Introduction
Cranial radiation therapy (CRT) is one of the most
effective treatment modalities of primary and secondary brain
tumors. However, the risk of CRT-induced injury to normal
surrounding brain tissues is also associated with adverse
side-effects. As many as 50% of CRT-treated brain cancer survivors
develop late-onset radiation-induced injury of normal brain
tissues, and pediatric cases account for a large portion of this
population. The recent development and application of 3D-conformal
and intensity-modulated radiation therapies, both of which deliver
radiation more precisely to the tumor, have helped to reduce the
amount of radiation-induced injury in surrounding normal tissues.
Nevertheless, radiation-induced brain injury (RIBI) remains a
common and severe side-effect of CRT. Survivors of irradiated
childhood brain tumors who develop late-delayed cognitive deficits
due to RIBI may present with learning disabilities as well as
growth and psychomotor retardation.
Although the precise pathogenic mechanisms involved
in the development and progression of RIBI have not yet been fully
elucidated, a variety of cell types within the brain, including the
microglia, astrocytes, oligodendrocytes, neurons and endothelial
cells, have been determined to contribute to this process (1). The microglia, in particular, represent
~10% of the total glial population in the central nervous system
(CNS), and are the macrophage equivalent of the CNS where they
serve as key mediators of neuroinflammation (2). Accumulating evidence suggests that
activated microglia may contribute to RIBI (3). Chiang et al reported that 20–45
Gray (Gy) radiation could increase glial fibrillary acidic protein
(GFAP) expression and induce astrocytic and microglial responses.
These responses were associated with reactive gliosis and
inflammation (4). In addition,
conditioned medium collected from irradiated microglial cells has
been shown to induce astrogliosis, which may contribute to
radiation-induced edema (5).
Under normal physiological conditions, microglial
cells react to a variety of stimuli, including lipopolysaccharide
and interferon-α. In vitro studies have shown that activated
microglia produce a variety of pro-inflammatory mediators and
cytokines, such as interleukin (IL)-1, reactive oxygen species,
nitric oxide (NO) and prostaglandin E, all of which are thought to
be responsible for inflammation-related diseases such as RIBI,
trauma, ischemia, Alzheimer’s disease and neural death. Similarly,
in vivo studies have shown that brain irradiation leads to a
marked increase in microglia activation and release of
pro-inflammatory cytokines associated with inhibition of
neurogenesis in hippocampus (6–9).
Although the acute mechanism of microglial activation during RIBI
remains unclear, it has been suggested that microglial activation
is closely correlated with inflammation. For example, irradiating
microglia leads to their activation and a marked elevation in the
expression of proinflammatory genes, including tumor necrosis
factor-α (TNF-α), IL-1β, IL-6 and cyclooxygenase-2 (COX-2)
(5,10,11).
DNA double-strand breaks (DSBs) are the most
deleterious form of DNA damage and numerous in vitro studies
have sought to elucidate the mechanism of the DSB repair system
that is activated upon exposure to ionizing radiation. It has been
determined that the presence of DNA DSBs can rapidly trigger
activation of nuclear factor (NF)-κB signaling pathway via the
NF-κB essential modulator (NEMO) (5,6). The
death-domain protein PIDD, which was originally identified as an
early p53-inducible gene and is implicated in p53-induced apoptosis
(5), also acts as a mediator of the
DNA-damage-activated stress response and is involved in genotoxic
stress-induced NF-κB activation (6,7). PIDD
expression enhances genotoxic-stress-induced NF-κB activation
through augmented sumoylation and ubiquitination of NEMO. Moreover,
the NF-κB signaling pathway and DNA DSBs have been linked to
microglial activation induced by various stimuli, such as the
inflammagen lipopolysaccharide (12,13).
Nevertheless, the involvement of NF-κB and DNA damage in
radiation-induced microglial activation remains to be
clarified.
In the present study, we investigated the release of
inflammatory factors, activation of NF-κB and DNA DSBs in microglia
BV-2 cells following irradiation. Our findings provide insight into
the mechanism of microglial activation-mediated brain injury during
radiation therapy.
Materials and methods
Reagents
RPMI-1640 culture medium and fetal bovine serum
(FBS) were purchased from Gibco (Grand Island, NY, USA). TRIzol
reagent was obtained from Invitrogen (Carlsbad, CA, USA). The
anti-Iba-1 rabbit antibody was purchased from Wako Chemical (Osaka,
Japan) and the NEMO and IκB-α primary antibodies were purchased
from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The
anti-γ-histone 2A, member X (γ-H2AX) mouse monoclonal antibody was
purchased from Abcam (Cambridge, MA, USA). The AlexaFluor 488
conjugated goat anti-mouse, donkey anti-goat secondary antibody and
the AlexaFluor 568 conjugated goat anti-rabbit secondary antibody
were purchased from Invitrogen. Vectashield mounting medium with
4′,6-diamidino-2-phenylindole (DAPI) was purchased from Vector
Laboratories (Burlingame, CA, USA). Mouse IL-1β, TNF-α and IL-6
Quantikine enzyme-linked immunosorbent assay (ELISA) kits were
purchased from R&D Systems (Minneapolis, MN, USA). The Griess
reagent NO assay kit was from Beyotime Biotech (Jiangsu, China).
M-MLV reverse transcriptase and SYBR-Green I were purchased from
Toyobo Company (Osaka, Japan). All primers and oligo(dT) were
synthesized by Shanghai Invitrogen (Shanghai, China). The dNTP mix
and Taq DNA polymerase were obtained from Fermentas
International (Burlington, ON, Canada). Nuclear and cytoplasmic
protein extraction kits were purchased from Bio-Rad Laboratories
(Hercules, CA, USA). The enhanced chemiluminescence western
blotting detection system was obtained from Millipore (Bedford, MA,
USA). The p65 rabbit polyclonal antibody was purchased from Santa
Cruz Biotechnology and the antibody against β-actin was from Sigma
(St. Louis, MO, USA).
Cell culture
The mouse microglial cell line BV-2 was maintained
in the Laboratory Center of Union Hospital, Tongji Medical College,
Huazhong University of Science and Technology. Cells were cultured
in RPMI-1640 culture medium supplemented with 10% FBS at 37°C in a
5% CO2-humidified incubator. Cells in the logarithmic
phase were used in the experiments.
Irradiation procedure
A cell suspension was prepared at a density of
2×105 cells/ml. A total of 10 ml of the cell suspension
was added to a 6-well plate, which was placed 100 cm away from the
radioactive source. Irradiation of the cells was performed with a
137Cs irradiator (Siemens, Munich, Germany) at a dose
rate of 2.0 Gy/min. After a single dose of radiation, cells were
returned to the 5% CO2 incubator. Control cells did not
receive radiation treatment.
Immunocytochemistry
At the indicated time points following irradiation,
cells were fixed, permeabilized and blocked with goat serum. Cell
samples were stained with anti-Iba-1 (1:200 dilution), anti-CD68
(1:800), anti-NEMO (1:200) or anti-IκB-α (1:200) primary antibodies
and then probed with AlexaFluor 568 or AlexaFluor 488-conjugated
secondary antibodies (both 1:200). For co-staining, cell samples
were stained with anti-γ-H2AX (1:400) and anti-p65 (1:400)
antibodies followed by incubation with secondary antibodies
(1:200). Cell nuclei were counterstained with Hoechst or DAPI.
Fluorescence intensity was examined using a confocal scanning
microscope (BX41F; Olympus, Tokyo, Japan).
ELISA
At the indicated time points post-irradiation, the
culture medium was collected and the levels of IL-1β, IL-6, TNF-α
and TGF-β were determined by ELISA.
RNA extraction and real-time reverse
transcriptase polymerase chain reaction (RT-PCR)
Total RNA was extracted from cultured BV-2 cells
using TRIzol reagent. The sequences of the specific primers used
were designed by Beacon software (Bio-Rad Laboratories) and were as
follows, with the forward primer followed by the reverse primer for
each target: mouse TNF-α 5′ primer, TTC TCA TTC CTG CTT GTG G and
3′ primer, CTT GGT GGT TTG CTA CGA C; mouse IL-1β5′ primer, AAA TCT
CGC AGC AGC ACA T and 3′ primer, CAC ACA CCA GCA GGT TAT CA; mouse
IL-6 5′ primer, TTG CCT TCT TGG GAC TGA T and 3′ primer, TTG CCA
TTG CAC AAC TCT T; mouse COX-2 5′ primer, GAG TGG GGT GAT GAG CAA
and 3′ primer, GCA ATG CGG TTC TGA TAC T; GAPDH 5′ primer, TCA CCA
CCA TGG AGA AGG C and 3′ primer, GCT AAG CAG TTG GTG GTG CA; mouse
toll-like receptor (TLR)-8, 5′ primer, GCC ACT GTG ACT AAT GGT CCT
AA and 3′ primer, CAC CAA CGC AAG CCA AAA TAA ATG. PCR
amplification was performed using a Stratagene Mx3000P QPCR System
(La Jolla, CA, USA). The thermocycling conditions were: 50 cycles
at 94°C for 30 sec, 57°C for 30 sec and then 72°C for 30 sec. The
relative expression from amplified RNA samples was calculated using
the 2−ΔΔCT method.
Immunoblotting
Nuclear or cytoplasmic protein was extracted from
BV-2 cells using lysis buffer. Protein concentrations were measured
using the bicinchoninic acid (BCA) assay. Equal amounts of protein
lysates were diluted in sample loading buffer at a ratio of 4:1,
heated to 95°C for 5 min, separated by 12% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred to a nylon membrane. Membranes were blocked with 5% w/v
non-fat dry milk in phosphate-buffered saline (PBS) plus Tween-20
(PBS-T) for 2 h and then incubated with an NF-κB p65 primary
antibody (1:200) at 4°C overnight. After washing with PBS-T,
membranes were incubated with HRP-conjugated secondary antibody for
1.5 h at room temperature. The results were detected using Kodak
film (Rochester, NY, USA).
Statistical analysis
Data were analyzed with the Statistical Program for
Social Sciences software (version 10.0; SPSS, Inc., Chicago, IL,
USA) and were expressed as the mean ± standard deviation (SD).
Differences between groups were determined using the analysis of
variance (ANOVA) or Student’s t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Microglial activation induced by
irradiation
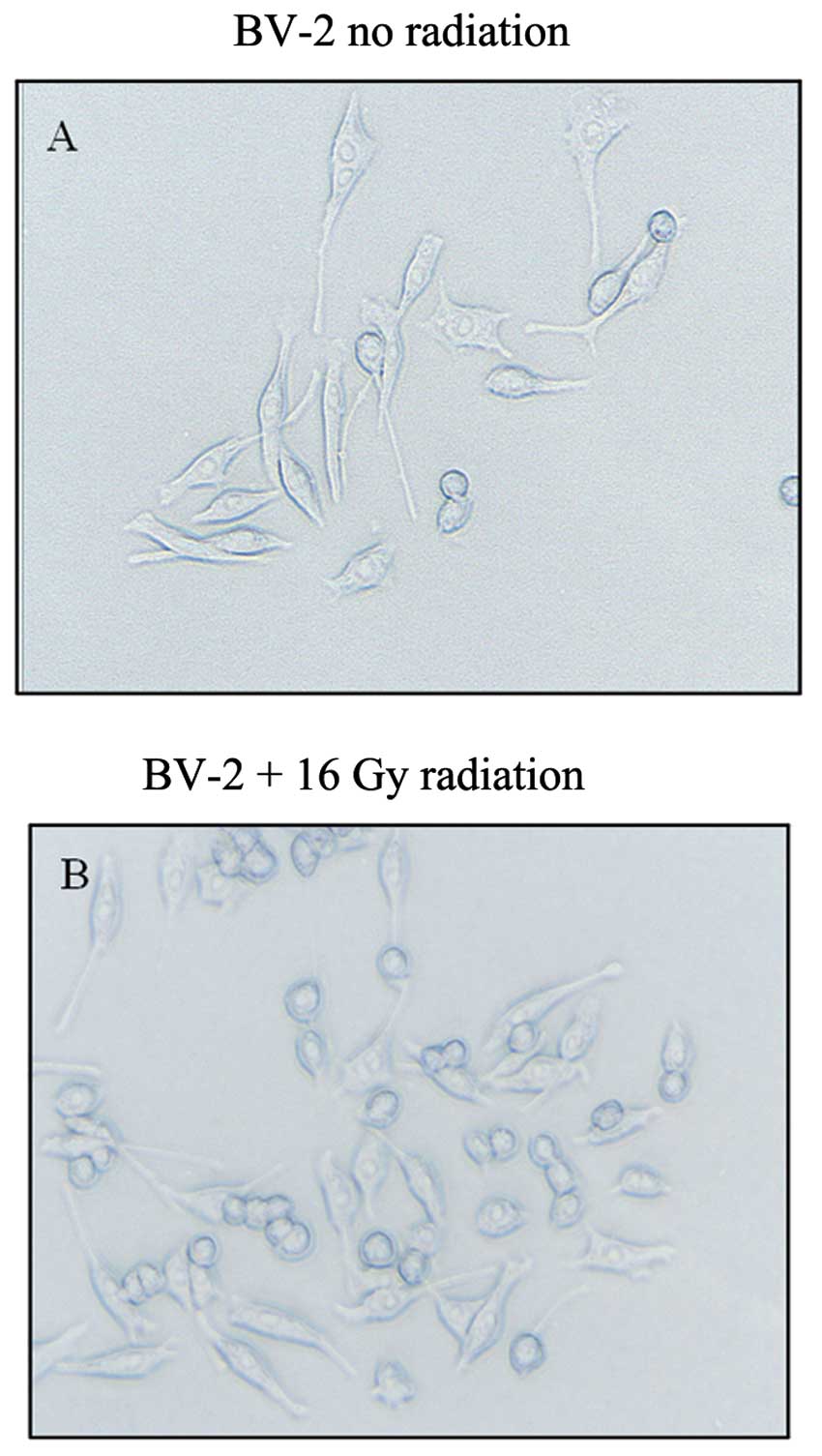
Control BV-2 microglial cells had a small cell body
and multiple long processes, showing a typical ramified structure
(Fig. 1A). Low-dose radiation
(<16 Gy) had no effect on the microglial morphology of these
cells (data not shown). However, 30 min following 16 Gy of
radiation, the microglia appeared to be activated, with cells
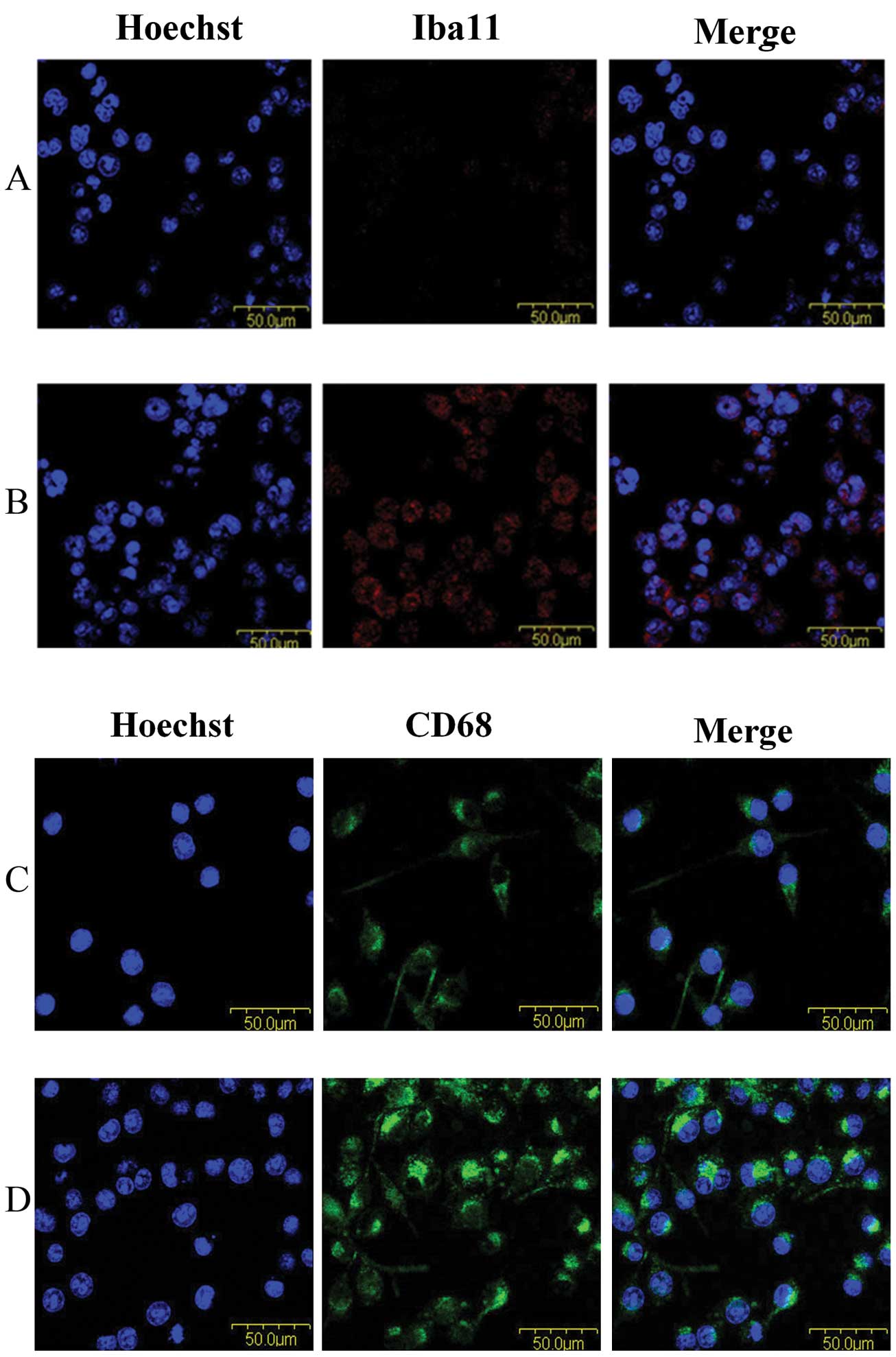
exhibiting a stouter spherical morphology (Fig. 1B). The levels of both Iba-1 and the
activation marker CD68 were greatly upregulated in cells following
irradiation (Fig. 2), indicating
microglia activation can be induced by radiation treatment.
Radiation-induced alteration of
inflammatory cytokinesis in microglia
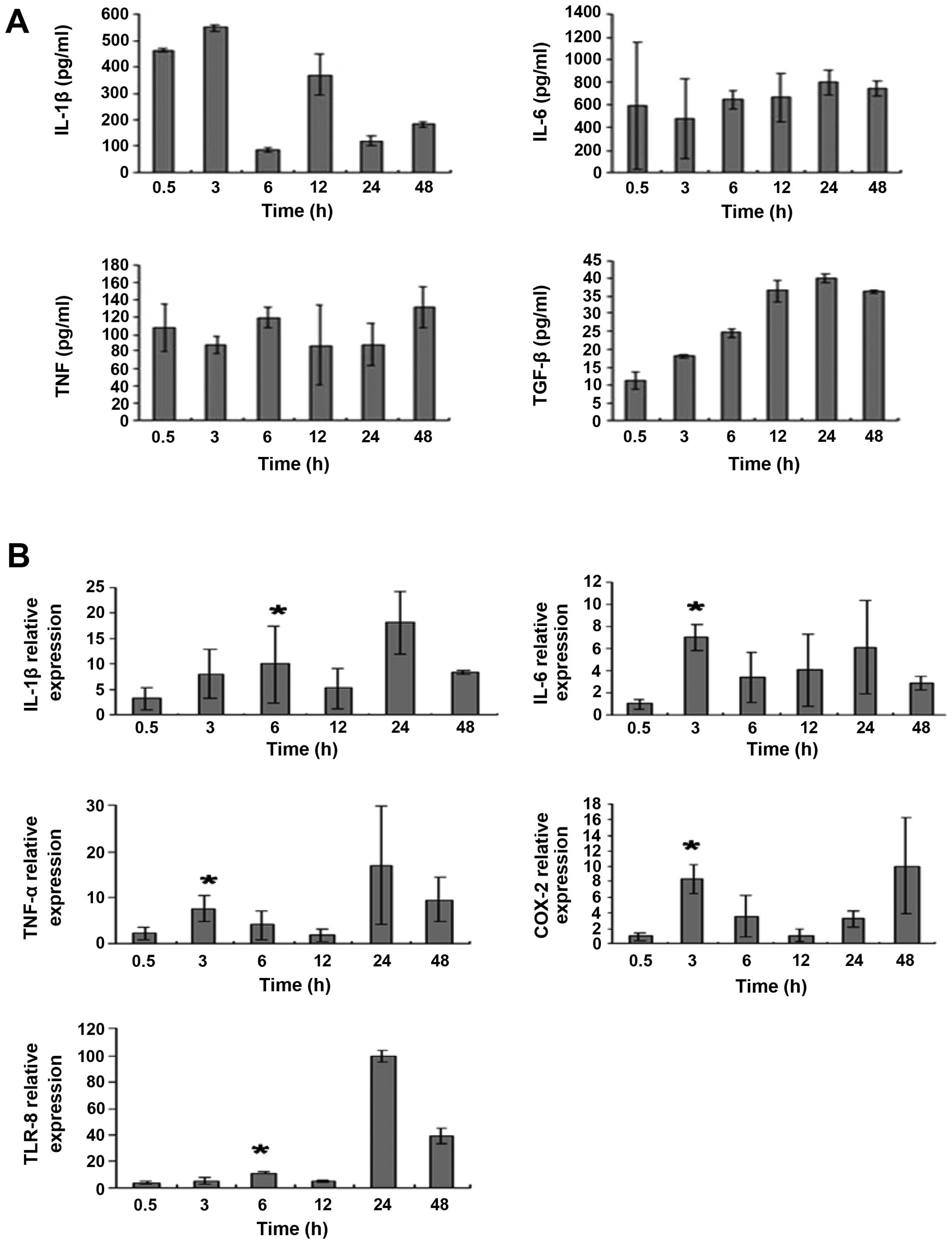
We next examined the levels of inflammatory factors,
including IL-1β, TNF-α, IL-6, TLR-8 and COX-2, following
irradiation. BV-2 microglial cells were irradiated with 32 Gy. As
revealed by the ELISA results in Fig.
3A and the real-time PCR results in Fig. 3B, the levels of IL-1β, TNF-α, IL-6,
TLR-8 and COX-2 in control microglia were relatively low. The
levels of these inflammatory factors increased post-irradiation,
peaking at 3 h (IL-6, TNF-α and COX-2) or 6 h (IL-1β and TLR-8)
(P<0.05 compared with control). Upregulation of these factors
was still detected at 24 or 48 h following irradiation.
Expression of γ-H2AX and NF-κB p65 in
irradiated BV-2 cells
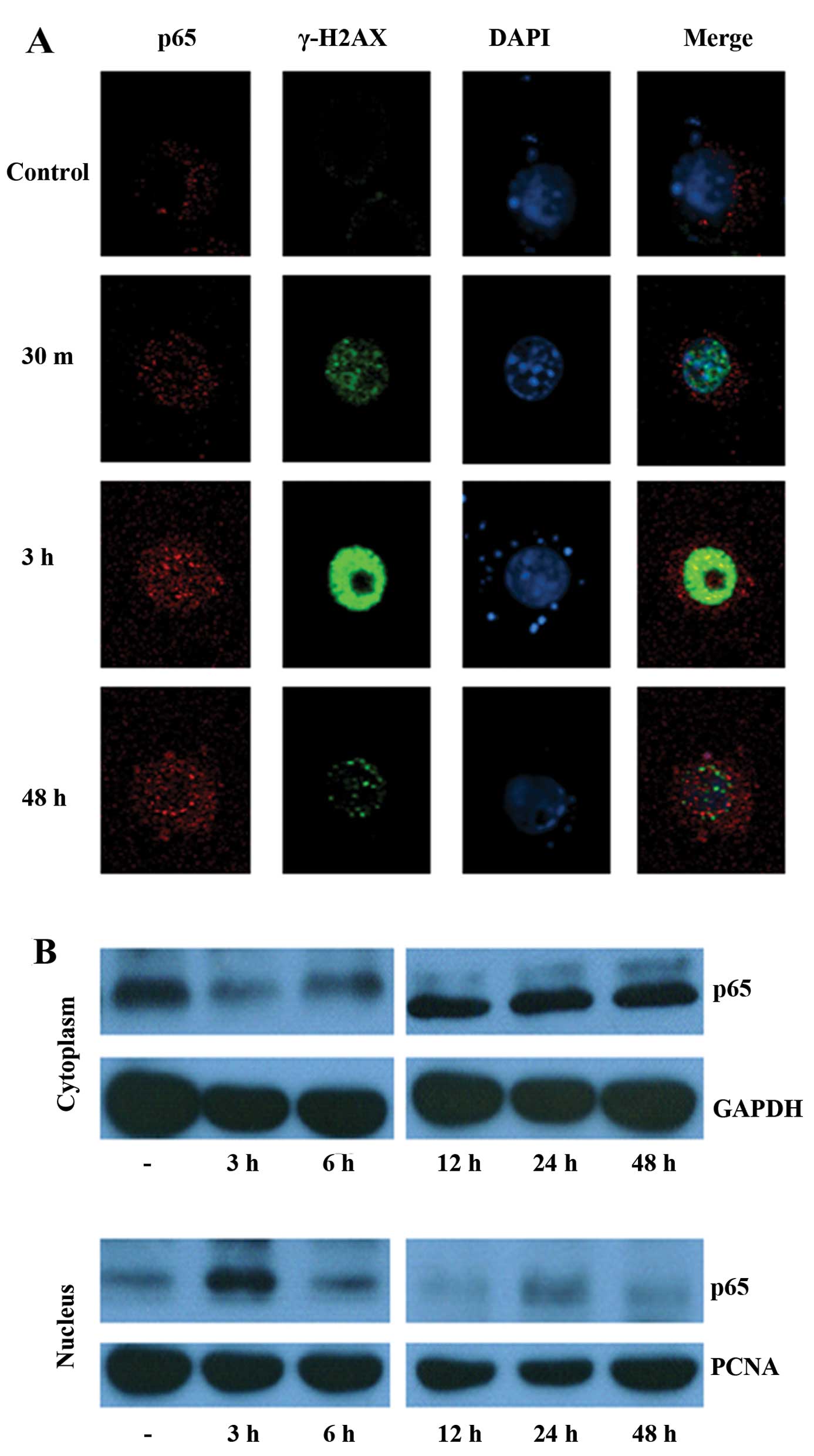
Since activated γ-H2AX that localizes to the nucleus
facilitates DNA DSBs, we determined the expression of γ-H2AX as
well as the activity of the transcription factor NF-κB. As shown in
Fig. 4A, very little γ-H2AX was
detected in non-irradiated BV-2 cells. However, 30 min following 32
Gy of radiation treatment, marked upregulation of γ-H2AX was
observed. In addition to this significant upregulation, γ-H2AX was
found to be localized to the nucleus, as can be seen from its
co-localization with the nuclear DAPI stain (Fig. 4A). Enhanced γ-H2AX expression was
detected 3 h post-irradiation but decreased after 48 h. In control
cells, the p65 subunit of NF-κB was primarily localized to the
cytoplasm. Nuclear translocation of p65 was detected 30 min
post-irradiation (32 Gy) and peaked at 3 h. However, 48 h after
radiation treatment, nuclear translocation of p65 was reduced.
Similar trends were observed in the cytoplasmic and nuclear
expression of p65 as determined by immunoblot analysis (Fig. 4B).
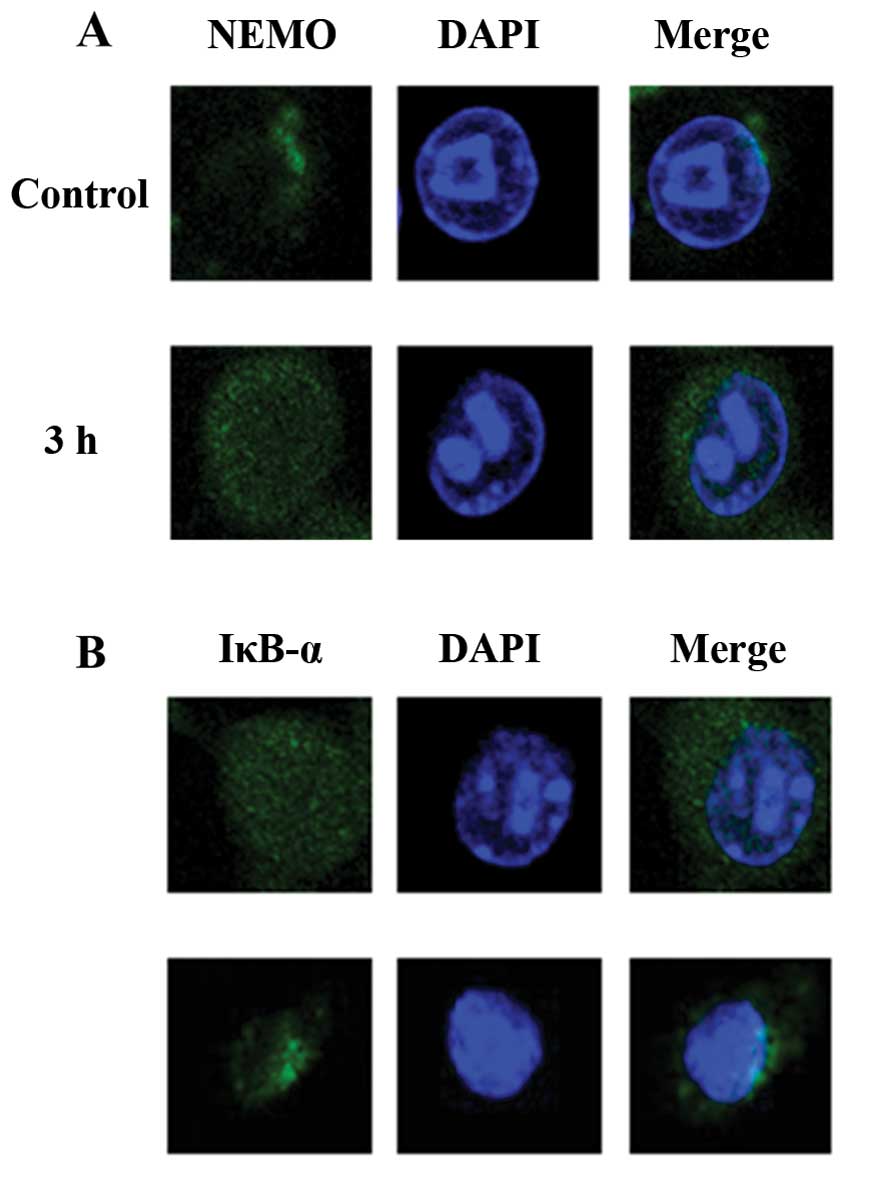
Expression of NEMO and IκB-α in
irradiated BV-2 cells
NEMO and IκB-α are both important mediators of the
NF-κB pathway. Immunocytochemical analysis indicated that NEMO
fluorescence was diffusely distributed in the cytoplasm at a
relatively low density under normal conditions (Fig. 5A). Three hours following 32 Gy
irradiation, the NEMO protein was markedly elevated. Moreover,
IκB-α levels were considerably decreased following irradiation
(Fig. 5B). This evidence suggests
that activation of NEMO and degradation of IκB-α occur after
radiation treatment of microglial cells.
Discussion
Partial or whole brain irradiation is a widely used,
effective treatment for primary and metastatic brain tumors.
However, it may increase the risk of radiation-induced brain
injury, including cognitive impairment with the manifestation of
functional deficits in memory, attention and executive function
that severely affect the patient’s quality of life (14). Therefore, it is critical to both
understand and minimize the side-effects of brain irradiation.
It has been reported that radiation may trigger
activation of the microglia, which releases pro-inflammatory
cytokines, subsequently inhibiting proliferation of neural
precursor cells and hippocampal neurogenesis (15,16).
Here, we observed that irradiation at doses over 16 Gy could
efficiently induce microglial activation and increase expression of
the microglial biomarkers Iba-1 and CD68. In addition, the
generation of pro-inflammatory factors, including IL-1β, TNF-α,
IL-6, TLR-8 and COX-2, was greatly upregulated following
irradiation. These results are consistent with previous reports
that showed enhanced production of pro-inflammatory mediators
within hours after brain irradiation (6,11,17).
Anti-inflammatory agents, including ramipril and indomethacin, have
been shown to decrease the number of activated microglia in the
hippocampus and/or perirhinal cortex and prevent radiation-induced
cognitive impairment in rodents (15,18).
Future studies will continue to explore the potential effects of
anti-inflammatory drugs on brain injury induced by irradiation.
DNA DSBs, which are closely associated with
phosphorylated γ-H2AX, represent a radiation-induced lesion
(19). Ataxia telangiectasia
mutated (ATM) is a nuclear protein kinase that mediates apoptosis
and cell cycle checkpoint responses after DNA DSBs (20,21).
Moreover, it also represents a crucial regulator of NF-κB signaling
pathway activation by mediating the activities of NEMO (which
modulates NF-κB) and IκB kinase (IKK) (which phosphorylates the
NF-κB inhibitor IκB) (22). In the
present study, we found that 3 h following irradiation, γ-H2AX was
upregulated and the NF-κB p65 subunit had undergone translocation
to the nucleus. Additionally, the NF-κB modulator NEMO was markedly
elevated, whereas the NF-κB regulatory inhibitor IκB-α was
considerably decreased. Collectively, this indicates that the NF-κB
signaling pathway is activated and may contribute to microglial
activation upon radiation treatment. Activation of NF-κB,
specifically nuclear translocation of the p65 subunit, is required
for the induction of several pro-inflammatory cytokines such as
TNF-α (23). Antisense p65
oligonucleotides have been shown to reduce proinflammatory cytokine
production (24). Based on these
observations, we hypothesize that radiation increases NEMO
activity, decreases the inhibitory effect of IκB, triggers the
nuclear translocation of p65 and subsequently increases the
upregulation of pro-inflammatory cytokines, such as IL-1β and
TNF-α.
In summary, we conclude that radiation at doses over
16 Gy can efficiently induce microglial activation through
activating the NF-κB signaling pathway and promoting inflammatory
factor release. Future studies will focus on exploring the precise
association between the NF-κB pathway and inflammation in activated
microglia.
Acknowledgements
This study was supported by grants from National
Natural Science Foundation of China (No. 30800283 and 81172595),
Postdoctor Foundation of China (No. 20100480905) and Postdoctor
Special Foundation of China (201104440).
References
|
1
|
Tofilon PJ and Fike JR: The radioresponse
of the central nervous system: a dynamic process. Radiat Res.
153:357–370. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
van Rossum D and Hanisch UK: Microglia.
Metab Brain Dis. 19:393–411. 2004.
|
|
3
|
Greene-Schloesser D, Robbins ME, Peiffer
AM, Shaw EG, Wheeler KT and Chan MD: Radiation-induced brain
injury: a review. Front Oncol. 2:732012. View Article : Google Scholar
|
|
4
|
Chiang CS, McBride WH and Withers HR:
Radiation-induced astrocytic and microglial responses in mouse
brain. Radiother Oncol. 29:60–68. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hwang SY, Jung JS, Kim TH, et al: Ionizing
radiation induces astrocyte gliosis through microglia activation.
Neurobiol Dis. 21:457–467. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee WH, Sonntag WE, Mitschelen M, Yan H
and Lee YW: Irradiation induces regionally specific alterations in
pro-inflammatory environments in rat brain. Int J Radiat Biol.
86:132–144. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pocock JM and Liddle AC: Microglial
signalling cascades in neurodegenerative disease. Prog Brain Res.
132:555–565. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim SU and de Vellis J: Microglia in
health and disease. J Neurosci Res. 81:302–313. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dropcho EJ: Neurotoxicity of radiation
therapy. Neurol Clin. 28:217–234. 2010. View Article : Google Scholar
|
|
10
|
Kyrkanides S, Olschowka JA, Williams JP,
Hansen JT and O’Banion MK: TNF-α and IL-1β mediate intercellular
adhesion molecule-1 induction via microglia-astrocyte interaction
in CNS radiation injury. J Neuroimmunol. 95:95–106. 1999.
|
|
11
|
Kyrkanides S, Moore AH, Olschowka JA, et
al: Cyclooxygenase-2 modulates brain inflammation-related gene
expression in central nervous system radiation injury. Brain Res
Mol Brain Res. 104:159–169. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Heese K, Fiebich BL, Bauer J and Otten U:
NF-κB modulates lipopolysaccharide-induced microglial nerve growth
factor expression. Glia. 22:401–407. 1998.
|
|
13
|
Liu B, Wang K, Gao HM, Mandavilli B, Wang
JY and Hong JS: Molecular consequences of activated microglia in
the brain: overactivation induces apoptosis. J Neurochem.
77:182–189. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Greene-Schloesser D, Moore E and Robbins
ME: Molecular pathways: radiation-induced cognitive impairment.
Clin Cancer Res. 19:2294–2300. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Monje ML, Toda H and Palmer TD:
Inflammatory blockade restores adult hippocampal neurogenesis.
Science. 302:1760–1765. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Monje ML, Mizumatsu S, Fike JR and Palmer
TD: Irradiation induces neural precursor-cell dysfunction. Nat Med.
8:955–962. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chiang CS, Hong JH, Stalder A, Sun JR,
Withers HR and McBride WH: Delayed molecular responses to brain
irradiation. Int J Radiat Biol. 72:45–53. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee TC, Greene-Schloesser D, Payne V, et
al: Chronic administration of the angiotensin-converting enzyme
inhibitor, ramipril, prevents fractionated whole-brain
irradiation-induced perirhinal cortex-dependent cognitive
impairment. Radiat Res. 178:46–56. 2012. View Article : Google Scholar
|
|
19
|
Lobrich M, Shibata A, Beucher A, et al:
γH2AX foci analysis for monitoring DNA double-strand break repair:
strengths, limitations and optimization. Cell Cycle. 9:662–669.
2010.
|
|
20
|
Abraham RT: Cell cycle checkpoint
signaling through the ATM and ATR kinases. Genes Dev. 15:2177–2196.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bakkenist CJ and Kastan MB: Initiating
cellular stress responses. Cell. 118:9–17. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu ZH, Shi Y, Tibbetts RS and Miyamoto S:
Molecular linkage between the kinase ATM and NF-κB signaling in
response to genotoxic stimuli. Science. 311:1141–1146. 2006.
|
|
23
|
Beg AA and Baltimore D: An essential role
for NF-κB in preventing TNF-α-induced cell death. Science.
274:782–784. 1996.
|
|
24
|
Tak PP and Firestein GS: NF-κB: a key role
in inflammatory diseases. J Clin Invest. 107:7–11. 2001.
|