Introduction
The phosphatase and tensin homologue (PTEN) gene is
registered with a mutation rate of 12.4% for colorectal cancer in
the COSMIC database (http://cancer.sanger.ac.uk/cancer-genome/projects/cosmic/).
This places the PTEN gene among the ‘hills’ of the colorectal
cancer genomic landscape (1). The
mutations are distributed fairly evenly over the gene’s 9 exons
which by recent concepts is a key molecular feature of
tumour-suppressor genes. In addition, losses of genomic material at
its locus 10p23.3 or epigenetic silencing of the gene promoter are
observed. Considering that the PTEN protein participates in cell
growth control by counteracting Akt signaling in the cytoplasm and
activity of the mitogen-activated protein kinase in the nucleus
(2), there seems to be a strong
case for PTEN as a tumour-suppressor gene. In fact, PTEN features
in the literature as a tumour-suppressor gene ever since its first
description (3).
For some types of cancers, most notably endometrial
adenocarcinoma, prostate cancer, gliomas and malignant melanoma,
the evidence for PTEN as a tumour-suppressor gene is strong indeed.
However, in colorectal carcinoma this may not be quite so. First,
PTEN gene mutations are particularly frequent among the ~15% of
colorectal carcinomas driven by microsatellite instability
(4), whereas the classical
tumour-suppressor gene mechanism that combines mutation of one
allele and loss of the other allele would rather be expected to
apply to colorectal carcinomas driven by chromosomal instability
(CIN). Second, published studies that have addressed allelic loss
usually employed microsatellite marker analyses using DNA from
tumour tissue homogenates, i.e. mixtures of the neoplastic cells
and the normal stroma. As a limitation of this technique, ‘loss of
heterozygosity’ (LOH) in most cases can only be scored according to
an arbitrary cut-off applied to the tumour/normal ratios of the PCR
products visualised in electropherograms or gel-autoradiographs. A
wise note of caution concerning this type of study has been made by
Devilee et al (5).
PTEN may be a colorectal carcinoma tumour-suppressor
gene by a less rigorous definition, i.e. it may be one of the
so-called ‘epi-driver genes’ (6).
In this type of tumour-suppressor genes, loss of expression (and
function) occurs e.g. by promoter methylation (epigenetic gene
silencing). PTEN promoter methylation has been reported in several
types of cancers, colorectal carcinoma among them (7).
In the present study, we investigated the mutational
status, LOH, promoter methylation, and, by immunohistochemistry,
expression of the PTEN gene in a series of 42 colorectal carcinomas
for a synoptic view on all molecular processes that by recent
concepts would identify PTEN as a tumour-suppressor gene in this
type of cancer. Importantly, we used tumour tissue from low-passage
xenotransplants or neoplastic glands isolated by laser-capture
microdissection for the LOH studies.
Materials and methods
Colorectal carcinoma specimens
In the present study, we used colorectal carcinoma
specimens collected at Rostock University as part of and according
to the guidelines and procedures of the ‘Norddeutsche Tumorbank’ as
has been described in detail in a previous publication (8). Briefly, surgical resection specimens
from patients with biopsy proven colorectal carcinoma
(adenocarcinoma not otherwise specified) were received fresh from
the operating theatre; patients had not received chemotherapy or
radiation prior to surgery. Small cubes (~3 × 3 × 3 mm) were
dissected from vital parts of the tumours by the senior author,
snap-frozen in liquid nitrogen and stored at −80°C; a strip of
normal mucosa was taken from near the resection margins. The
present series consisted of 22 cases for which subcutaneous
xenotransplantation into athymic nude mice was successful or for
which a primary colorectal carcinoma cell line was generated (19
and 3, respectively; subset 1), the techniques used have been
previously described (9); 20
additional tumours were selected from the tumour bank for which
xenografting had not been attempted or that had failed to grow as
xenotransplants (subset 2). The animal studies were registered
officially and received formal ethical approval (ref. LALLF
M-V/TSD/7221.3-1.1-071/10). After overnight-fixation in buffered
aqueous formaldehyde solution (4% w/v) the remaining major portions
of the surgical specimens were dissected, slices of the tumours
blocked in paraffin, and a surgical pathology report was issued
that typed, graded and staged the tumours according to the TNM
system of the UICC (7th edition). Information on clinical staging
and the patient’s personal and family history (as recorded on
first-contact interviews) were available from the clinical charts.
Prior written informed consent was obtained from all patients, and
all procedures were approved by the Ethics Committee of Rostock
University (ref. II HV 43/2004).
Molecular studies for classification of
the tumours
Molecular classification of the tumours was carried
out according to the results of molecular studies as previously
published (10). Briefly, DNA was
extracted from the tumour xenografts (passages 1–3) (subset 1); or
from the tumour specimens stored in the tumour bank (subset 2) in
which case cryosections were taken and examined histologically to
ascertain that the tumour was well represented.
Microsatellite instability was assayed by the
Bethesda panel and CAT26 as an additional quasi-monomorphic
mononucleotide marker. Tumours were classified as high-degree
microsatellite instable (MSI-H) if positive with three or more of
the Bethesda markers; CAT26 was positive in all of these cases.
Methylations were assessed by quantitative real-time
PCR using MethyLight technology as published by Ogino et al
(11) using the following marker
panel: CACNA1G, CDKN2A, CRABPB1, MLH1 and NEUROG. COL2A1 was used
for normalization of the input DNA. The EpiTect Bilsulfite kit
(Qiagen, Hilden, Germany) was used for bisulfite treatment. A locus
was classified as methylated when the percentage of methylated
reference exceeded 4 (PMR >4).
K-Ras codon 12/13 and B-raf V600E mutations were
tested as previously described (10).
Molecular analyses of PTEN
PTEN gene sequencing
PTEN gene exons 1–9 were amplified by PCR from
genomic DNA extracted from xenotransplants or from the tumour
tissues retrieved from the tumour bank. PCR primers and
amplification conditions are documented in Table I. PCR products were purified with
alkaline phosphatase (Fermentas, Schwerte, Germany) and Exonuclease
I (Fermentas). Subsequently, the sequencing reactions were
performed using the BigDye Terminator v3.1 cycle sequencing kit
(Applied Biosystems, Darmstadt, Germany) with each pair of forward
and reverse primers. The sequence data were compared with the
published PTEN sequence (Genbank GI: 4240386) using
SeqScape® Software v2.7 (Applied Biosystems) and Chromas
Lite (Technelysium Pty. Ltd., South Bristol, Australia). Sequencing
of non-tumour DNA (from normal mucosa) was used to ascertain that
PTEN mutations found in the tumours actually were somatic.
Mutations were confirmed by independent repeat analyses.
Furthermore, to determine if the mutations were monoallelic or
biallelic for positive cases from subset 2, sequencing was repeated
with DNA from neoplastic glands that were isolated from cryostat
sections using a PALM laser-capture device (Carl Zeiss, Göttingen,
Germany).
 | Table IPrimers and PCR conditions for PTEN
sequencing. |
Table I
Primers and PCR conditions for PTEN
sequencing.
| Exon | Primer sequences | AT (°C) | Product size
(bp) |
|---|
| 1 |
5′-CAGCCGTTCGGAGGATTA-3′
5′-ATATGACCTAGCAACCTGACCA-3′ | 60 | 484 |
| 2 |
5′-GTACTTTAGTTCTGTGATGTATAAACCGT-3′
5′-CTGAAGTCCATTA-3′ | 60 | 509 |
| 3 |
5′-ATGTTTGTGAGGGTCGAATG-3′
5′-GGACTTCTTGACTTAATCGGTTTAG-3′ | 60 | 726 |
| 4 |
5′-TGAAAAAGGTGATCGTTGGC-3′
5′-ATTGTTATGACAGTAAGATACAGTCTATCG-3′ | 60 | 657 |
| 5 |
5′-TTCTGAGGTTATCTTTTTACCACAG-3′
5′TCCAGGAAGAGGAAAGGAAAA--3′ | 60 | 303 |
| 6 |
5′-AATGTATATATGTTCTTAAATGGCTACGA-3′
5′-TCATAAATATAATTTGGCTTCGACTAC-3′ | 60 | 484 |
| 7 |
5′-TGACAGTTTGACAGTTAAAGG-3′
5′-GATATTTCTCCCAATGTAAAGT-3′ | 55 | 262 |
| 8 |
5′-GCAACAGATAACTCAGATTGCC-3′
5′-TCAAGCAAGTTCTTCATCAGC-3′ | 54 | 514 |
| 9 |
5′-TGTTCATCTGCAAAATGGAAT-3′
5′-CAAGTGTCAAAACCCTGTGG-3′ | 54 | 469 |
PTEN allelotyping
Allelotyping of the PTEN gene was carried out with
DNA extracted from the tumour xenografts/primary cell lines (subset
1), or with DNA obtained by laser-capture microdissection of the
neoplastic glands (subset 2). Tumour and non-tumour DNA was
amplified by PCR with published polymorphic microsatellite markers
located close to the PTEN gene (D10S541, D10S579 and D10S1765)
(12,13). Each sense primer was fluorescence
labelled with FAM at the 5′ end. PCR was carried out as follows.
Reactions were started at 95°C for 10 min, and this was followed by
30 cycles at 94°C for 30 sec, 60°C for 30 sec and 72°C for 45 sec
and finally a 10-min extension at 72°C. PCR products were separated
by capillary gel electrophoresis and detected on an automated ABI
3500 Genetic Analyzer (Applied Biosystems). Fragment length and
fluorescence intensity were evaluated by GeneMapper software. The
GeneScan™-500LIZ™ size marker (Applied Biosystems) served as an
internal standard. A tumour was considered to have PTEN allelic
imbalance (AI) if the tumour to normal ratios were ≤0.5 or ≥2.0.
Only in the case of a complete loss of one allele, was the case
considered positive for LOH of the PTEN gene.
PTEN gene promoter methylation
assays
A primer/probe combination specific for the
methylated PTEN promoter sequence was used (forward,
5′-GGTGATGTGGCGGGATT TT-3′ and reverse, 5′-CGCCTCACAACGACTCAAC-3′;
probe: 5′-6FAM-TGCGGTAGGATACGCGTTCGGC-TM R-3′) with the SensiFast
Probe kit (Bioline, Luckenwalde, Germany). CpG Methylase
(SssI)-treated DNA served as a calibrator, since it is considered
to be fully methylated. The collagenase gene 2A1 (COL2A1) was used
as endogenous control (forward, 5′-TCTAACAATTATAAACTCCAACCAC CAA-3′
and reverse, 5′-GGGAAGATGGGATAGAAGGGAA TAT-3′; probe,
5′-6FAM-CCTTCATTCTAACCCAATACCT ATCCCACCTCTAAA-TMR-3′). Quantitative
PCR was performed using the StepOne Plus System (Applied
Biosystems). The PMR value was calculated by dividing the gene of
interest/COL2A1 ratio of the sample by the gene of interest/COL2A1
ratio of the SssI-treated DNA, and multiplying by 100. Samples with
a PMR value >4 were considered as methylated. All reactions were
performed in triplicates.
PTEN immunohistochemistry
A paraffin block representing a full cross-section
through the tumour was selected from the archived material for each
case. Anti-PTEN monoclonal antibody 6H2.1 was applied at a titre of
1:100 to 4-μm sections after heat-induced antigen retrieval and
stained on an autoimmunostainer (both from Dako, Glostrup, Denmark)
according to the manufacturer’s standard procedures. Mab 6H2.1 has
been previously well characterised for its specificity (13); furthermore, strong nuclear and
cytoplasmic immunostaining of endothelia provides a convenient
internal control of the immunoreactions and gives a standard to
compare to when scoring tumour tissues. In the present study,
nuclear and cytoplasmic immunostaining was scored as strong (score
2) when comparable to the staining of endothelia, as moderate
(score 1) when observed to be unequivocally present but weaker than
endothelia, or absent (score 0). Heterogeneity of the
immunoreactions was recorded, but the predominant pattern was used
for the final results.
Results
PTEN molecular analyses
A total of 42 primary colorectal carcinomas were
included in the present study, 19 of which were successfully
propagated as subcutaneous xenotransplants in athymic nude mice
generated in our laboratory and 3 were available as primary tumour
cell lines (subset 1), whereas xenotransplantation had not been
attempted/successful for the remaining 20 tumours (subset 2).
Clinicopathological features and the molecular data that formed the
basis for the molecular classifications of these tumours are
provided in Table II. The tumours
were grouped according to the criteria set out in Ostwald et
al (10). Subset 1 consisted of
5 sporadic high-degree microsatellite instable colorectal
carcinomas with extensive and strong methylations that included
(and silenced) the MLH1 gene (spMSI-H type), 3 microsatellite
stable colorectal carcinomas of the CpG island methylator phenotype
(CIMP-H/non-MSI-H type), and 14 colorectal carcinomas of the
sporadic standard type (spSTD type). A similar composition was
found for subset 2: 4 tumours were of the spMSI-H type; 2 of the
CIMP-H/non-MSI-H type and 14 of the spSTD type.
| Table IIClinicopathological and molecular
characteristics of the tumours included in the present study. |
Table II
Clinicopathological and molecular
characteristics of the tumours included in the present study.
| | | | | MethyLight |
|---|
| | | | |
|
|---|
| Tumour ID (molecular
type) | Histotype
(grade) | TNM | K-Ras (G12, G13) | B-Raf (V600E) | MLH1 | Othersa |
|---|
| Subset 1 |
| HROC 24
(spMSI-H) | ADCb (G2) | T2N0M0 | Wt | Mutated | Positive | 5 |
| HROC 48
(spMSI-H) | ADC (G3) | T2N1M0 | Wt | Wt | Positive | 5 |
| HROC 50
(spMSI-H) | ADC (G2) | T4N0M0 | Wt | Mutated | Positive | 5 |
| HROC 53
(spMSI-H) | ADC (G3) | T3N0M0 | Wt | Wt | Positive | 2 |
| HROC 87
(spMSI-H) | ADC (G3) | T3N0M0 | Wt | Mutated | Positive | 5 |
| HROC 40
(CIMP-H/non-MSI-H) | ADC (G3) | T3N1M0 | Mutated | Wt | Negative | 3 |
| HROC 60
(CIMP-H/non-MSI-H) | ADC (G2) | T2N0M0 | Wt | Wt | Negative | 4 |
| HROC 54
(CIMP-H/non-MSI-H) | ADC (G2) | T3N2M0 | Mutated | Wt | Negative | 3 |
| Cell line HROC 18
(spSTD) | ADC (G2) | T2N0M0 | Wt | Wt | Negative | 0 |
| HROC 32 (spSTD) | ADC (G2) | T4N2M0 | Mutated | Wt | Negative | 0 |
| HROC 39 (spSTD) | ADC (G3) | T4N0M0 | Wt | Wt | Negative | 1 |
| HROC 46 (spSTD) | ADC (G3) | T3N0M1 | Mutated | Wt | Negative | 0 |
| HROC 59 (spSTD) | ADC (G2) | T3N1M1 | Wt | Wt | Negative | 1 |
| HROC 62
(spSTD) | ADC (G3) | T4N2M0 | Mutated | Wt | Negative | 1 |
| HROC 65
(spSTD) | ADC (G2) | T3N2M1 | Mutated | Wt | Negative | 1 |
| HROC 68
(spSTD) | ADC (G2) | T4N2M1 | Mutated | Wt | Negative | 0 |
| HROC 69
(spSTD) | ADC (G3) | T3N0M0 | Wt | Wt | Negative | 1 |
| HROC 70
(spSTD) | ADC (G2) | T4N1M0 | Mutated | Wt | Negative | 2 |
| HROC 80
(spSTD) | ADC (G2) | T3N2M0 | Mutated | Wt | Negative | 1 |
| Cell line HROC 85
(spSTD) | ADC (G2) | T3N1M0 | Wt | Wt | Negative | 1 |
| Cell line HROC 86
(spSTD) | ADC (G3) | T3N1M0 | Mutated | Wt | Negative | 1 |
| HROC 107
(spSTD) | ADC (G2) | T3N2M1 | Mutated | Wt | Negative | 0 |
| Subset 2 |
| HROC 21
(spMSI-H) | ADC (G3) | T2N0M0 | Wt | Mutated | Positive | 3 |
| HROC 35
(spMSI-H) | ADC (G3) | T4N1M0 | Wt | Mutated | Positive | 5 |
| HROC 55
(spMSI-H) | ADC (G3) | T2N0M0 | Wt | Mutated | Positive | 5 |
| HROC 146
(spMSI-H) | ADC (G3) | T3N0M0 | Wt | Mutated | Positive | 3 |
| HROC 1
(CIMP-H/non-MSI-H) | ADC (G3) | T3N0M0 | Mutated | Wt | Negative | 5 |
| HROC 30
(CIMP-H/non-MSI-H) | ADC (G2) | T2N0M0 | Mutated | Wt | Negative | 4 |
| HROC 2
(spSTD) | ADC (G3) | T3N0M0 | Mutated | Wt | Negative | 1 |
| HROC 3
(spSTD) | ADC (G1) | T3N0M0 | Wt | Wt | Negative | 0 |
| HROC 4
(spSTD) | ADC (G2) | T2N0M0 | Wt | Wt | Negative | 0 |
| HROC 7
(spSTD) | ADC (G2) | T1N0M0 | Mutated | Wt | Negative | 1 |
| HROC 17
(spSTD) | ADC (G2) | T3N0M0 | Wt | Wt | Negative | 2 |
| HROC 26
(spSTD) | ADC (G3) | T4N2M0 | Wt | Wt | Negative | 2 |
| HROC 52
(spSTD) | ADC (G2) | T2N0M0 | Wt | Wt | Negative | 1 |
| HROC 64
(spSTD) | ADC (G2) | T2N0M0 | Wt | Wt | Negative | 0 |
| HROC 67
(spSTD) | ADC (G2) | T3N1M0 | Wt | Wt | Negative | 0 |
| HROC 82
(spSTD) | ADC (G2) | T3N0M0 | Wt | Wt | Negative | ND |
| HROC 105
(spSTD) | ADC (G2) | T4N2M1 | Wt | Wt | Negative | 0 |
| HROC 122
(spSTD) | ADC (G3) | T4N0M0 | Wt | Wt | Negative | 0 |
| HROC 125
(spSTD) | ADC (G2) | T3N1M0 | Wt | Wt | Negative | 2 |
| HROC 176
(spSTD) | ADC (G2) | T3N2M0 | Mutated | Wt | Negative | 0 |
The results of the molecular analyses of the PTEN
gene are summarised in Table III.
Sequencing of exons 1 to 9 of the PTEN gene revealed a total of 8
somatic mutations in 5 tumours. Colorectal carcinoma HROC 69
(spSTD, subset 1) harboured three different mutations: one stop
mutation at codon 7 (E7*), and 2 point mutations in exon 5 (R130Q
and R142Q). Furthermore, a monoallelic point mutation at codon 60
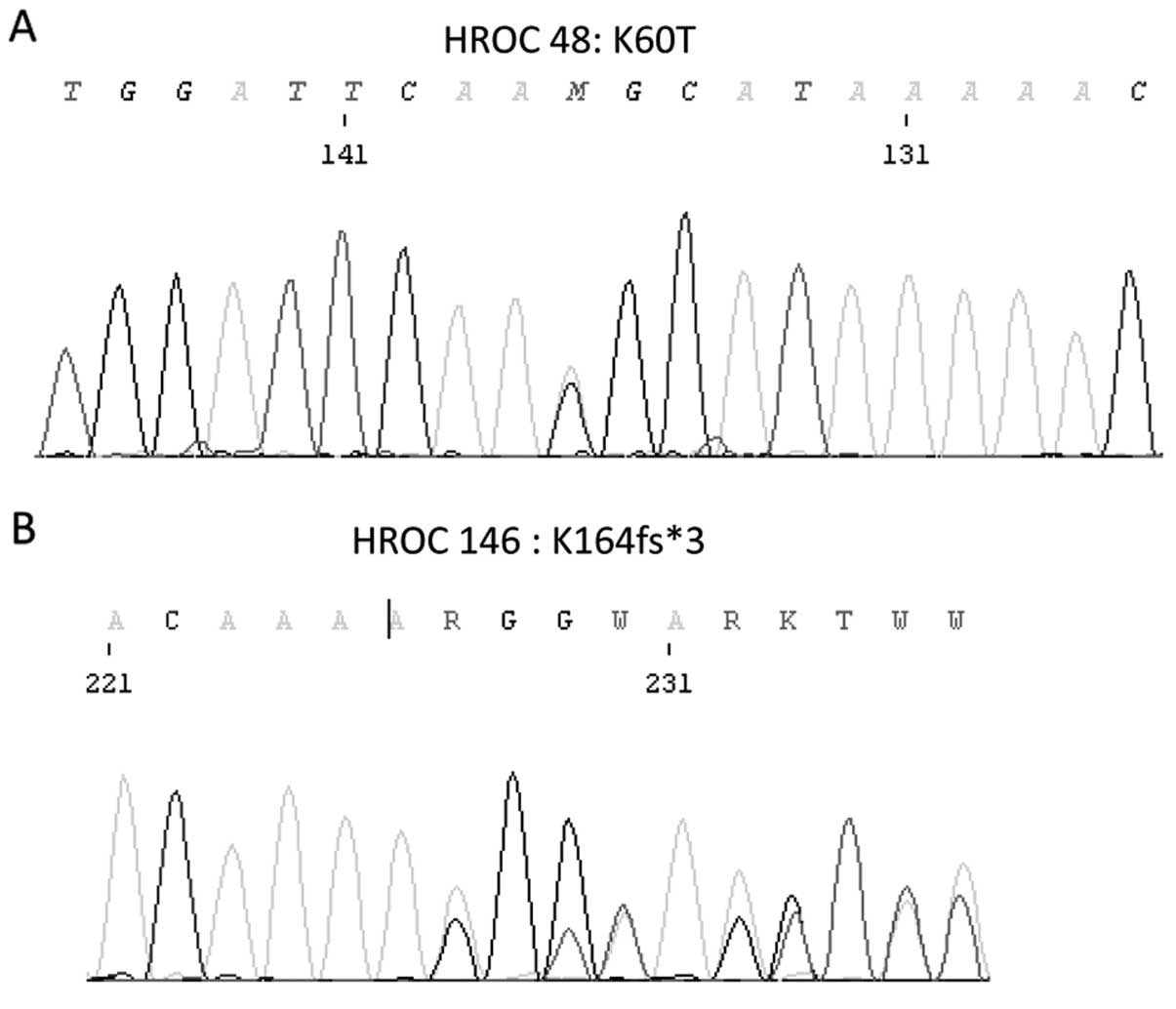
(K60T) was found in HROC 48 (spMSI-H, subset 1), and as another
monoallelic mutation a frameshift mutation at codon 267 (K267fs*9)
was observed in HROC 53 (spMSI-H, subset 1) (Fig. 1). Three mutations were found in two
tumours of subset 2: one monallelic point mutation in one tumour
(R14G; HROC 30, CIMP-H/non-MSI-H) and two monoallelic frameshift
mutations in another tumour (K164fs*1 and E288fs*1; HROC 146,
spMSI-H); to discriminate between monoallelic and biallelic
mutations, the DNAs from tumours of subset 2 were from neoplastic
glands isolated by laser-capture microdissection.
| Table IIIResults of the molecular and
immunohistochemical studies of the PTEN gene. |
Table III
Results of the molecular and
immunohistochemical studies of the PTEN gene.
| Mutation | Allelotype
analyses | Methylation
(PMR) |
Immunohistochemistry (expression) |
|---|
|
|---|
| D10S541 | D10S579 | D10S1765 |
|---|
| Subset 1 |
| HROC 24
(spMSI-H) | Wt | MSIa | 0b | MSI | 1 | Retained |
| HROC 48
(spMSI-H) | K60T (new)c | MSI | LOHd | MSI | 0 | Retained |
| HROC 50
(spMSI-H) | Wt | MSI | MSI | N.i.e | 0 | Retained |
| HROC 53
(spMSI-H) | K267fs*9 (24×) | MSI | AI (0.5)f | MSI | 0 | Reduced/loss |
| HROC 87
(spMSI-H) | Wt | MSI | 0 | MSI | 0 | Retained |
| HROC 40
(CIMP-H/non-MSI-H) | Wt | AI (0.5) | AI (0.5) | 0 | 0 | Retained |
| HROC 60
(CIMP-H/non-MSI-H) | Wt | N.i. | 0 | AI (0.4) | 100 | Retained |
| HROC 54
(CIMP-H/non-MSI-H) | Wt | 0 | 0 | 0 | 0 | Retained |
| Cell line HROC 18
(spSTD) | Wt | 0 | 0 | 0 | 0 | Retained |
| HROC 32
(spSTD) | Wt | AI (0.2) | N.i. | 0 | 0 | Reduced/loss |
| HROC 39
(spSTD) | Wt | 0 | N.i. | 0 | 0.7 | Reduced/loss |
| HROC 46
(spSTD) | Wt | 0 | 0 | 0 | 0 | Reduced/loss |
| HROC 59
(spSTD) | Wt | LOH | N.i. | LOH | 0 | Reduced/loss |
| HROC 62
(spSTD) | Wt | 0 | N.i. | 0 | 0 | Reduced/loss |
| HROC 65
(spSTD) | Wt | N.i. | AI (2.1) | 0 | 0 | Retained |
| HROC 68
(spSTD) | Wt | LOH | N.i. | LOH | 0 |
Reduced/lossg |
| HROC 69
(spSTD) | E7* (5×)
R130Q (7×)
R142Q (new) | 0 | N.i. | 0 | 0 | Reduced/loss |
| HROC 70
(spSTD) | Wt | 0 | 0 | 0 | 0 | Reduced/loss |
| HROC 80
(spSTD) | Wt | 0 | N.i. | 0 | 0 | Retained |
| Cell line HROC 85
(spSTD) | Wt | 0 | 0 | 0 | ND | Reduced/loss |
| Cell line HROC 86
(spSTD) | Wt | 0 | 0 | 0 | ND | Retained |
| HROC 107
(spSTD) | Wt | N.i. | 0 | N.i | 0 | Retained |
| Subset 2 |
| HROC 21
(spMSI-H) | Wt | MSI | 0 | 0 | 0 | Retained |
| HROC 35
(spMSI-HI) | Wt | MSI | AI (0.37) | 0 | 0 | Retained,
strong |
| HROC 55
(spMSI-H) | Wt | N. ampl.h | 0 | 0 | 0 | Reduced/loss |
| HROC 146
(spMSI-H) | K164fs*3
(new)
E288fs*3 (1×) | MSI | N.i. | MSI | 0 | Reduced/loss |
| HROC 1
(CIMP-H/non-MSI-H) | Wt | N. ampl. | 0 | 0 | 0 | Retained |
| HROC 30
CIMP-H/non-MSI-H) | R14G (2×) | 0 | 0 | 0 | 0 | Reduced/loss |
| HROC 2
(spSTD) | Wt | 0 | 0 | 0 | 0 | Retained,
strong |
| HROC 3
(spSTD) | Wt | N.i. | 0 | 0 | 0 | Retained |
| HROC 4
(spSTD) | Wt | N. ampl. | 0 | 0 | 0 | Retained |
| HROC 7
(spSTD) | Wt | 0 | 0 | 0 | 0 | Reduced/loss |
| HROC 17
(spSTD) | Wt | 0 | 0 | N.i. | 0 | Retained |
| HROC 26
(spSTD) | Wt | N.i. | 0 | 0 | 0 | Reduced/loss |
| HROC 52
(spSTD) | Wt | 0 | 0 | 0 | 0 | Retained,
strong |
| HROC 64
(spSTD) | Wt | 0 | 0 | AI (0.5) | 0 | Retained |
| HROC 67
(spSTD) | Wt | N.i. | N.i. | N.i. | 0 | NDi |
| HROC 82
(spSTD) | Wt | 0 | N.i. | 0 | 0 | Reduced/loss |
| HROC 105
(spSTD) | Wt | 0 | N.i. | N.i. | 0 | Retained |
| HROC 122
(spSTD) | Wt | 0 | N.i. | 0 | 0 | Reduced/loss |
| HROC 125
(spSTD) | Wt | 0 | 0 | MSI | 0 | Reduced/loss |
| HROC 176
(spSTD) | Wt | 0 | N.i. | AI (2.2) | 0 | Retained |
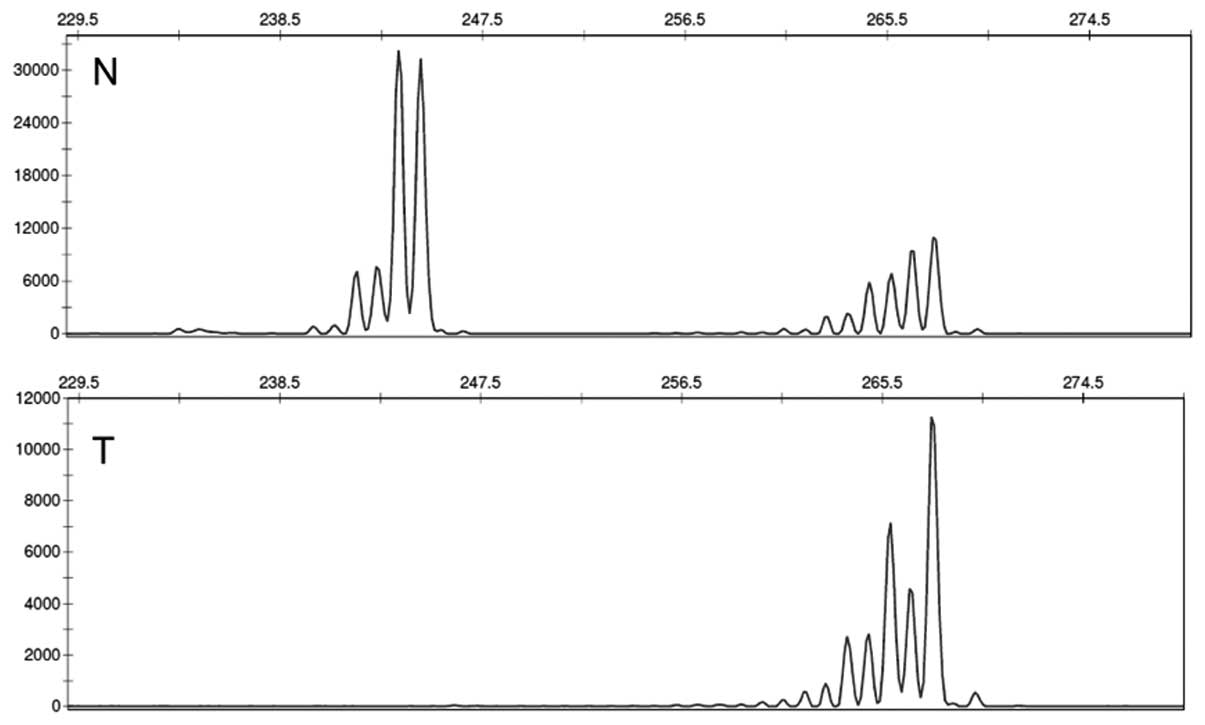
LOH at the PTEN gene locus 10p23 was addressed with
the polymorphic microsatellite markers D10S541, D10S579 and
D10S1765. Importantly, in order to be able to demonstrate actual
complete loss of one allele, we used DNA extracted from the
xenotransplants (subset 1), or DNA from neoplastic glands that had
been separated from the stroma by laser-capture microdissections
(subset 2). By this approach, we were able to distinguish between
AI and LOH sensu stricto: complete loss of one allele was
noted in 3 tumours (two spSTD and one spMSI-H, all of subset 1; see
Fig. 2 for an exemplary
electropherogram). AI was more frequent, as it was found in 8
additional tumours. HROC 48 was the only tumour observed to combine
LOH and PTEN gene mutation. Results of the microsatellite marker
analyses are documented in detail in Table III.
Quantitative real-time PCR using the MethyLight
technology was performed to assay for methylations of CpGs in the
PTEN gene promoter. Strong methylation (PMR=100) was observed in
only 1 case (HROC 60), a colorectal carcinoma of the
CIMP-H/non-MSI-H type.
PTEN immunohistochemistry
PTEN immunohistochemistry with mAb 6H2.1 worked
reasonably well with the paraffin sections from the archived
blocks. Endothelia and, if present, nerves and colonocytes of the
adjacent normal mucosa conveniently provided internal controls of
the immunoreactions and a standard for the evaluations. Strong
immunostaining of the nuclei and the cytoplasm of endothelia was
noted without heterogeneity in all sections. Immunostaining of
colonocytes was weaker; the former was taken as the standard for
score 2, the latter for score 1. In most cases, although
convincingly specific, PTEN immunostaining was fairly weak in the
cytoplasm and/or nuclei of tumour cells. Furthermore, it was
somewhat heterogeneous in several of the cases; however, regional
differences did not exceed one score-point, and the final
classification was made according to the dominant pattern. By this
approach, 18 tumours were observed to have reduced or loss of PTEN
expression, and retention of PTEN expression (i.e. immunostaining
similar to colonocytes) was recorded for 20 tumours (one tumour was
not amenable to immunostaining for technical reasons).
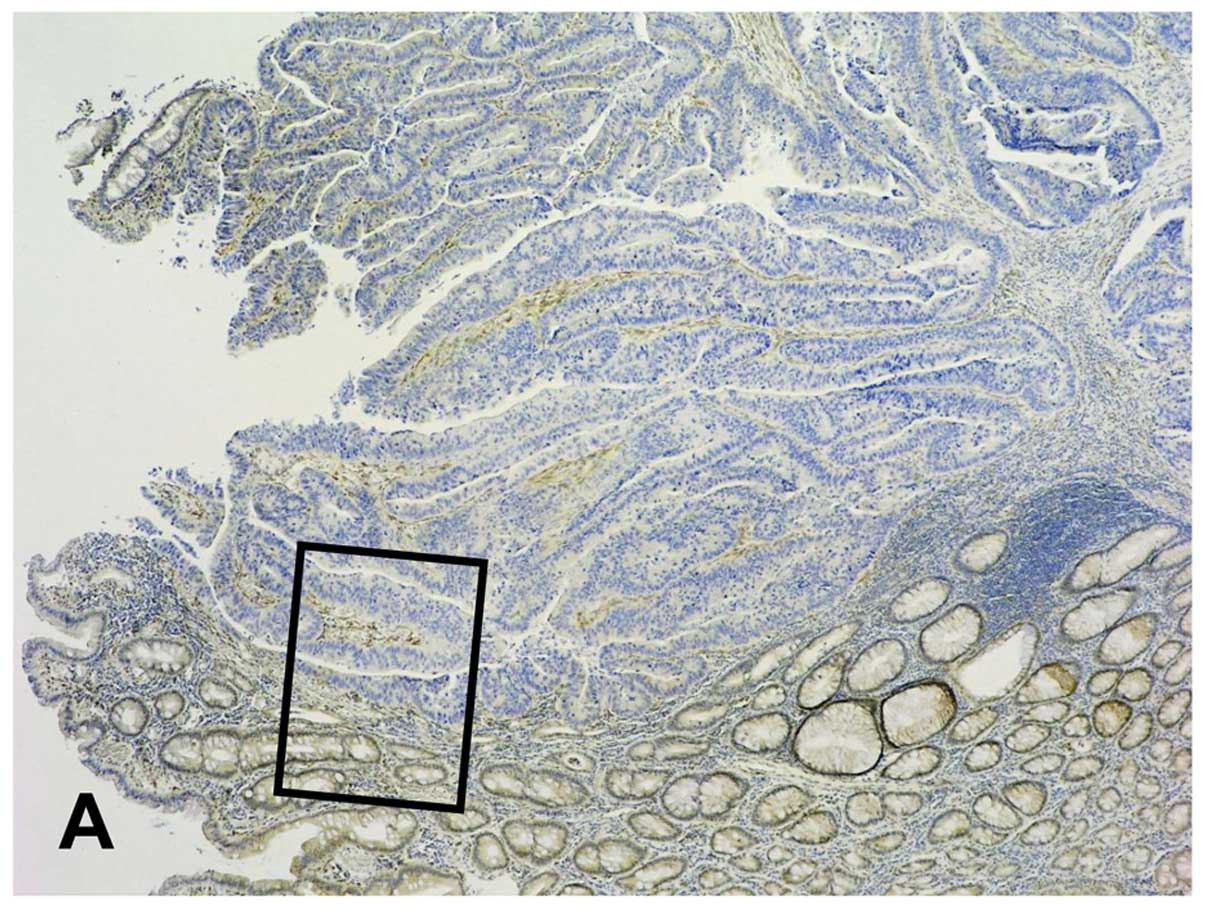
Overexpression was noted in 3 cases only. Representative images of
PTEN immunohistochemistry are provided in Fig. 3, and the results are summarised in
Table III. Importantly, in a
single case (HROC 68, subset 1, spSTD) strong immunostaining of the
cytoplasm (cytoplasmic score 2) was observed neatly restricted to a
minority of tumour cells at the invasive margin (Fig. 3) while the bulk of the tumour had
lost PTEN expression. As far as we are aware, this pattern of PTEN
immunohistochemistry has not been previously reported.
Discussion
The numbers of PTEN gene mutations in the present
study align quite well with the published data; in the COSMIC
database 349 mutations were reported for 2,807 colorectal
carcinomas analyzed (12.4%) while we found mutations in 5 of 42
colorectal carcinomas (11.9%). Five mutations from the present
study are on record in the COSMIC database but 3 have not
previously been reported (annotated in Table III). Similar to published series
(4,13), we observed that mutations were more
frequent among colorectal carcinomas with microsatellite
instability (3 of the 9 spMSI-H in this series). This is well
explained by the incapacity for mismatch repair that causes
microsatellite instability, including insertions/deletions in
nucleotide repeats that are frequent in downstream coding regions
of the PTEN gene.
However, the present study was not limited to
mutational analyses. Specifically, we addressed whether molecular
aberrations of the PTEN gene found in a series of colorectal
carcinoma that encompassed the major molecular types would fit with
its universally presumed role of a tumour-suppressor gene. As far
as we are aware, this is the only study of colorectal carcinoma
using DNA from tumour cells isolated from the stroma to which
simultaneously allelotyping and mutational as well as methylation
analysis were applied, allowing a synoptic assessment. While PTEN
gene mutations and complete loss of one allele (i.e. LOH sensu
stricto) of the PTEN gene were observed in 5 and 3 tumours,
respectively, a combination of mutation and LOH was found in one of
the tumours only (HROC 48), and the mutation was missense. This
largely contradicts what would be expected of a tumour-suppressor
gene in the classical sense, where gene function is lost by an
inactivating (often truncating) mutation of one allele, and loss of
the genomic material that harbours the other allele. Another tumour
in our series, however, was observed to have mutations of both
alleles (one truncating and two missense), a constellation that may
be taken to emulate the classic combination.
Our observation of only 3 cases of PTEN LOH in a
total of 42 tumours analyzed (7.1%) is considerably lower than the
19.0–34.7% reported in other studies (12,13,15).
However, it must be remembered that the usual technique of using
DNA from whole tumour homogenates includes DNA from stromal cells,
thus considerably contaminating the ‘tumour’ DNA with normal DNA.
To circumvent this potential source of error, we used tumour DNA
derived from low-passage xenografts or from neoplastic glands of
primaries that were isolated from the stroma by laser-capture
microdissection. Accordingly, so-called LOH in other studies in the
majority of cases amounts to what in our study was scored as
allelic imbalance (AI); this was found in 8 additional tumours.
Thus, apparently the majority of so-called PTEN LOHs reported in
the literature ‘ever since Knudson’ (5) may well be spurious and not play a role
functionally, particularly not as one of the ‘hits’ of a
tumour-suppressor gene mechanism involving the PTEN gene. The
reason for AI is well explained when taking into account the
results from a different methodological approach; some of our
xenotransplants have previously been analyzed by single-nucleotide
polymorphism (SNP)-arrays [HROCs 18, 32, 39, 40, 46, 59, 60, 80;
published in (16)]. These
investigations showed that the AI is in fact the result of
unbalanced large-scale genomic amplification (trisomy 10 or p-arm
amplification).
To address whether alternatively PTEN gene silencing
may play a role, we also assayed promoter methylation. This was
carried out by real-time PCR [MethyLight technology, (11)], a quantitative approach that, as
opposed to methylation-specific PCR, allows an objective and
functionally meaningful classification of cases. While other CpG
loci tested for the molecular classification of our tumours
frequently had strong methylations (Table II), PTEN gene promoter methylation
was noted in only one case, not combined with mutation or LOH. The
results from our series, therefore, do not provide evidence that
PTEN in colorectal carcinoma may function as an ‘epi-driver’ gene
instead of as a tumour-suppressor gene in the classical sense.
PTEN expression in our tumours was studied by
immunohistochemistry using the monoclonal antibody 6H2.1. This
antibody’s specificity has been shown in previous studies of
colorectal carcinoma (13,14). By immunohistochemistry, reduction or
loss of PTEN expression was a frequent event, noted in 18 of the 41
tumours amenable to this study (43.9%). Considering the
well-established function of PTEN in cell cycle control and cell
migration, this observation points to a definite role of PTEN in
colorectal carcinoma tumour biology, although apparently by
mechanisms different from what is usually meant when referring to
PTEN as a tumour-suppressor gene; the most plausible mechanism to
explain reduction or even loss of PTEN expression in colorectal
carcinoma cells at this juncture is (dys)regulation by unknown
factors from the microenvironment that act on the tumour cells.
This hypothetical explanation also allows for plasticity, i.e. it
can explain fluctuations in downregulation and re-expression, and
thus accounts for the somewhat inhomogenous patterns of expression
observed by immunohistochemistry in many of the cases; in fact, the
unusual very focal overexpression at the invasive margin noted in
one of our tumours (HROC 68, Fig.
3) would be difficult to explain if PTEN were thought to be
compromised by genetic defects.
Reduced PTEN expression has been reported as a
negative predictor for EGF-R blocking agents such as cetuximab or
panitumumab in the setting of metastasizing colorectal carcinoma
(17,18), underscoring its functional
importance. Given the difficulties that surgical pathologists
usually encounter when trying to adapt
quantitative/semi-quantitative immunohistochemistry for routine
use, testing the gene by molecular pathology instead would have
been desirable; but alterations of PTEN gene expression are not
appreciably mirrored in genomic aberrations, precluding this
approach.
We conclude that downregulation of the PTEN gene is
frequent in colorectal carcinoma, and is likely to have a
functional role. However, two hits on the PTEN gene, by mutation
and LOH or epigenetic silencing, respectively, is a rare event.
Acknowledgements
This study was substantially supported by grant no.
108919 from the Deutsche Krebshilfe (http://www.northgermantumorbank-crc.de).
References
|
1
|
Wood LD, Parsons DW, Jones S, et al: The
genomic landscapes of human breast and colorectal cancers. Science.
318:1108–1113. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Planchon SM, Waite KA and Eng C: The
nuclear affairs of PTEN. J Cell Sci. 121:249–253. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Steck PA, Pershouse MA, Jasser SA, et al:
Identification of a candidate tumour suppressor gene, MMAC1,
at chromosome 10q23.3 that is mutated in multiple advanced cancers.
Nat Genet. 15:356–362. 1997.PubMed/NCBI
|
|
4
|
Shin KH, Park YJ and Park JG: PTEN
mutations in colorectal cancers displaying microsatellite
instability. Cancer Lett. 174:189–194. 2001. View Article : Google Scholar
|
|
5
|
Devilee P, Cleton-Jansen AM and Cornelisse
CJ: Ever since Knudson. Trends Genet. 17:569–573. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vogelstein B, Papadopoulos N, Velculescu
V, Zhou S, Diaz LA Jr and Kinzler KW: Cancer genome landscapes.
Science. 339:1546–1558. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Goel A, Arnold CN, Niedzwiecki D, et al:
Frequent inactivation of PTEN by promoter hypermethylation in
microsatellite instability-high sporadic colorectal cancers. Cancer
Res. 64:3214–3221. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Oberländer M, Linnebacher M, König A, et
al: The ‘North German Tumor Bank of Colorectal Cancer’: status
report after the first 2 years of support by the German Cancer Aid
Foundation. Langenbecks Arch Surg. 398:251–258. 2013.
|
|
9
|
Linnebacher M, Maletzki C, Ostwald C,
Klier U, Krohn M, Klar E and Prall F: Cryopreservation of human
colorectal carcinomas prior to xenografting. BMC Cancer.
10:3622010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ostwald C, Linnebacher M, Weirich V and
Prall F: Chromosomally and microsatellite stable colorectal
carcinomas without the CpG island methylator phenotype in a
molecular classification. Int J Oncol. 35:321–327. 2009.
|
|
11
|
Ogino S, Cantor M, Kawasaki T, Brahmandam
M, Kirkner GJ, Weisenberger DJ, Campan M, et al: CpG island
methylator phenotype (CIMP) of colorectal cancer is best
characterised by quantitative DNA methylation analysis and
prospective cohort studies. Gut. 55:1000–1006. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nassif NT, Lobo GP, Wu X, Henderson CJ,
Morrison CD, Eng C, Jalaludin B and Segelov E: PTEN
mutations are common in sporadic microsatellite stable colorectal
cancer. Oncogene. 23:617–628. 2004. View Article : Google Scholar
|
|
13
|
Zhou XP, Loukola A, Salovaara R,
Nystrom-Lahti M, Peltomäki P, de la Chapelle A, Aaltonen LA and Eng
C: PTEN mutational spectra, expression levels, and
subcellular localization in microsatellite stable and unstable
colorectal cancers. Am J Pathol. 161:439–447. 2002. View Article : Google Scholar
|
|
14
|
Perren A, Weng LP, Boag AH, et al:
Immunohistochemical evidence of loss of PTEN expression in primary
ductal adenocarcinomas of the breast. Am J Pathol. 155:1253–1260.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Garcia JM, Rodriguez R, Silva J, et al:
Intratumoral heterogeneity in microsatellite alterations in BRCA1
and PTEN regions in sporadic colorectal cancer. Ann Surg Oncol.
10:876–881. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Linnebacher M, Ostwald C, Koczan D, et al:
Single nucleotide polymorphism array analysis of
microsatellite-stable, diploid/near-diploid colorectal carcinomas
without the CpG island methylator phenotype. Oncol Lett. 5:173–178.
2013.
|
|
17
|
Perrone F, Lampis A, Orsenigo M, et al:
PIK3CA/PTEN deregulation contributes to impaired responses
to cetuximab in metatatic colorectal cancer patients. Ann Oncol.
20:84–90. 2009. View Article : Google Scholar
|
|
18
|
Razis E, Briasoulis E, Vrettou E, et al:
Potential value of PTEN in predicting cetuximab response in
colorectal cancer: an exploratory study. BMC Cancer. 8:2342008.
View Article : Google Scholar : PubMed/NCBI
|