes involves mitochondrial
pathway, activation of caspase 3 and G2/M phase cell cycle arrest.
BMC Complement Altern Med. 13:41–68. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ning Y, Riggins RB, Mulla JE, et al:
Interferon α restores breast cancer sensitivity to fulvestrant by
regulating STAT1, IRF1, NF-κB, BCL2 family members, and signaling
to caspase-dependent apoptosis. Mol Cancer Ther. 9:1274–1285.
2010.
|
|
32
|
Liang Y and Sundberg JP: SHARPIN regulates
mitochondria-dependent apoptosis in keratinocytes. J Dermatol Sci.
63:148–153. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Joshi S, Braithwaite AW, Robinson PJ and
Chircop M: Dynamin inhibitors induce caspase-mediated apoptosis
following cytokinesis failure in human cancer cells and this is
blocked by Bcl-2 overexpression. Mol Cancer. 10:78–103. 2011.
View Article : Google Scholar
|
|
34
|
Favaloro B, Allocati N, Graziano V, et al:
Role of apoptosis in disease. Aging. 4:330–349. 2012.
|
|
35
|
Merhi F, Tang R, Piedfer M, et al:
Hyperforin inhibits Akt1 kinase activity and promotes
caspase-mediated apoptosis involving Bad and Noxa activation in
human myeloid tumor cells. PLoS One. 6:e259632011. View Article : Google Scholar : PubMed/NCBI
|