Introduction
Gastric cancer is the second most common cause of
cancer-related mortality (1,2), and
the most common malignancy in Korea. Accumulated data have
established that carcinogenesis is a multi-step process associated
with alterations in cellular ontogenesis and tumor suppressor genes
necessary for malignant transformation (3,4).
Cancer cell metastasis and invasion are considered to represent
uncontrolled tissue remodeling and degradation of the extracellular
matrix (ECM) involved in the events of cancer progression (4,5).
Several ECM degradation models involving matrix metalloproteinases
(MMPs) and the plasminogen activators (PAs) proteolytic axis have
been shown in various cell types (6). Tumor cell invasion and the metastatic
process have been associated with elevated levels of cell surface
urokinase plasminogen activator (uPA)-mediated plasminogen
activation, while in vitro, in vivo and clinical
studies have suggested that inhibition of cell surface uPA
expression is associated with reduced tumor cell invasion,
metastasis and improved clinical outcome (7,8).
Hepatocyte growth factor (HGF), which is produced by
surrounding stromal cells, including fibroblasts and endothelial
cells, has been shown to be a significant factor responsible for
cancer cell invasion mediated by tumor stromal interaction. In our
previous study, we reported that HGF increased the expression of
uPA in gastric cancer cells (9).
Pro-inflammatory cytokines, such as IL-1, potently
induce expression of proteases of the MMP and plasmin families in
astrocytes in vitro (10,11).
IL-1 gene polymorphisms are associated with the development of
gastric atrophy and increased risk of gastric carcinoma. Several
lines of evidence have shown that the concentration of IL-1β in
plasma of patients with lung cancer is significantly elevated
(12) and is linked to the risk of
lung cancer (13). Moreover, IL-1β
has been reported to regulate uPA expression in various cancer
cells (14,15).
However, the mechanism by which IL-1β activates the
metastatic phenotype in stomach cancer is unknown. Since uPA has a
well-established role in tumor cell invasion and metastases, we
undertook the present study to determine whether or not the
expression of uPA is regulated by IL-1β, and to determine whether
or not ERK and NF-κB are the predominant pathways for uPA
regulation.
Materials and methods
Cell culture
We used two human gastric cancer cell lines [poorly
differentiated adenocarcinoma (NUGC-3) and moderately
differentiated tubular adenocarcinoma (MKN-28)], which were
obtained from the Korean Cell Line Bank (Seoul, Korea). The cells
were maintained in Dulbecco’s modified Eagle’s medium (DMEM)
supplemented with 10% fetal bovine serum (FBS), 1 mM sodium
pyruvate, 0.1 mM non-essential amino acids, 2 mM L-glutamine, 2X
vitamin solution, and 50 U/ml penicillin/streptomycin (Life
Technologies, Inc., Gaithersburg, MD, USA). Unless otherwise noted,
cells underwent passage and were removed from flasks when 70–80%
confluent.
Reagents and antibodies
The reagents and antibodies used in the experiments
were purchased from the following sources: horseradish
peroxidase-conjugated anti-mouse and anti-rabbit antibodies
(Bio-Rad Laboratories, Philadelphia, PA, USA); recombinant human
HGF (R&D Systems, Inc., Minneapolis, MN, USA); rabbit
polyclonal antibody against human IL-1β, Cell Signaling Technology,
Inc. (Beverly, MA, USA); human recombinant HGF, Becton-Dickinson
Lab (Beverly, MA, USA); uPA, American Diagnostica (Greenwich, CT,
USA); NF-κB, Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA);
PDTC, Calbiochem, Inc. (San Diego, CA, USA); PD098059, Biomol
Research Laboratories, Inc. (Butler Pike, PA, USA) and LY294002 was
from Calbiochem, Inc.
Northern blot analysis
Total RNA was extracted by acid-phenol-guanidium
thiocyanate-chloroform extraction. Total RNA (10 μg) was separated
on a 1% formaldehyde agarose gel and transferred to a Hybond
N+ nylon membrane by the capillary method. RNA was
cross-linked by UV irradiation (1,400 μJ/cm2) using a UV
cross-linker (Uvp, Inc., Upland, CA, USA). The membrane was
hybridized with a 32P-labeled c-fos or
c-jun probe overnight at 42°C, then washed in 2X SSC for 5
min at room temperature, 2X SSC/0.1% SDS at 42°C for 30 min, and
0.5X SSC/0.1% SDS at 42°C for 30 min. The membranes were exposed to
X-ray films at −70°C. Equal loading of the RNAs was confirmed by
hybridization with a 32P-labeled GAPDH probe.
cDNA microarray analysis
The cDNA microarray containing a set of 17,448
sequence-verified human cDNA clones was provided by Genomictree,
Inc. (Daejeon, Korea). cDNA microarray experiments were performed
as described by Yang et al (16). Briefly, total RNA (100 μg) was
reverse-transcribed in the presence of Cy3-dUTP or Cy5-dUTP (25 mM
stock; NEN Life Science Products, Boston, MA, USA) at 42°C for 2 h.
The labeled cDNA was then hybridized with the cDNA microarray at
65°C for 16 h. The hybridized slides were washed, scanned with an
Axon 4000B scanner (Axon Instruments), and analyzed using GenePix
Pro 4.0 (Axon Instruments). Raw data were normalized and analyzed
using GeneSpring 6.0 (Silicon Genetics). Genes were filtered based
on intensity in the control channel. When the control channel
values were <80 in all of the samples, we considered the results
to be unreliable genes. Intensity-dependent normalization (LOWESS)
was performed in which the ratio was reduced to the residual of the
LOWESS fit of the intensity vs. ratio curve. Average normalized
ratios were calculated by dividing the average normalized signal
channel intensity by the average normalized control channel
intensity. Welch’s ANOVA test was performed for P-values ≤0.1 of
0.05 to identify sample genes differentially expressed. Correlation
analysis was performed using Pearson correlation (−1 to 1). Spots
showing changes ≥2-fold were considered significant.
Semi-quantitative reverse
transcription-polymerase chain reaction (RT-PCR)
Complementary DNA (cDNA) was synthesized from total
RNA using MMLV reverse transcriptase (Promega Corp., Madison, WI,
USA) by the oligo (dT) priming method in a 10 μl reaction mixture.
PCR was performed in 10 μl reaction volume containing 10 mM
Tris-HCl (pH 8.5), 50 mM KCl, 1 μl cDNA, 200 μM dNTPs, 1 mM
MgSO4, 1U of platinum Pfx Taq polymerase, and 2 μM
primers. The reactions were as follows: the initial denaturation at
95°C for 4 min, 27 cycles at 94°C for 15 sec, 60°C for 15 sec, and
72°C for 30 sec and the final extension at 72°C for 10 min. The PCR
products were separated on a 1.5% agarose gel containing ethidium
bromide and visualized on a UV transilluminator.
Western blot analysis
Cells were harvested and incubated with a lysis
buffer [50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA, 1% Triton
X-100, 10% glycerol, 1 mM PMSF, 1 mM sodium vanadate, and 5 mM NaF)
with protease inhibitors and centrifuged at 15,000 rpm for 10 min
at 4°C. Proteins (50 μg) were separated on 10% SDS-polyacrylamide
gels and transferred to nitrocellulose membranes. The membranes
were soaked with 5% non-fat dried milk in TTBS [10 mM Tris-HCl (pH
7.5), 150 mM NaCl, and 0.05% Tween-20] for 30 min, then incubated
overnight with a primary antibody at 4°C. After washing 6 times
with TTBS for 5 min, membranes were incubated with a horseradish
peroxidase-conjugated secondary antibody for 1 h 30 min at 4°C. The
membranes were rinsed 3 times with TTBS for 30 min and the
antigen-antibody complex was detected using an enhanced
chemiluminescence detection system.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
The cells and IL-1β shRNA (1,500/well) were seeded
in 96-well plates in DMEM supplemented with 5% FBS and incubated
for 24 h. Cells were then serum-starved for 24 h and treated for 72
h with or without HGF (10 ng/ml). At the end of this incubation
period, 50 μl of a 2 mg/ml MTT solution was added and the cells
were allowed to incubate for 3 h at 37°C. The supernatant was
carefully removed by aspiration, and convert dye was dissolved with
100 μl of DMSO. The plates were placed in a microplate shaker for 5
min, and the absorbance was measured at 570 nm using a Biorad
Multiskan plate reader.
IL-1β knockdown with short hairpin RNA
(shRNA)
The human IL-1β-specific shRNA expression vector
(IL-1β shRNA, RHS4533-NM_000576) containing an IL-1β-targeted shRNA
sequence (AAACCCAGGGCTGCCTTGGAAAAG) was purchased from Open
Biosystems (Huntsville, AL, USA). Cells were transfected with IL-1β
shRNA using Lipofectamine (Life Technologies, Inc., Gaithersburg,
MD, USA). Clonal selection was conducted by culturing with
puromycin (10 μg/ml) followed by serial dilution of the cells.
Stable transfectant clones with low expression of the target genes
were identified by western blot analysis.
Standard two-chamber invasion assay
Control and transfected cells (1×104)
were placed in the upper chamber of a Matrigel migration chamber
with 0.8-μm pores (Thermo Fisher Scientific, Houston, TX, USA) in
media containing 5% FBS with or without HGF (10 ng/ml). Following
incubation for 48 h, cells were fixed and stained using a HEMA 3
stain set (Curtis Matheson Scientific, Houston, TX, USA) according
to the manufacturer’s instructions. The stained filter membrane was
cut and placed on a glass slide. The migrated cells were counted
under light microscopy (10 fields at ×200 power).
Chromatin immunoprecipitation (ChIP)
assay
The ChIP assay was performed using a ChIP assay kit
(Upstate Biotechnology, Waltham, MA, USA) following the
manufacturer’s directions. Briefly, cells were fixed with 1%
formaldehyde at 37°C for 10 min. Cells were washed twice with
ice-cold PBS with protease inhibitors (1 mM phenylmethylsulphonyl
fluoride, 1 mg/ml of aprotinin, and 1 mg/ml of pepstatin A),
scraped and pelleted by centrifugation at 48°C. Cells were
resuspended in a lysis buffer [1% SDS, 10 mM EDTA, and 50 mM
Tris-HCl (pH 8.1)], incubated for 10 min on ice, and sonicated to
shear DNA. After sonication, lysate was centrifuged for 10 min at
13,000 rpm at 48°C. The supernatant was diluted in ChIP dilution
buffer [0.01% SDS, 1% Triton X-100, 2 mM EDTA, 16.7 mM Tris-HCl (pH
8.1), 167 mM NaCl, and protease inhibitors]. Primary antibodies
were added and incubated overnight at 48°C with rotation. The
immunocomplex was collected by protein A/G agarose beads and washed
with low-salt washing buffer [0.1% SDS, 1% Triton X-100, 2 mM EDTA,
200 mM Tris-HCl (pH 8.1), and 150 mM NaCl], high-salt buffer [0.1%
SDS, 1% Triton X-100, 2 mM EDTA, 200 mM Tris-HCl (pH 8.1), and 500
mM NaCl], LiCl washing buffer [0.25 M LiCl, 1% NP40, 1%
deoxycholate, 1 mM EDTA, and 10 mM Tris-HCl (pH 8.1)], and TE
buffer [10 mM Tris-HCl and 1 mM EDTA (pH 8.0)]. The immunocomplex
was then eluted using elution buffer (1% SDS, 0.1 M
NaHCO3, and 200 mM NaCl) and the cross-links were
reversed by heating at 65°C for 4 h. After reaction, the samples
were adjusted to 10 mM EDTA, 20 mM Tris-HCl (pH 6.5), and 40 mg/ml
of proteinase K, and incubated at 45°C for 1 h. DNA was recovered
and subjected to PCR amplification of the uPA promoter region
(−1747 to −2042) using 5′-GAGGGGGCGGAAGGGGAGAA-3′
(forward) and 5′-TGTGGTCAGTTTTGTTTGGATTTG-3′
(reverse).
uPA promoter analysis
The transcriptional regulation of NF-κB by HGF was
examined using transient transfection with a uPA promoter
luciferase reporter construct (uPA-pMetLuc reporter). Cell
transfection was performed using Lipofectamine™ 2000 (Invitrogen,
Carlsbad, CA, USA) according to the manufacturer’s instructions.
For the luciferase reporter gene assay, control cells and shIL-1β
expression cells were co-transfected with 1 μg of
uPA-pMetLuc-reporter plamids and 0.05 μg of pHYK plasmid, which was
used as an internal transfection-efficiency control. Transfected
cells were stimulated with or without 10 ng/ml of HGF for 1 h. The
promoter activity was analyzed in each well of the cultured medium
using a Dual Glo® luciferase assay system with a Turner
Designs instrument luminometer (Turner Designs, Inc., Sunnyvale,
CA, USA). The measured luminescence of firefly luciferase was
divided by Renilla luciferase and the resulting quotient
corresponded to the relative amounts of luciferase.
Statistical analysis
Values are expressed as means ± SD. The Student’s
t-test was employed for the analyses. A P-value of <0.05 was
considered to indicate a statistically significant difference.
Results
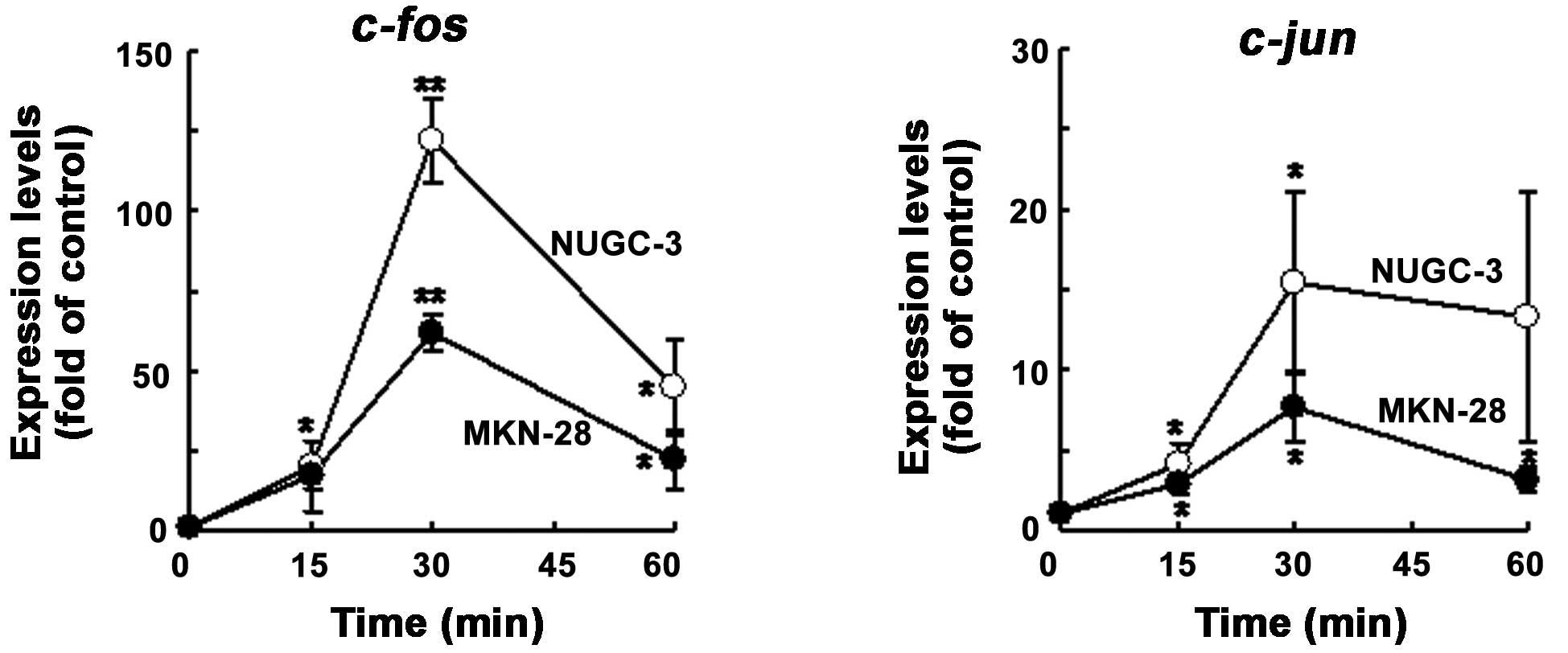
Induction of c-fos and c-jun by HGF in
NUGC-3 and MKN-28 cells
As it is well-known that HGF induces c-fos
and c-jun in a variety of cells, we tested whether or not
NUGC-3 and MKN-28 cells also showed HGF-mediated c-fos and
c-jun induction by northern blot analysis. As expected, the
levels of expression of c-fos mRNA were increased with HGF
at an early phase (to 30 min), then decreased in both cell lines;
the c-jun mRNA levels gradually increased until 1 h after
treatment with HGF in both cell lines (Fig. 1).
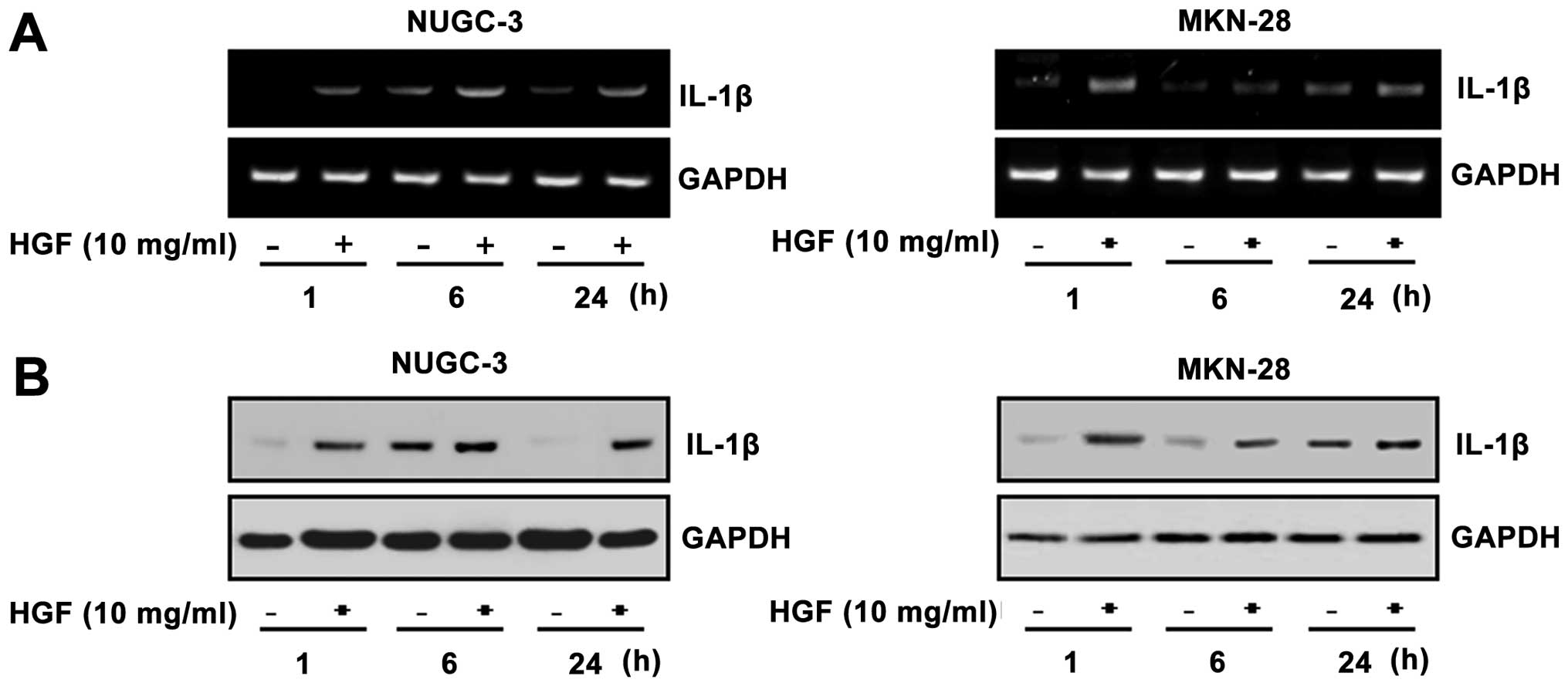
Identification of HGF-responsive genes by
cDNA microarray in NUGC-3 cells
In an attempt to explore differentially-expressed
genes in NUGC-3 cells treated with HGF, we used 17k human cDNA
microarrays. The initial analysis of the cDNA microarray expression
data indicated that the presence of 26 genes changed by ≥2-fold
after HGF treatment. A variety of genes were found to be
differentially expressed. The genes were selected and the
expressions were confirmed by RT-PCR. RT-PCR showed that the level
of expression of IL-1β was increased after HGF-treatment (Fig. 2A). The level of IL-1β protein was
also enhanced by HGF treatment, as confirmed by western blot
analysis (Fig. 2B).
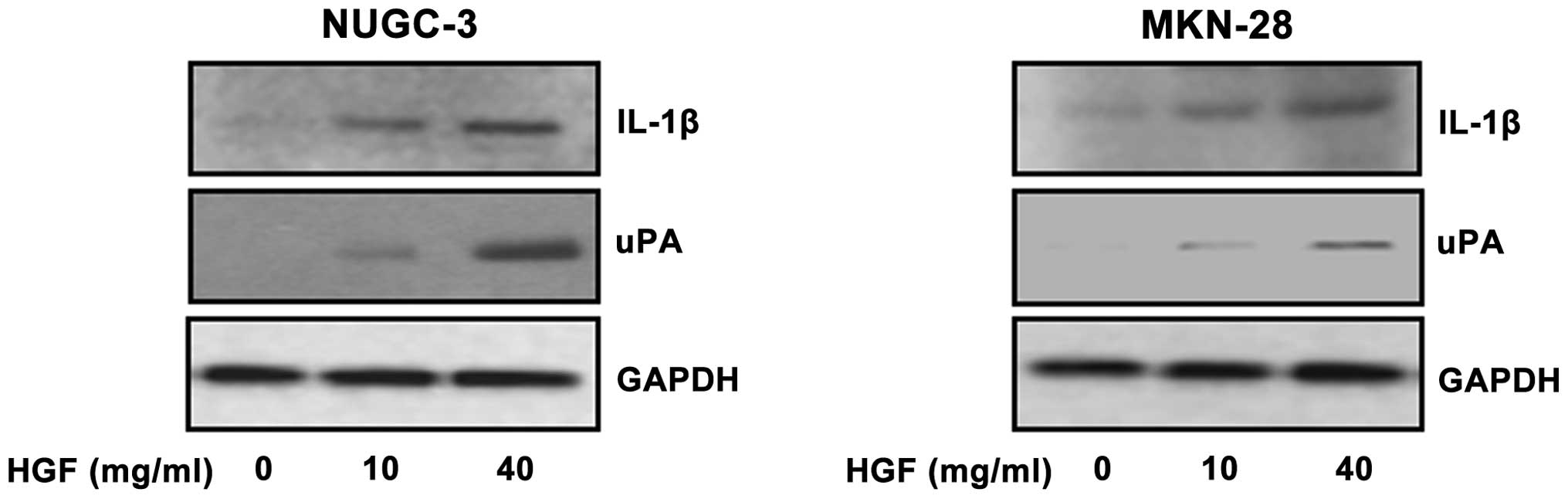
Activation of IL-1β and uPA following
treatment with HGF
To elucidate the IL-1β and uPA expression following
HGF treatment, we analyzed the expression after treatment with 0,
10 and 40 μg/ml of HGF in NUGC-3 and MKN-28 gastric cancer cell
lines. IL-1β expression was increased in HGF-treated cells in a
dose-dependent manner and uPA protein also was increased similar to
IL-1β in a dose-dependent manner (Fig.
3).
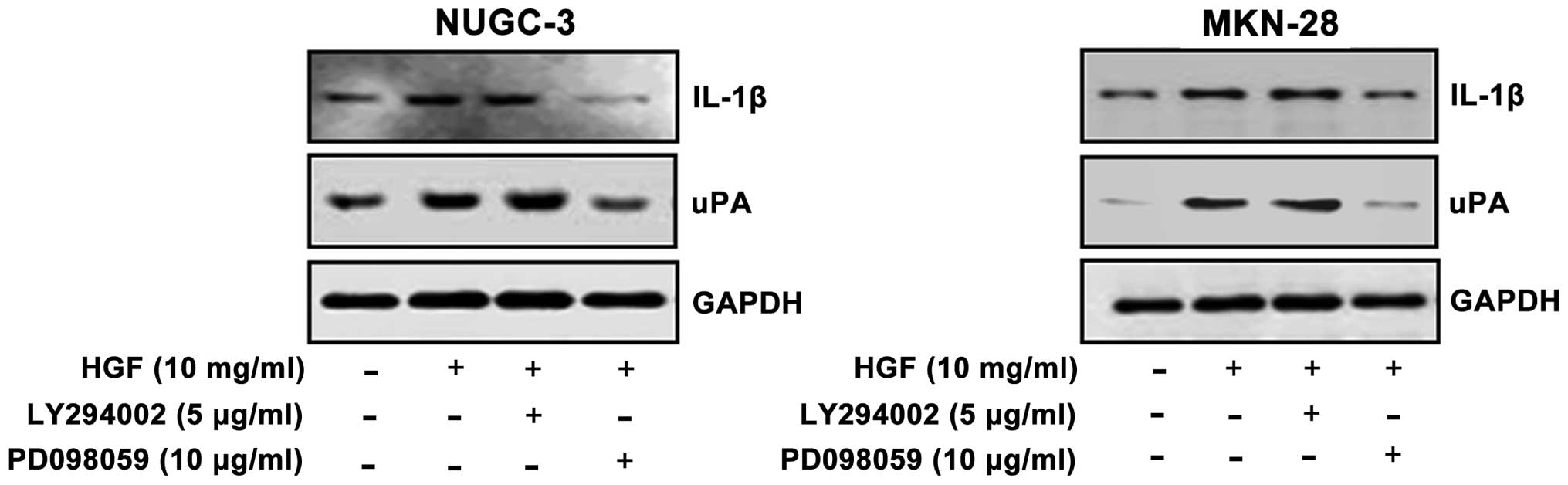
Effects of PD098059 and LY294002 on IL-1β
and uPA expression
To test whether or not ERK and PI3-kinase activation
is involved in HGF-induced IL-1β and uPA expression, the cells were
pre-treated with a MEK inhibitor (PD098059) or a PI3-kinase
inhibitor (LY294002) and measured by western blotting. The cells
showed that HGF-mediated IL-1β expression and uPA was decreased
with PD098059. Densitometric analysis indicated that pre-treatment
of PD098059 resulted in 2–4-fold decrements of IL-1β expression and
2–3-fold decrements of uPA expression in both cell lines. In
contrast, pre-treatment of LY294002 showed no change in IL-1β and
uPA expression. These results suggested that HGF-mediated IL-1β and
uPA expression is regulated by ERK, and not PI3-kinase (Fig. 4).
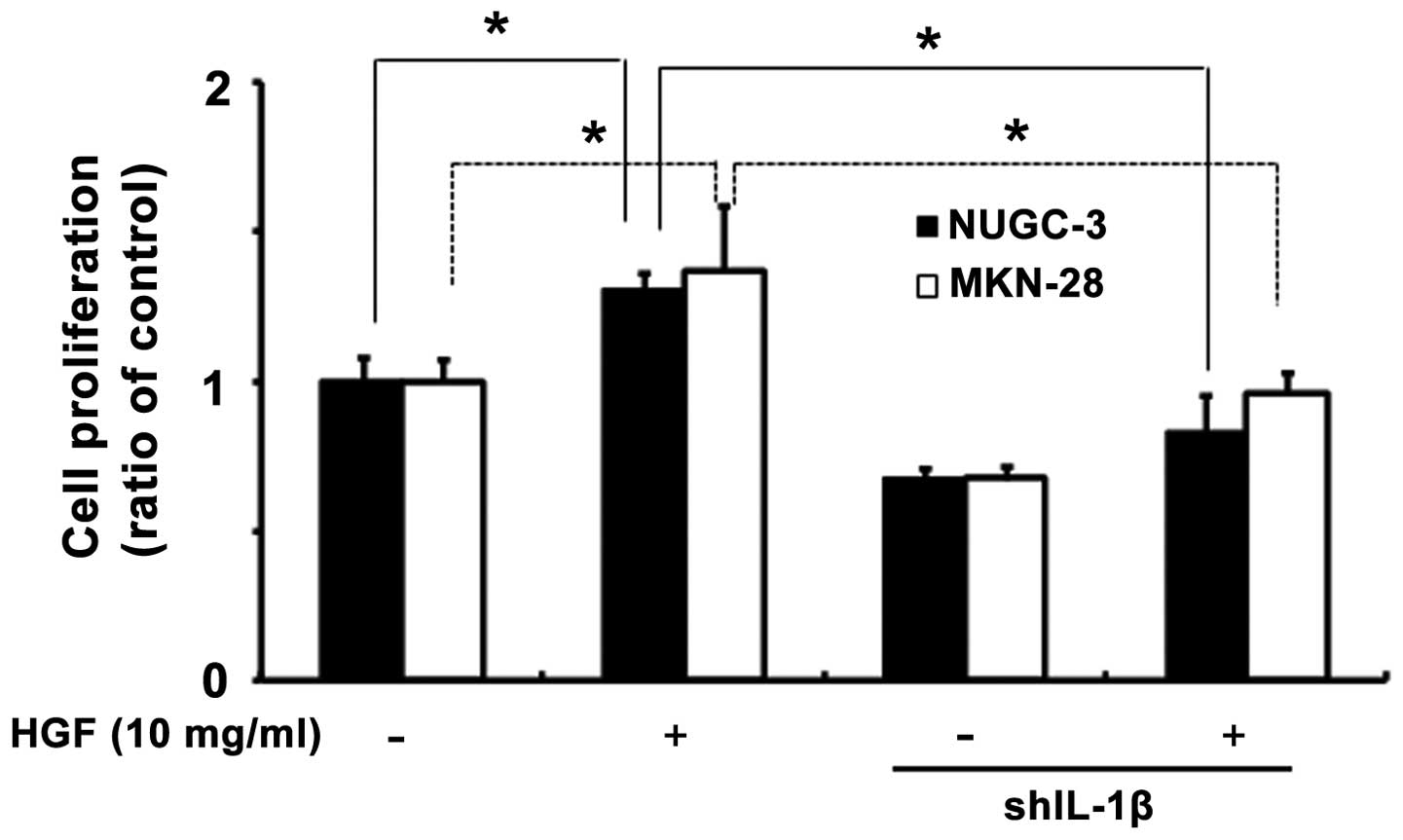
Effects of IL-1β on cell viability with
IL-1β shRNA stable cells
To determine whether or not IL-1β plays a role in
cell viability, we generated IL-1β shRNA stable cells. Knockdown
IL-1β shRNA stable cells was confirmed by RT-PCR (data not shown).
An MTT assay was performed after treatment of cells with HGF in
both cell lines. Following 72 h of treatment, IL-1β had cell
viability effects induced by HGF (Fig.
5).
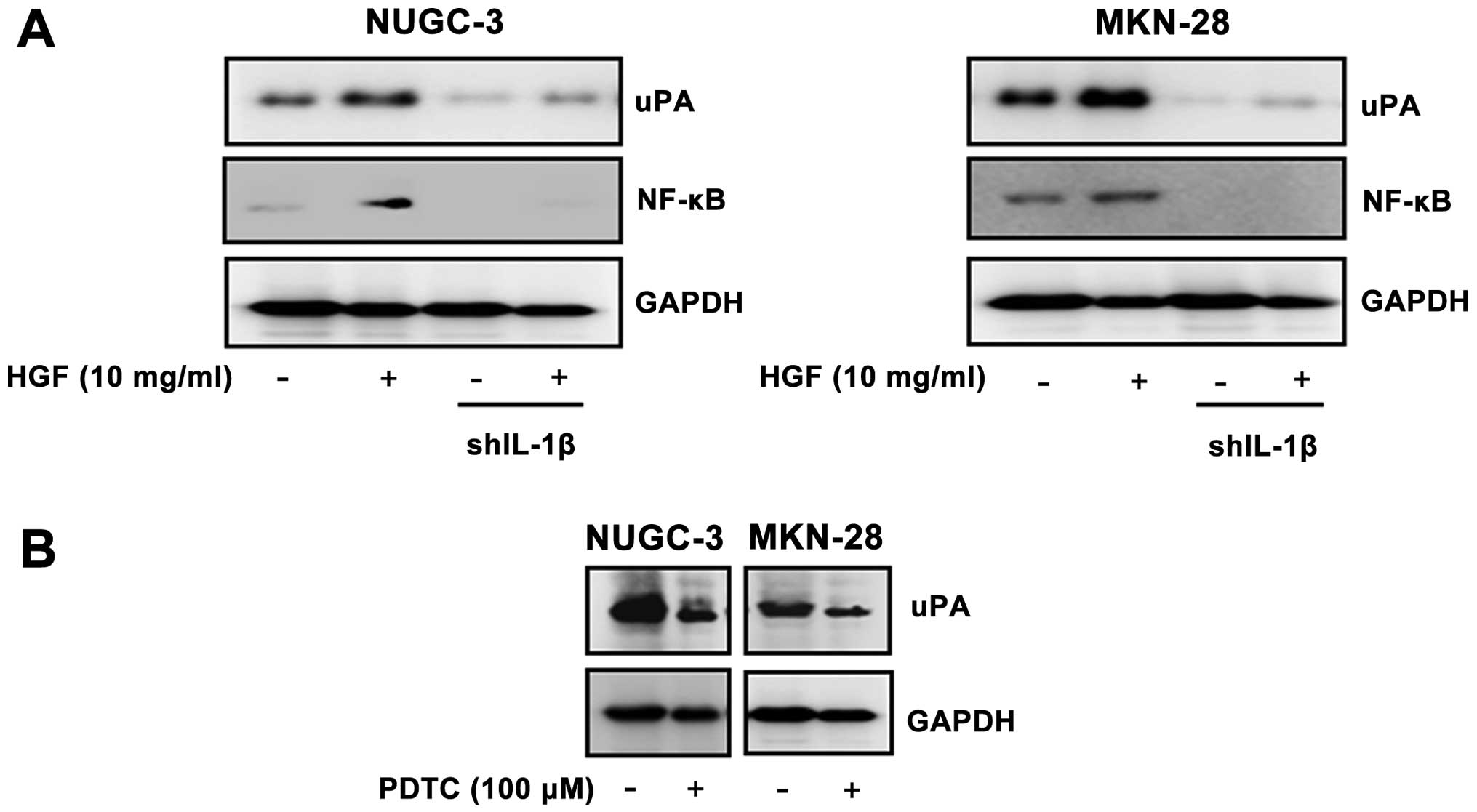
Effects of IL-1β shRNA stable cells on
regulation of uPA and a role of NF-κB
Following selection, cloning, and amplification of
stable cells, we generated IL-1β stable cells. When the cell lines
were treated with HGF to determine the effect of IL-1β knockdown
cells on regulation of uPA and NF-κB, a decrease in IL-1β mediated
uPA and NF-κB expression occurred in both gastric cancer cell lines
(Fig. 6A). To assess the role NF-κB
activity in HGF-mediated uPA regulation, we analyzed the level of
expression of uPA after treatment with the NF-κB inhibitor, PDTC
(100 mM). Expression of uPA was decreased in the PDTC treated cells
(Fig. 6B).
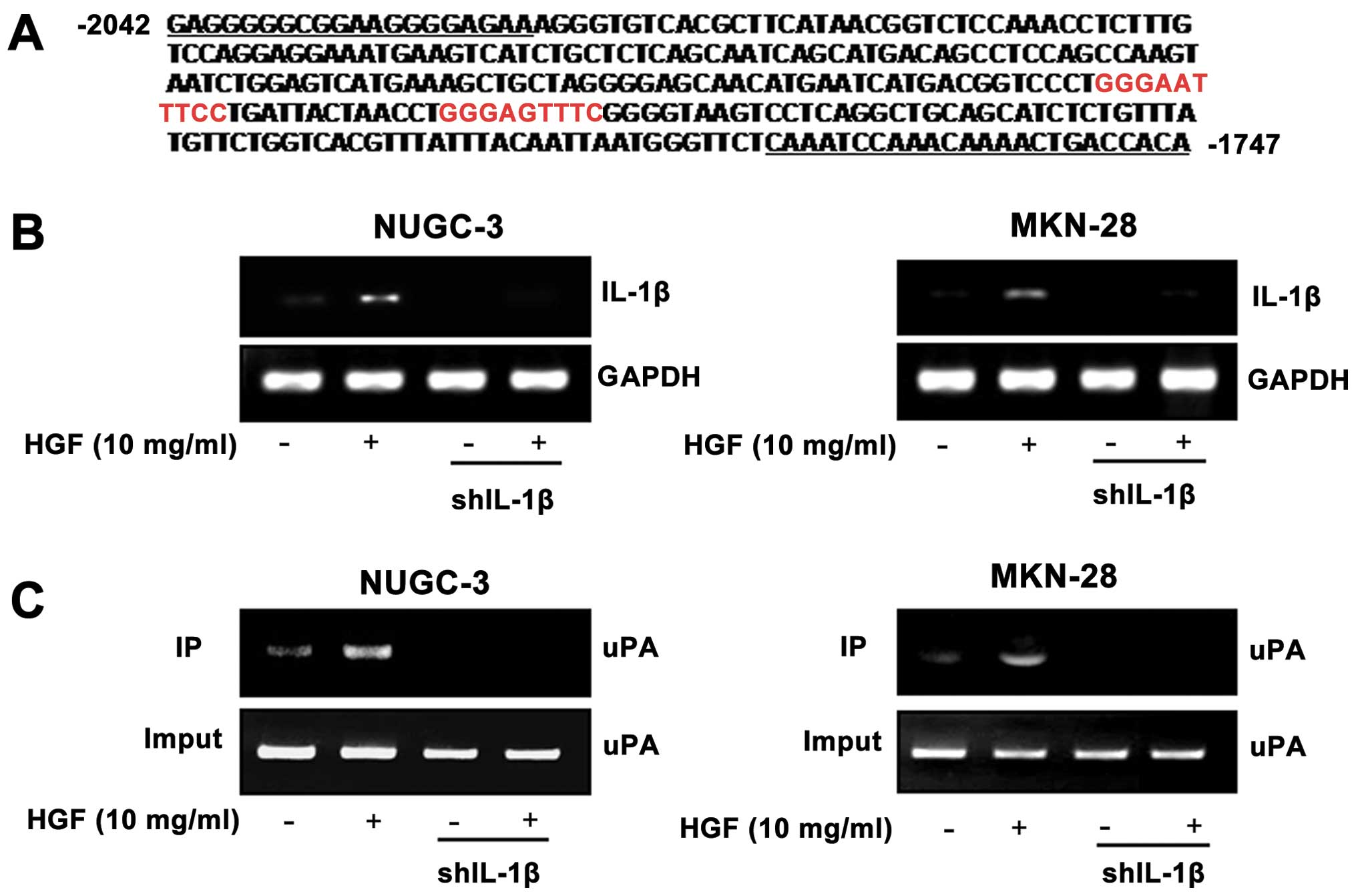
Binding of NF-κB to a uPA promoter in
both IL-1β shRNA cells and the luciferase reporter gene assay
We analyzed the promoter sequence of uPA genes to
identify the putative NF-κB binding sequence using the TESS program
(17). Two putative NF-κB binding
sites were identified within the uPA promoter. The NF-κB
transcription factor binding site for the uPA promoter was located
within the proximal promoter regions upstream of the
transcriptional start site (Fig.
7A). IL-1β shRNA-stable cells showed a decrease in the levels
of IL-1β RNA as compared with control cells (Fig. 7B). To demonstrate a comparable NF-κB
binding site function in the uPA promoter, IL-1β shRNA and control
cells were treated with 10 μg/ml of HGF and binding of NF-κB to
putative NF-κB binding sites was measured by the ChIP assay. HGF
enhanced the binding activity of NF-κB to the uPA promoter with
relatively strong constitutive activity in control cells, but not
in the IL-1β shRNA cells (Fig. 7C).
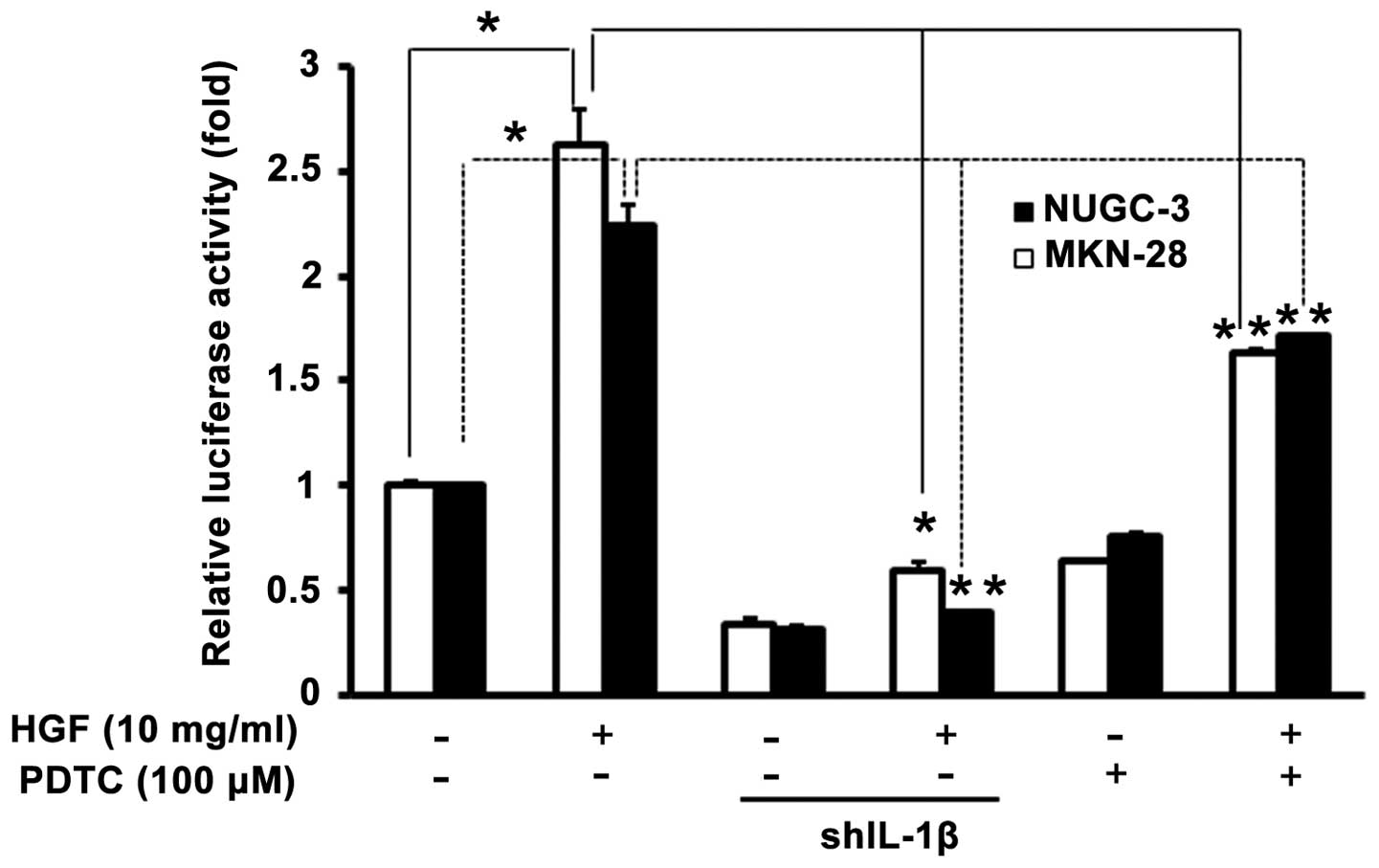
To further confirm the functional role of HGF in the activation of
the promoter of genes identified by ChIP analysis, both cells were
co-transfected with uPA promoter, then cultured with or without
additional HGF, NF-κB inhibitor (PDTC). Knockout of the IL-1β gene
decreased the basal and HGF-induced uPA promoter activity in both
cells (Fig. 8). These findings
provide direct evidence that the portion of the uPA promoter
containing NF-κB sites is optional and activated by HGF-induced
IL-1β.
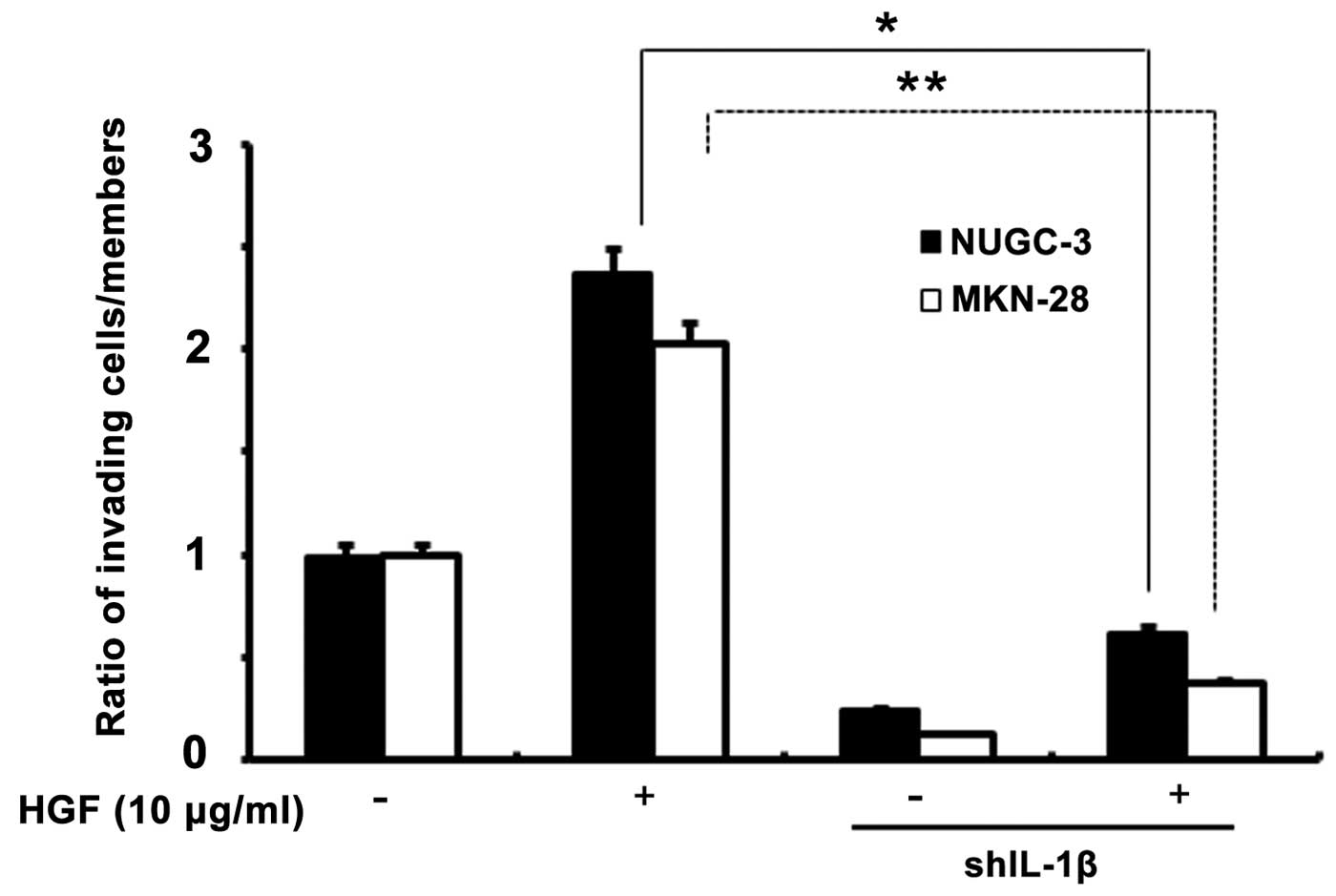
Effects of IL-1β knockdown cells on tumor
invasion
To assess the role of IL-1β in HGF-mediated cell
invasion, an in vitro invasion using a Matrigel migration
chamber assay was used in both transfected cells. IL-1β shRNA cells
had a decrease in HGF-mediated cell invasion compared to the
control cells, suggesting that IL-1β may play an important role in
HGF-mediated cell invasion through NF-κB and uPA (Fig. 9).
Discussion
The lack of a reduction in gastric cancer mortality
rates indicates that there is an urgent need for the identification
of novel targets that prevent or inhibit invasion and metastasis.
Tumor invasion and metastasis are the major characteristics of
aggressive phenotypes of various human cancers, and therefore the
major causes of cancer mortality. Cancer cells must acquire several
properties to disseminate from the primary tumor, including the
ability to degrade and migrate through the ECM, a process called
invasion (18). Tumor invasion and
metastasis are often associated with increased expression of
ECM-degrading proteases, among which uPA is of central importance
(19).
IL-1 is a potent pro-inflammatory cytokine that is
upregulated in the presence of Helicobacter pylori and is
important in initiating and amplifying the inflammatory response to
infection (20,21). The IL-1 receptor antagonist (K-1ra)
is a naturally-occurring anti-inflammatory cytokine that binds
competitively to IL-1 receptors, thus blunting the potentially
injurious effects of IL-1. Polymorphisms of IL-1β and IL-1 RN
(encoding IL-1ra) have been reported to be associated with the risk
for gastric carcinoma (22).
Another report showed that the plasma level of IL-1β is
significantly elevated in patients with lung cancer (12). Although uPA is regulated by
cytokines, such as IL-1β, the intracellular signaling pathway
leading to uPA expression remains largely unknown. Much effort has
been directed at defining the signal transduction pathways induced
by IL-1β. Several studies have documented that MAPKs have roles in
IL-1β-induced signal transduction, but the profiles of
IL-1β-induced kinase activation appear to vary in a cell
type-dependent fashion (23).
Cheng et al (24) investigated whether or not
IL-1β-induced expression of uPA was involved in lung cancer
progression. In their study, IL-1β significantly induced uPA
expression and activity via PKCα-dependent JNK1/2 and NIK cascades,
linking to IKKα/β activation and p65 translocation and
transcription activity using pharmacologic inhibitors and
transfection with dominant-negative mutants and siRNAs. In an
attempt to explore differentially-expressed genes in stomach cancer
cells (NUGC-3) treated with growth factor, such as HGF, we found
that IL-1β had a 3.26-fold upregulation using human cDNA
microarrays. Although IL-1β-induced uPA regulation was similar to
other studies (15,24), the mechanism by which the pathway
appears to be the predominant pathway for uPA regulation was shown
differently. Our results are consistent with the report showing
that activation of p42/p44 MAPK is involved in the expression of
uPA in different cell types (25,26).
These differences may be due to cell types and different
experimental conditions used in these assays. In addition, we found
that pre-treatment with LY294002 could not enhance uPA expression.
In the subsequent experiments, we characterized the sites in the
uPA promoter that were required for IL-1β-induced uPA gene
expression in NUGC-3 and MKM-28 cells.
As shown in Fig. 7,
regions of the promoter containing candidate-binding sites for
NF-κB at (−1880 to −1871, and −1857 to −1849) were required for
IL-1β-mediated activation of the full-length (295 bp) uPA promoter.
To further confirm the functional role of HGF in the activation of
the promoter of uPA identified by ChIP analysis, we found that the
portion of the uPA promoter containing NF-κB sites is optimal and
is activated by HGF/IL-1β-induced NF-κB. This may be the first
report to provide evidence that ERK plays an important role in the
regulation of IL-1β-induced NF-κB activation in human gastric
NUGC-3 and MKN-28 cells.
While the pathway by which tumor cells acquire
invasive and metastatic capacity is probably cell type-dependent,
little is known regarding this important biological process. The
NF-κB transcription factor family is one of the major mediators of
the intracellular function of IL-1β and the uPA promoter has NF-κB
binding sites (27,28).
In our additional results, we reported that IL-1β
induced IL-8 overexpression in IL-1β shRNA gastric cancer cell
lines. Although it is well-known that IL-1β upregulates IL-8
expression in various cells, such as endothelial cells, epithelial
cells, and smooth muscle cells (29), the molecular mechanism for
IL-1β-induced IL-8 expression in gastric cancer is not known.
Kitadai et al (30)
demonstrated that human gastric carcinomas overexpressed IL-8 and
the IL-8 mRNA level directly correlated with the vascularity of the
gastric tumor. The process of angiogenesis is essential for tumor
growth. In the search for a better understanding of the process of
tumor angiogenesis, it is necessary to acknowledge that the
development of new blood vessels is dependent on the function and
activity of tumor cells in the vascular microenvironment.
Furthermore, HGF-induced upregulation of IL-8 might be more complex
and could possibly be controlled by multiple transcriptional and/or
post-transcriptional mechanisms. It is conceivable that a cytokine
network exists between inflammatory cells producing cytokines that
can initiate signaling in tumor cells.
In summary, we identified IL-1β-induced uPA
expression via activation of the ERK1/2 and NF-κB pathways, which
results in invasion of gastric cancer cells. These interactions
might be promising targets for the development of future treatment
strategies to improve the response rate and overall survival after
treatment of gastric cancer.
Acknowledgements
The present study was supported by a grant from the
Yeungnam University Medical Center (2012).
References
|
1
|
Pisani P, Parkin DM, Bray F and Ferlay J:
Estimates of the worldwide mortality from 25 cancers in 1990. Int J
Cancer. 83:18–29. 1999. View Article : Google Scholar
|
|
2
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
3
|
Fearon ER and Vogelstein B: A genetic
model for colorectal tumorigenesis. Cell. 61:759–767. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kinzler KW and Vogelstein B: Life (and
death) in a malignant tumour. Nature. 379:19–20. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Romer J: Skin cancer and wound healing.
Tissue-specific similarities in extracellular proteolysis. APMIS.
111(Suppl 107): 1–36. 2003.
|
|
6
|
Pepper MS: Role of the matrix
metalloproteinase and plasminogen activator-plasmin systems in
angiogenesis. Arterioscler Thromb Vasc Biol. 21:1104–1117. 2001.
View Article : Google Scholar
|
|
7
|
Foekens JA, Peters HA, Look MP, et al: The
urokinase system of plasminogen activation and prognosis in 2780
breast cancer patients. Cancer Res. 60:636–643. 2000.PubMed/NCBI
|
|
8
|
Giavazzi R and Taraboletti G: Preclinical
development of metalloproteasis inhibitors in cancer therapy. Crit
Rev Oncol Hematol. 37:53–60. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee KH, Choi EY, Hyun MS, et al:
Hepatocyte growth factor/c-met signaling in regulating urokinase
plasminogen activator in human stomach cancer: a potential
therapeutic target for human stomach cancer. Korean J Intern Med.
21:20–27. 2006. View Article : Google Scholar
|
|
10
|
Falsig J, Porzgen P, Lund S, Schrattenholz
A and Leist M: The inflammatory transcriptome of reactive murine
astrocytes and implications for their innate immune function. J
Neurochem. 96:893–907. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gottschall PE and Yu X: Cytokines regulate
gelatinase A and B (matrix metalloproteinase 2 and 9) activity in
cultured rat astrocytes. J Neurochem. 64:1513–1520. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
De Vita F, Orditura M, Auriemma A,
Infusino S and Catalano G: Serum concentrations of proinflammatory
cytokines in advanced non small cell lung cancer patients. J Exp
Clin Cancer Res. 17:413–417. 1998.PubMed/NCBI
|
|
13
|
Asada M, Yasuda H, Ebihara S, et al:
Interleukin-1β gene polymorphisms associated with risk of lung
cancer in Japanese. Lung Cancer. 54:261–263. 2006.
|
|
14
|
Niiya K, Shinbo M, Ozawa T, Hayakawa Y and
Sakuragawa N: Modulation of urokinase-type plasminogen activator
gene expression by inflammatory cytokines in human pre-B lymphoma
cell line RC-K8. Thromb Haemost. 74:1511–1515. 1995.PubMed/NCBI
|
|
15
|
Tran-Thang C, Kruithof E, Lahm H, Schuster
WA, Tada M and Sordat B: Modulation of the plasminogen activation
system by inflammatory cytokines in human colon carcinoma cells. Br
J Cancer. 74:846–852. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang SH, Kim JS, Oh TJ, et al:
Genome-scale analysis of resveratrol-induced gene expression
profile in human ovarian cancer cells using a cDNA microarray. Int
J Oncol. 22:741–750. 2003.PubMed/NCBI
|
|
17
|
Schug J and Overton GC: Technical Report
CBIL-TR-1997-1001-v0. 0. Computational Biology and Informatics
Laboratory, School of Medicine, University of Pennsylvania;
1997
|
|
18
|
Rao JS: Molecular mechanisms of glioma
invasiveness: the role of proteases. Nat Rev Cancer. 3:489–501.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sidenius N and Blasi F: The urokinase
plasminogen activator system in cancer: recent advances and
implication for prognosis and therapy. Cancer Metastasis Rev.
22:205–222. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Noach LA, Bosma NB, Jansen J, Hoek FJ, van
Deventer SJ and Tytgat GN: Mucosal tumor necrosis factor-alpha,
interleukin-1 beta, and interleukin-8 production in patients with
Helicobacter pylori infection. Scand J Gastroenterol.
29:425–429. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Basso D, Scrigner M, Toma A, et al:
Helicobacter pylori infection enhances mucosal
interleukin-1β, interleukin-6, and the soluble receptor of
interleukin-2. Int J Clin Lab Res. 26:207–210. 1996. View Article : Google Scholar
|
|
22
|
El-Omar EM, Carrington M, Chow WH, et al:
Interleukin-1 polymorphisms associated with increased risk of
gastric cancer. Nature. 404:398–402. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guan Z, Baier LD and Morrison AR: p38
mitogen-activated protein kinase down-regulates nitric oxide and
up-regulates prostaglandin E2 biosynthesis stimulated by
interleukin-1β. J Biol Chem. 272:8083–8089. 1997.PubMed/NCBI
|
|
24
|
Cheng CY, Hsieh HL, Sun CC, Lin CC, Luo SF
and Yang CM: IL-1β induces urokinse-plasminogen activator
expression and cell migration through PKCα, JNK1/2, and NF-κB in
A549 cells. J Cell Physiol. 219:183–193. 2009.
|
|
25
|
Lee D, Yang Y, Lee S, et al: Macrophage
inhibitory cytokine-1 induces the invasiveness of gastric cancer
cells by up-regulating the urokinase-type plasminogen activator
system. Cancer Res. 63:4648–4655. 2003.PubMed/NCBI
|
|
26
|
Estrella VC, Eder AM, Liu S, et al:
Lysophosphatidic acid induction of urokinase plasminogen activator
secretion requires activation of the p38MAPK pathway.
Int J Oncol. 31:441–449. 2007.PubMed/NCBI
|
|
27
|
Reuning U, Guerrini L, Nishiguchi T, et
al: Rel transcription factors contribute to elevated urokinase
expression in human ovarian carcinoma cells. Eur J Biochem.
259:143–148. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kamio N, Hashizume H, Nakao S, Matsushima
K and Sugiya H: IL-1β stimulates urokinase-type plasminogen
activator expression and secretion in human dental pulp cells.
Biomed Res. 28:315–322. 2007.
|
|
29
|
Jung YD, Fan F, McConkey DJ, et al: Role
of P38 MAPK, AP-1, and NF-κB in interleukin-1β-induced IL-8
expression in human vascular smooth muscle cells. Cytokine.
18:206–213. 2002.
|
|
30
|
Kitadai Y, Takahashi Y, Haruma K, et al:
Transfection of interleukin-8 increases angiogenesis and
tumorigenesis of human gastric carcinoma cells in nude mice. Br J
Cancer. 81:647–653. 1999. View Article : Google Scholar : PubMed/NCBI
|