Introduction
The number of new cases of melanoma was estimated at
76,250, and 9,180 patients died of melanoma of the skin in the
United States in 2012 (1). Despite
a relatively low incidence of melanoma accounting for only 4% of
all dermatological cancers, metastatic melanoma is responsible for
~80% of deaths caused by skin cancer due to its aggressive
properties and drug-resistance (2,3). When
patients are diagnosed with primary melanoma early, surgical
resection is the best option for the majority in order to reduce
the risk of mortality. However, a recent report revealed that the
1-year overall survival rate in phase II trials for patients with
metastatic melanoma was 25%, with a median survival time of 6.2
months (4). Therefore, the search
for effective agents to overcome this devastating disease is still
the top priority in melanoma therapy.
Dihydromyricetin (DHM) is a flavonoid compound
isolated from the classical Chinese herb Ampelopsis
grossedentata widely distributed in South China (5–7). DHM
is also called ampelopsin
[(2R,3R)-3,5,7-trihydroxy-2-(3,4,5-trihydroxyphenyl)-2,3-dihydrochromen-4-one],
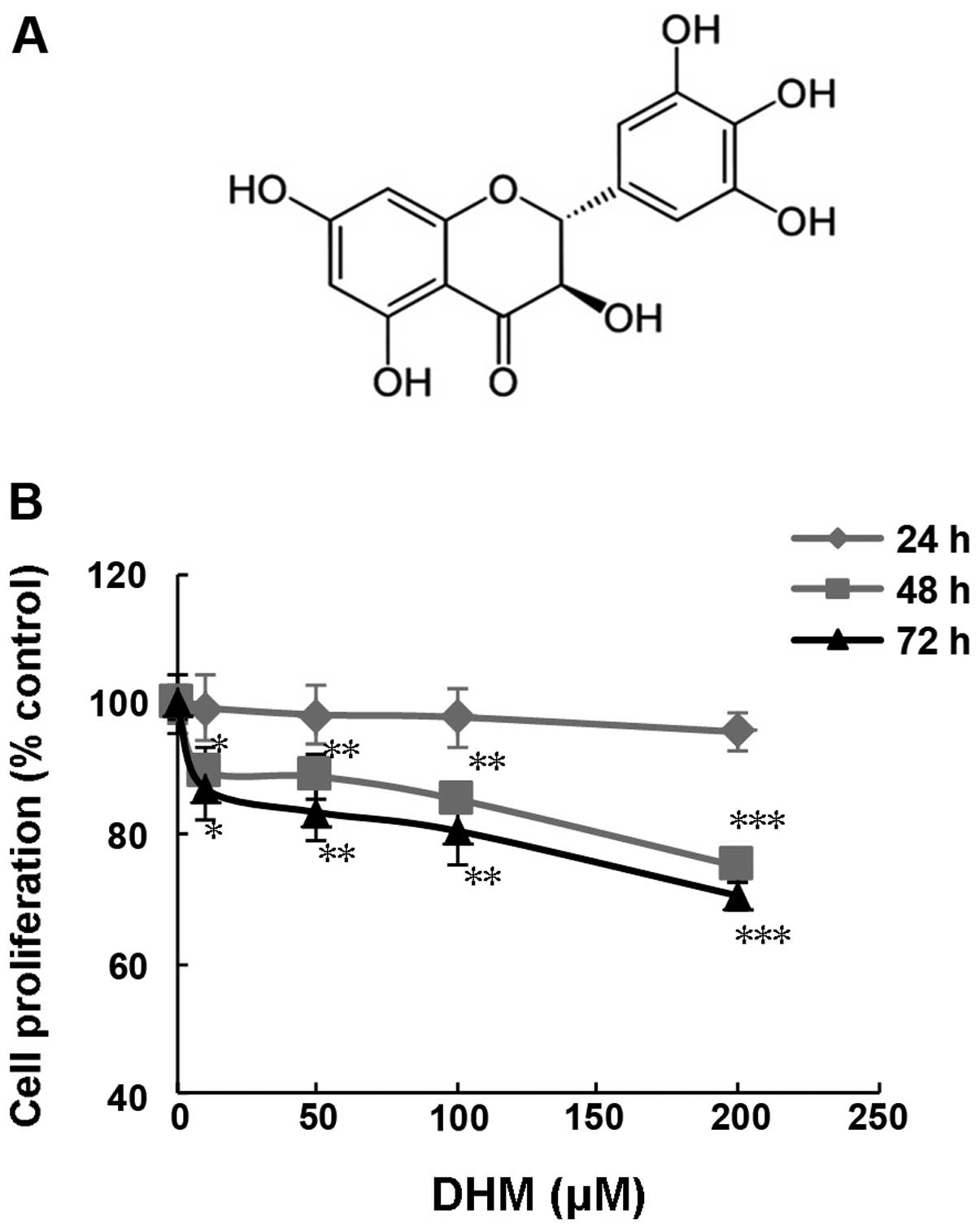
and its chemical structure is similar to myrecetin (Fig. 1A). DHM was previously reported to
exert multiple bioactivities including hepatic protection (8,9),
antioxidation (10,11), hypoglycemic activity (12) and antiinflammation (13).
In recent years, researchers have had great interest
in exploring the anticancer effects of DHM. A previous study
confirmed that DHM inhibits the cell proliferation and metastasis
of prostate cancer in vitro and in vivo (14). DHM sodium was shown to inhibit the
proliferation of bladder carcinoma, and its molecular mechanism was
partially attributed to cell cycle arrest (15). DHM was found to suppress the growth
of transplanted tumors derived from human lung cancer GLC-82 cells
in nude mice (16). In addition,
DHM suppressed the proliferation of hepatoma cells by inhibiting
angiogenesis via downregulation of vascular endothelial growth
factor and basic fibroblast growth factor expression (17,18).
Additionally, from the PubMed Database, we found
that only several articles in regard to the effects of DHM on
melanoma have been reported in the past two decades, and those
reports only showed DHM-inhibited migration, invasion and adhesion
of mouse melanoma B16 cells in vitro, and decreased
pulmonary metastasis of B16 cells in mice (19–21).
However, the molecular mechanisms remain unclear. Therefore, in
light of the previous studies, we speculated that DHM may be an
effective therapeutic candidate for melanoma therapy. In the
present study, we investigated the inhibitory effects of DHM in
human melanoma SK-MEL-28 cells and the underlying cellular and
molecular mechanisms. We demonstrated that DHM inhibited the
proliferation of SK-MEL-28 cells in a concentration- and
time-dependent manner and induced cell cycle arrest and promoted
the apoptosis of SK-MEL-28 cells.
Materials and methods
Cell line and chemicals
The human melanoma SK-MEL-28 cell line was purchased
from the Shanghai Cell Bank of the Chinese Academy of Sciences, and
was cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco,
Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS)
(Gibco) in a humidified incubator at 37°C with 5% CO2.
Cells were passed by dissociation with 0.25% trypsin-EDTA solution
(Gibco) for 1–2 min after having grown to 80–90% confluency.
Dihydromyricetin was purchased from Sigma-Aldrich Biotechnology
(St. Louis, MO, USA) and dissolved in dimethyl sulfoxide (DMSO) for
the stock solution. It was then diluted to a final concentration in
culture medium for the following studies, and the final
concentration of DMSO was <0.3%. TUNEL assay kit for detecting
apoptosis was obtained from Promega Corporation (Madison, WI, USA).
All primary antibodies used for p53, p21, Cdc25A, Cdc2, P-Cdc2,
Bax, IKK-α, NF-κB p65, p38 and P-p38 were purchased from Cell
Signaling Technology (Beverly, MA, USA). Cell cycle staining
solution was obtained from Multisciences Biotechnology, Co., Ltd.
(Hangzhou, China) (cat# CCS012A).
Growth inhibitory assay
The effect of DHM treatment on the inhibition of
SK-MEL-28 cells was measured by 3-(4,
5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
(Sigma-Aldrich Biotechnology) assay according to a previously
published study with minor modifications (22). Briefly, SK-MEL-28 cells were
cultured in 96-well plates at 1×104 cells/well in 1640
culture medium with 10% FBS. DHM was added after 10 h starting from
inoculation of SK-MEL-28 cells. After incubation for 24, 48 and 72
h, 20 μl of MTT solution (5 mg/ml) in 1X phosphate-buffered saline
(PBS) was directly added into each well and incubation was
continued for 4 h at 37°C. Thereafter, the medium was removed, and
150 μl of DMSO was added to the wells. The absorbance was measured
at a wavelength of 570 nm with an automated spectrophotometric
plate reader (Perkin-Elmer, Waltham, MA, USA). The experiments were
independently performed at least three times.
DHM regulates cell cycle progression in
SK-MEL-28 cells
To determine the effects of DHM treatment on cell
cycle distribution in SK-MEL-28 cells, the previously described
procedure was performed with slight modification (14). Briefly, cells were treated without
or with 50 and 100 μM DHM for 24, 48 and 72 h. Subsequently, the
cells were digested with 0.25% trypsin-EDTA solution and
centrifuged at 1×1,000 rpm. The harvested cells were pipetted into
75% ethanol for 1 h, and were centrifugated to discard the 75%
ethanol. The pellet of cells was tapped to be loosened and 2–3 ml
PBS was added to let the cells re-hydrate for 15 min at room
temperature. Subsequently, the cell suspension was centrifugated to
discard the supernatant. The cells were incubated with cell cycle
staining solution containing propidium iodide for 30 min at room
temperature according to the operating instructions. Then, stained
cells were measured by FACScan (Becton-Dickinson, Franklin Lakes,
NJ, USA) for cell cycle distribution, and the experiments were
carried out in duplicate for three times.
TUNEL assay for detecting apoptosis
induced by DHM
The fragmentation of nuclear chromatin is one of the
important hallmarks of late-stage apoptosis. Thus, in order to
detect the apoptosis of SK-MEL-28 cells treated by DHM, TUNEL assay
was used to measure nuclear DNA fragmentation according to the
instructions provided in the kit. Briefly, SK-MEL-28 cells were
treated with various final concentrations of 0 (served as control),
50 and 100 μM DHM after cell inoculation and were seeded in glass
bottom microwell dishes (MatTek Corp., Ashland, MA, USA). After 48
h of DHM treatment, the culture medium was removed, and the cells
were washed twice with PBS and fixed in 4% paraformaldehyde for 30
min at 4°C. Subsequently, the cells were pretreated with 0.2%
Triton X-100 in PBS for 5 min, and stained for 1 h at 37°C, with
compound solution, consisting of fluorescein-12-dUTP, dATP,
Tris-HCl, EDTA and terminal deoxynucleotidyl transferase according
to the instructions, and then were counterstained with DAPI (blue)
for 5 min at room temperature. The cells were observed under a
laser confocal scanning microscope (Leica TCS SP8; Leica
Microsystems Mannheim, Germany) using a standard parameter for
green fluorescence of fluorescein. To measure the apoptotic cells,
at least five images were randomly captured for the control and
treated cells, respectively.
Western blot analysis
Human melanoma SK-MEL-28 cells were treated with 50
and 100 μM DHM for 24 and 48 h, and cell lysates were prepared in
lysis buffer [0.5 M Tris-HCl, pH 7.4, 1.5 M NaCl, 1% NP-40, 0.02%
NaN3, 1 mM sodium orthovanadate, 2.5% deoxycholic acid,
10 mM ethylenediaminetetraacetate (EDTA), 1 mM phenylmethylsulfonyl
fluoride (PMSF) and a protease inhibitor cocktail set (Roche,
Penzberg, Germany)]. The lysates were centrifuged at 12,000 × g at
4°C for 10 min to obtain supernatant soluble proteins, and the
protein concentrations were determined using the BCA assay
(Beyotime Institute of Biotechnology, Beijing, China). Equal
amounts of protein (10 μg) from each sample were separated by
electrophoresis on 10% SDS-polyacrylamide gel, and transferred to
polyvinylidene difluoride (PVDF) membranes, and then the PVDF
membranes were blocked with 5% skim milk in 1X TBST containing 0.1%
Tween-20 at room temperature for 1 h. The membranes were probed
with primary antibodies including p53, p21, Cdc25A, Cdc2, P-Cdc2,
Bax, IKK-α, NF-κB p65, p38 and P-p38 as well as β-tubulin as a
loading control. The membranes were incubated for 1 h with
horseradish peroxidase-conjugated anti-rabbit secondary antibodies.
The bands were detected by an enhanced chemiluminescence reagent
and system (Amersham Biosciences Corp., Piscataway, NJ, USA).
Statistical analysis
Data are expressed as means ± SD. Statistical
analysis was performed by the Student’s t-test, and a probability
(P) value of <0.05 was considered to indicate a statistically
significant result.
Results
DHM exerts a growth inhibitory effect on
SK-MEL-28 cells
The inhibitory effect of DHM on cell proliferation
was evaluated by MTT assay after incubation with several
concentrations of DHM for 24, 48 and 72 h. The results indicated
that DHM treatment for 48 and 72 h, had a strong growth inhibitory
effect on SK-MEL-28 cells in a concentration- and time-dependent
manner (Fig. 1B).
DHM induces cell cycle arrest in
SK-MEL-28 cells
It is well known that the most common phenotype
observed in cancer cells is their rapid proliferation rate, which
causes the cell cycle to be susceptible to modulation. In order to
determine whether DHM has any effects on cell cycle distribution of
SK-MEL-28 cells, we tested the cell cycle status by using flow
cytometry after cells were treated with 50 and 100 μM DHM for 24,
48 and 72 h. The results showed that DHM treatment for 24 h did not
significantly change the cell cycle profile in the SK-MEL-28 cells,
whereas DHM treatment induced a significant increase in the
percentage of cells in the G1 phase with a parallel
significant reduction in cells in the S and G2/M phase
following 48 h in SK-MEL-28 cells, compared to the control
(Table I). Furthermore, continuous
DHM treatment for up to 72 h led to a high increase in the
proportion of G1 phase cells, causing a reduction in
cells in the S and G2/M phase of the cell cycle in
SK-MEL-28 cells. These results suggest a potential blockade of cell
cycle progression induced by DHM at the G1/S checkpoint
in SK-MEL-28 cells.
 | Table IEffects of DHM treatment on cell
cycle distribution of SK-MEL-28 cells. |
Table I
Effects of DHM treatment on cell
cycle distribution of SK-MEL-28 cells.
| Time | Concentration | G1
(%) | S (%) | G2/M
(%) |
|---|
| 24 h | μM (DHM) | | | |
| 0 | 53.23±0.56 | 32.93±0.68 | 13.84±0.72 |
| 50 | 53.12±0.54 | 32.21±0.39 | 14.67±0.55 |
| 100 | 52.73±0.91 | 32.52±0.77 | 14.75±0.74 |
| 48 h | 0 | 54.18±1.32 | 30.68±0.89 | 15.14±0.43 |
| 50 | 59.69±1.65a | 26.82±1.52a | 13.49±0.66a |
| 100 | 61.34±0.74a | 25.71±0.52a | 12.94±0.54a |
| 72 h | 0 | 59.29±2.58 | 27.73±1.40 | 12.98±1.22 |
| 50 | 66.40±3.10a | 21.33±2.50a | 12.27±0.79 |
| 100 | 73.50±3.05a,b | 18.13±2.46a | 8.37±0.60a,b |
Since it has been well recognized that the
tumor-suppressor gene p53 regulates the expression and activity of
downstream genes that control cell cycle progression, we decided to
determine the expression level of p53 and the cell cycle-related
biomarkers, p21, cell division cycle 25A (Cdc25A), cell division
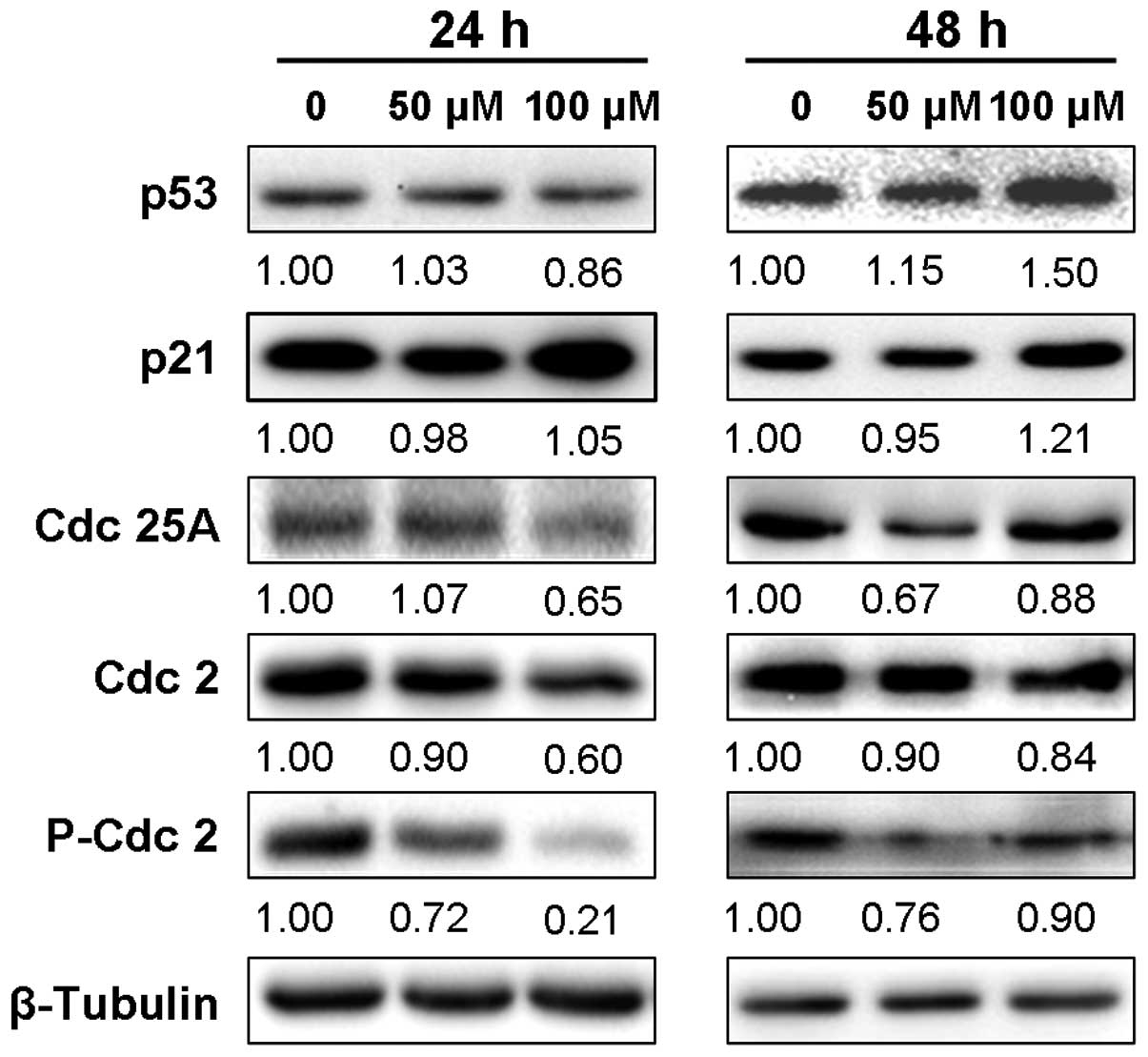
cycle 2 (Cdc2) and P-Cdc2. We found that immunoblotting of
SK-MEL-28 cell lysates after treatment with 50 or 100 μM DHM for 48
h demonstrated an apparent increase in p53 protein, although DHM
did not appear to upregulate p53 expression within 24 h of
treatment (Fig. 2). As p21 is one
of the most vital downstream transcriptional targets of p53, we
also detected the production of p21 protein. The results showed
that the expression level of p21 was evidently increased after 48 h
of incubation with 100 μM DHM in melanoma SK-MEL-28 cells, which
indicated that DHM modulated p53-mediated cell cycle arrest via
upregulation of the protein level of p21. In addition, the
expression levels of Cdc25A, Cdc2 and P-Cdc2 were clearly
downregulated by DHM treatment within 24 or 48 h (Fig. 2). Therefore, DHM treatment
upregulated the production of p53 and p21 proteins. Meanwhile, it
downregulated the expression of Cdc25A, Cdc2 and P-Cdc2 related
with cell cycle arrest in SK-MEL-28 cells.
DHM induces apoptosis in SK-MEL-28
cells
Cell cycle checkpoint is an important intersection
for cell survival or cell death. If conditions where cells live are
favorable for successful interphase and mitosis, cells can divide
and maintain growth or cells may ‘switch on’ a death procedure.
Thus, according to the above results showing cell cycle arrest
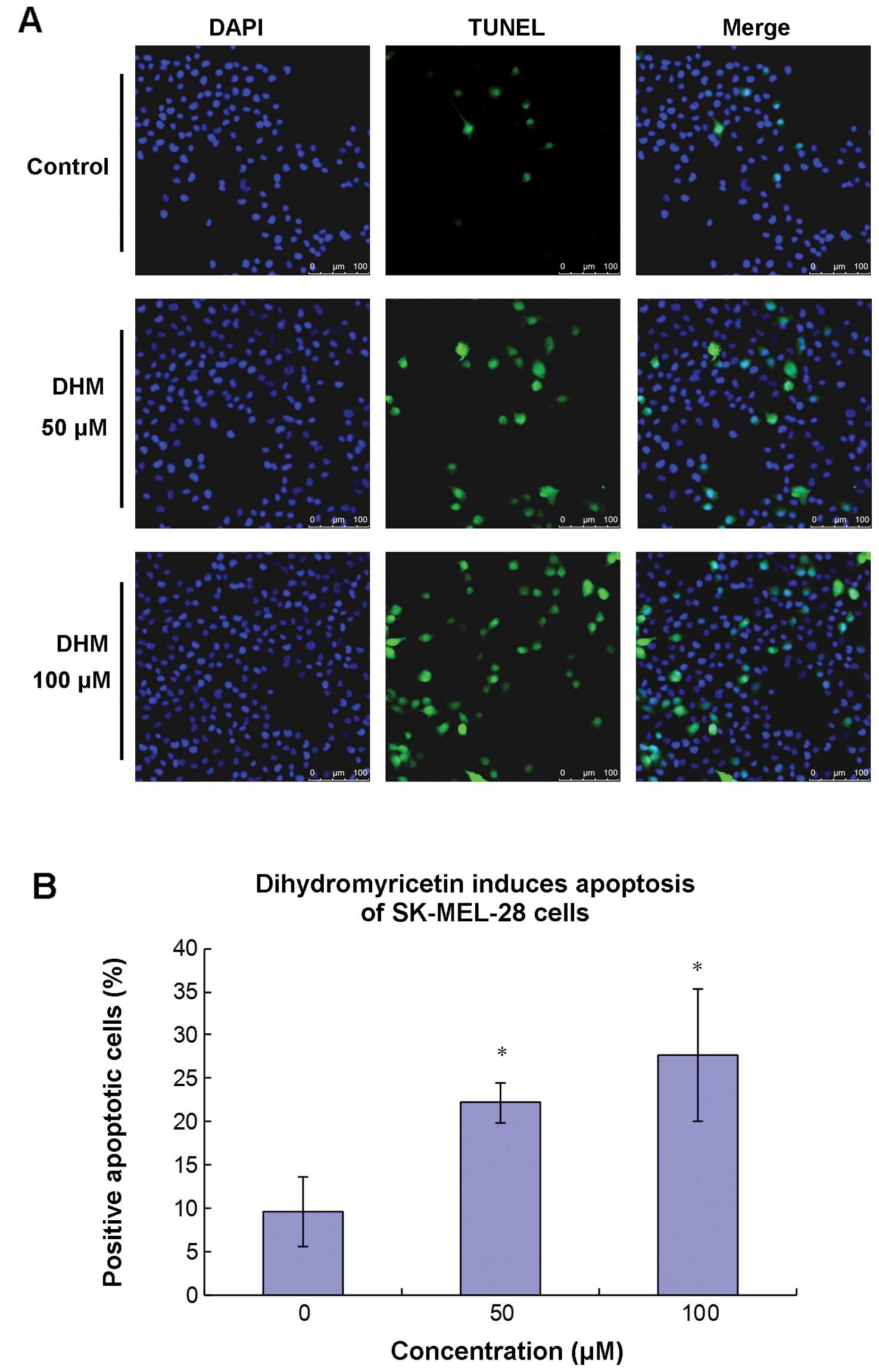
mediated by DHM, we applied TUNEL analysis to detect the effects of
DHM treatment on apoptotic induction in SK-MEL-28 cells. The cells
were treated for 48 h at a concentration of 50 or 100 μM DHM,
respectively. Following the procedure for TUNEL assay, the stained
cells were observed by laser confocal scanning microscopy (Fig. 3A). The results demonstrated that the
number of TUNEL-positive cells (apoptotic cells) was significantly
increased after 50 and 100 μM DHM treatment for 48 h. Compared to
the control, the percentage of positive apoptotic cells increased
from 9.50±4.04 to 22.20±2.35 and 27.65±7.65%, respectively after 48
h of DHM treatment at 50 and 100 μM (Fig. 3B). Additionally, the data indicate
that nuclear chromatin in SK-MEL-28 cells was fragmented, which is
an apoptotic characteristic.
DHM induces apoptosis by regulating the
expression levels of p53, Bax, NF-κB p65, IKK-α and P-p38
p53 is a critical mediator of cell death and its
role in apoptosis is well established (23). Therefore, we assessed the expression
levels of p53 downstream genes such as Bax and NF-κB p65. The
immunoblot results showed that the expression level of Bax protein
in SK-MEL-28 cells was increased after 48 h of DHM treatment,
although 24 h of incubation with 50 or 100 μM DHM barely affected
its expression level (Fig. 4). In
addition, the protein expression level of NF-κB p65 was decreased
within 24 or 48 h of DHM treatment. We also assessed the expression
levels of upstream protein, IKK-α. The results showed that IKK-α
protein was apparently downregulated between 24 and 48 h of
incubation with 100 μM DHM. Unexpectedly, DHM treatment for 24 h
decreased its protein expression, but continuous treatment for up
to 48 h upregulated its expression. Moreover, we found that
although the protein level of p38 was slightly affected, its
phosphorylated form, P-p38, was clearly downregulated in a
concentration- and time-dependent manner (Fig. 4). Taken together, DHM treatment led
to upregulation of p53 and Bax protein expression, and decreased
expression levels of NF-κB p65, IKK-α and P-p38, which were
associated with DHM-induced apoptosis in SK-MEL-28 cells.
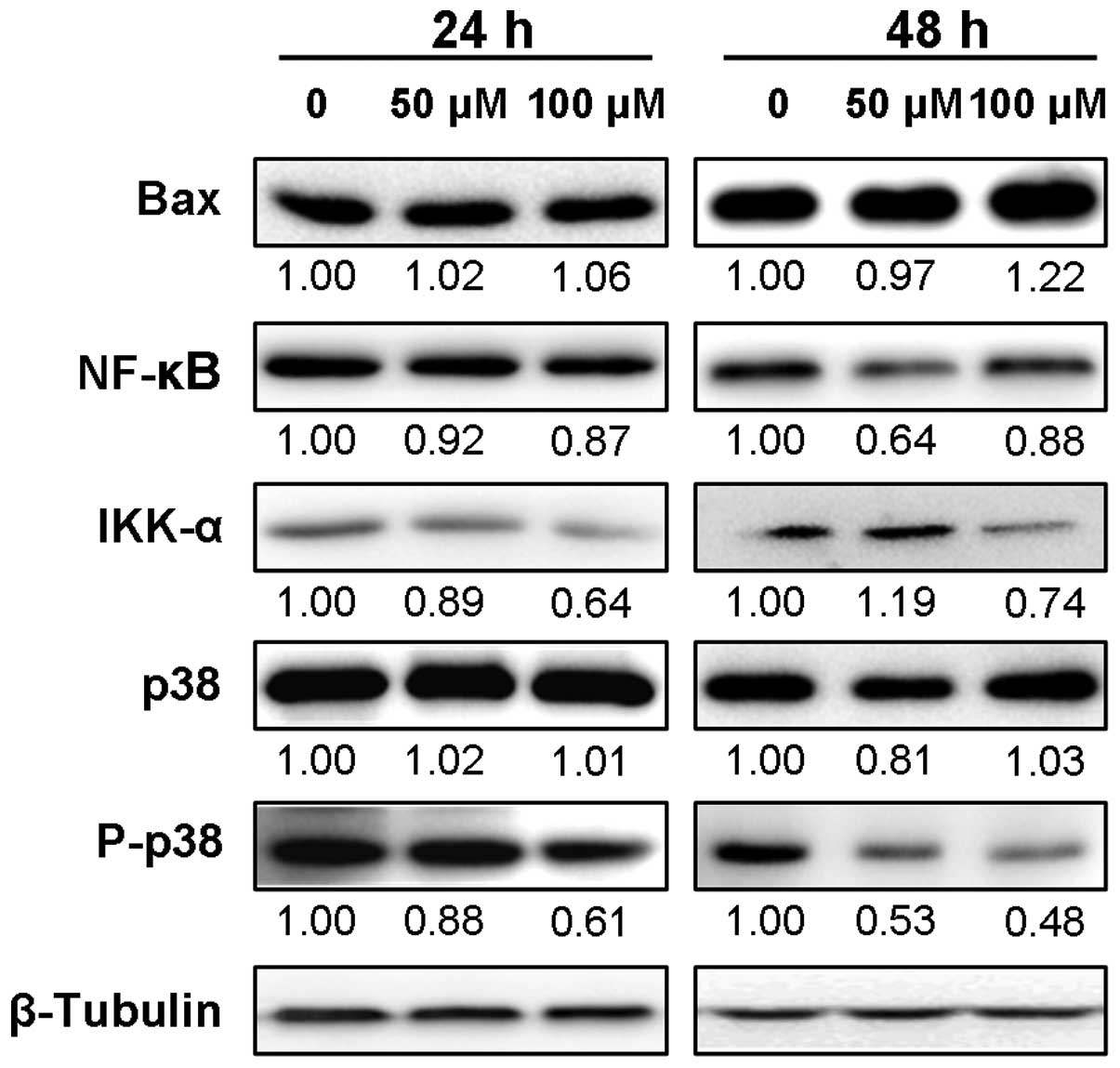 | Figure 4Protein expression levels of Bax,
NF-κB p65, IKK-α, p38 and P-p38 are associated with apoptosis
induced by DHM. Proteins (10 μg) of total cell lysates from each
sample were resolved by 10% acrylamide SDS-PAGE, blotted on
membrane and probed with specific antibodies containing Bax, NF-κB
p65, IKK-α, p38 and P-p38. The number under each lane of the
representative western blot images indicates the relative
expression level of targeted protein (Bax, NF-κB p65, IKK-α, p38
and P-p38)/β-tubulin (% control), assuming the control targeted
protein in SK-MEL-28 cells untreated by DHM to be 1.00. β-tubulin
was used as the loading control. |
Discussion
Melanoma is one of the most aggressive forms of
cancer with increasing incidence rates and high resistance to
current therapies (1,24). Until recently, no agent has been
developed for the effective long-term treatment of patients with
metastatic skin cancer (25,26).
With so few treatment options available, new therapeutic agents are
urgently needed to prevent and overcome this aggressive cancer.
Therefore, to investigate a possible anti-melanoma effect of DHM as
a first step toward the development of a novel putative anticancer
agent, we studied DHM for its capability to inhibit cell
proliferation in the melanoma SK-MEL-28 cell line.
Previous reports showed that DHM is able to inhibit
cell growth in a panel of cancer cell lines, such as human prostate
cancer cell lines PC-3 and LNCaP (14), human bladder carcinoma EJ cells and
murine sarcoma 180 cells (15), and
hepatoma HepG2 cell line (17). In
the present study, we firstly found that DHM significantly
inhibited the proliferation of the human melanoma cell line
SK-MEL-28 (Fig. 1B). Furthermore,
our data demonstrated that the antiproliferation effect of DHM in
SK-MEL-28 cells was associated with cell cycle arrest and induction
of apoptosis.
We confirmed that DHM treatment arrested
SK-MEL-cells at the G1 phase, as well as decreased the
proportion of cells at the S and G2/M phase (Table I). Notably, a previous study
reported that DHM induced cell cycle arrest at the S phase in LNCaP
cells or at the S and G2/M phase in PC-3 cells (14). We estimated that the mechanism
involved in cell cycle arrest caused by DHM varied in different
cancer cells. Furthermore, to clarify the mechanism of DHM-mediated
cell cycle arrest in melanoma SK-MEL-28 cells, we confirmed the
hypothesis that DHM modulates the p53 signaling pathway to affect
cell cycle status. The results revealed a significant increase in
p53 protein expression after 48 h of DHM treatment (Fig. 2). p53 functions as a node for
organizing whether cells respond to various types and levels of
stress with apoptosis, cell cycle arrest, senescence, DNA repair,
cell metabolism or autophagy (27).
Serving as one of the most important p53 target genes inducing
growth arrest, expression level of p21 protein in SK-MEL-28 cells
was obviously upregulated after 48 h of DHM treatment (Fig. 2). Recruitment of coactivators
including CBP/p300 and Tip 60/hMOF in a p53-dependent manner is
absolutely required for p21 activation (28,29).
p21 has been shown to contribute to the regulation of cell cycle
progression in G1/S phase transition (30). Thus, our data showed that DHM
treatment led to cell cycle arrest at G1 phase and
decreased the proportion of S and G2/M phase cells via
upregulation of the p53 and p21 protein expression in SK-MEL-28
cells. However, it is still not clear whether DHM treatment causes
translocation or a change in the subcellular localization of p21,
and this issue requires further study. The primary substrate of
Cdc25A is cyclin-dependent kinase 2 which allows cell cycle
progression through the G1/S checkpoint when active. Our
data indicated that expression levels of Cdc25A, Cdc2 and P-Cdc2
were decreased, similar to previous studies (31–33).
The induction of apoptosis is considered to be a
useful approach for cancer therapy. The MTT assay initially
indicated that DHM acts as an antiproliferation agent against
melanoma SK-MEL-28 cells (Fig. 1B).
Therefore, we investigated whether DHM-mediated growth arrest was
due to induction of apoptosis apart from cell cycle arrest. In our
experiments, significant apoptosis following DHM treatment in
SK-MEL-28 cells was observed by laser confocal scanning microscopy
(Fig. 3). Further investigation
indicated that apoptosis induced by DHM was associated with the
upregulation of expression levels of p53 and Bax protein (Fig. 4). p53-mediated apoptosis has been
intensively studied since it was first demonstrated (34). Numerous publications have confirmed
the importance of p53 transcriptional regulation in apoptosis. Bax
is one of the p53 target genes inducing apoptosis, and so is an
important regulator of apoptosis (35). p53 interacts with Bcl-XL or Bcl-2 to
promote the oligomerization of Bak and Bax that assemble in the
mitochondrial membrane to form pores, resulting in the release of
cytochrome c and other apoptotic activators from the
mitochondria (5,36). Overexpression of Bcl-2 blocks
p53-mediated apoptosis, whereas, Bax binds to the Bcl-2 protein and
abolishes its anti-apoptotic ability. In brief, the p53-dependent
increase in Bax leads to apoptosis. Moreover, we did not detect the
expression level of Bcl-2 protein in the SK-MEL-28 cells by western
blot analysis. It has been reported that the expression level of
Bcl-2 protein in SK-MEL-28 cells was barely detectable compared
with the induction of Mcl-1S and Bak protein after 18 h of exposure
to bortezomib, a proteasome inhibitor (37). In the present study, the expression
levels of NF-κB p65 and its upstream IKK-α protein were
downregulated following DHM treatment. NF-κB regulates cell
proliferation and survival in eukaryotic cells. Active NF-κB turns
on the expression of genes which promotes cell proliferation and
protects cells from apoptosis. It was found that blockage or
defective NF-κB can make tumor cells stop proliferating and enhance
the susceptibility to apoptosis (38). We hypothesized that downregulation
of NF-κB p65 probably increased the sensitivity of SK-MEL-28 cells
to DHM and contributed to induction of apoptosis. IKK-α is an
upstream regulatory component for the NF-κB signaling pathway
(38). A decline in the expression
level of IKK-α possibly boosts the downregulation of NF-κB p65 and
helps to strengthen anticancer ability. In the present study, we
found that the expression level of p38 in SK-MEL-28 cells was
hardly affected after 24 or 48 h of incubation, while the
expression level of P-p38 was unexpectedly decreased (Fig. 4). The signaling of mitogen-activated
protein kinases (MAPKs) regulates various cellular functions such
as cell differentiation, proliferation and cell death. p38 is one
component of the MAPK family, and p38 activation generally
possesses a pro-apoptotic function. A previous study showed that
Cdc25B degradation was dependent mainly on c-Jun N-terminal kinase
(JNK) and partially on p38 (39).
We hypothesized that downregulation of P-p38 in SK-MEL-28 cells
probably contributed to pro-apoptotic activity and cell cycle
arrest. In addition, we also found that the phosphorylated form of
p38 was decreased in hepatoma HepG2 cells (unpublished data). The
effects of DHM treatment on the signaling of MAPKs consisting of
c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK),
p38 and extracellular signal-regulated kinase (ERK1/2) still
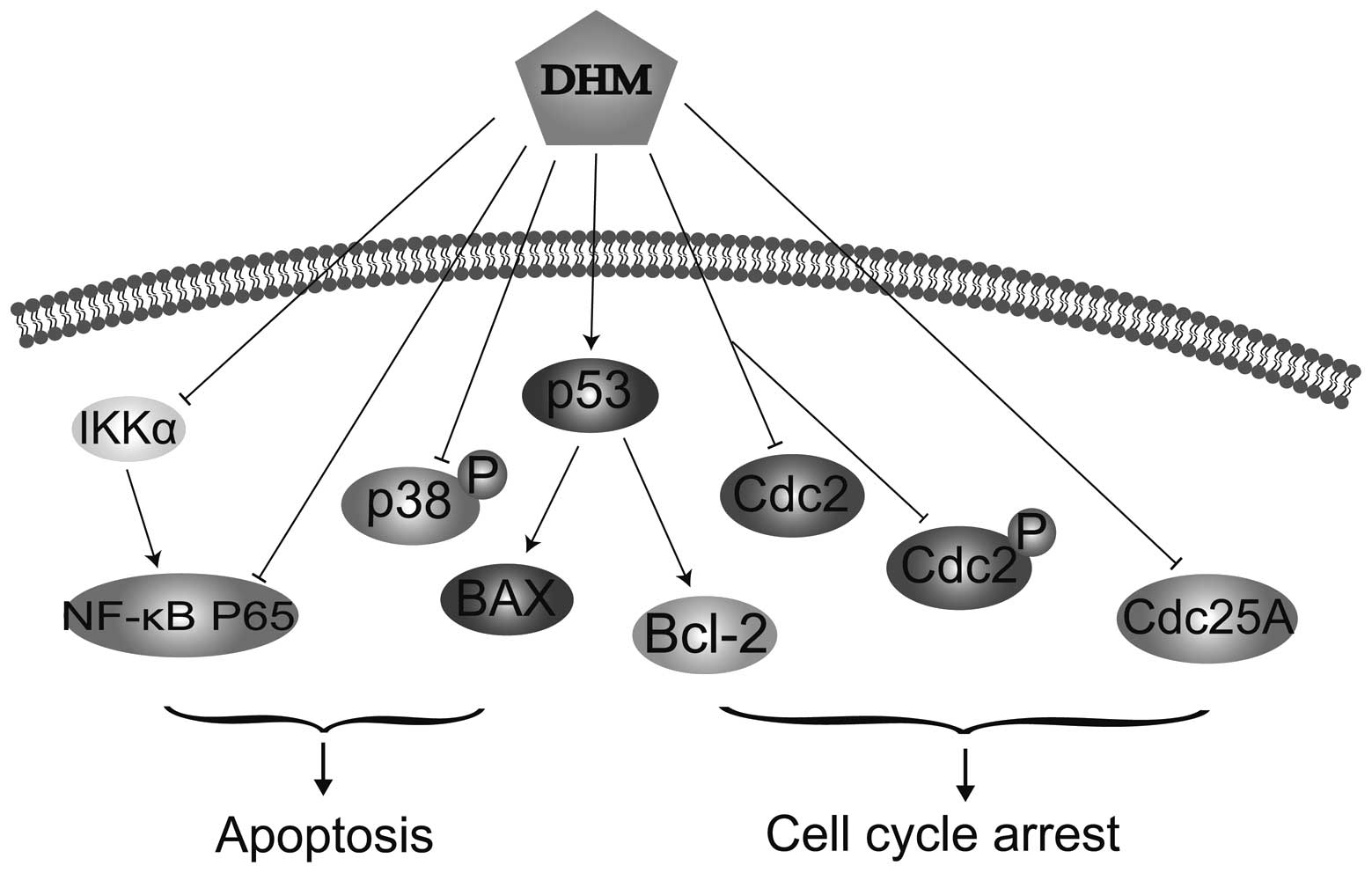
require further clarification. Taken together, the mechanisms of
DHM that induced cell cycle arrest and apoptosis in SK-MEL-28 cells
are possibly complex (Fig. 5).
In conclusion, our results demonstrated the
inhibitory effects and induction of cell cycle arrest at the
G1 phase and apoptosis in SK-MEL-28 cells in response to
DHM treatment. Within the above indicated dosage range, DHM
treatment upregulated the production of p53 protein, including its
downstream transcriptional target genes such as p21 and Bax protein
in SK-MEL-28 cells. Moreover, DHM also downregulated the protein
level of NF-κB p65, IKK-α and P-p38. Although, these data
apparently imply a pivotal role of p53 in DHM-mediated cell cycle
arrest and apoptosis, additional research must be performed to
clarify the role of p53 upstream and downstream signal networks. In
summary, our results imply that DHM treatment may be a new
therapeutic strategy against the growth of melanoma.
Acknowledgements
The present study was supported by the National
Natural Science Funds (grant no. 81041099) and Guangdong Province
Natural Science Funds, China (grant no. S2011010003750).
Abbreviations:
|
DHM
|
dihydromyricetin
|
|
DMSO
|
dimethyl sulfoxide
|
|
IKK-α
|
IκB kinase-α
|
|
TUNEL assay
|
terminal deoxynucleotidyl
transferase-mediated dUTP nick end labeling assay
|
|
MTT
|
3-(4, 5-dimethylthiazol-2-yl)-2,
5-diphenyltetrazolium bromide
|
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar
|
|
2
|
Gray-Schopfer V, Wellbrock C and Marais R:
Melanoma biology and new targeted therapy. Nature. 445:851–857.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Miller AJ and Mihm MC Jr: Melanoma. N Engl
J Med. 355:51–65. 2006. View Article : Google Scholar
|
|
4
|
Korn EL, Liu PY, Lee SJ, et al:
Meta-analysis of phase II cooperative group trials in metastatic
stage IV melanoma to determine progression-free and overall
survival benchmarks for future phase II trials. J Clin Oncol.
26:527–534. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
He GX, Pei G, Zhou TD and Zhou XX:
Determination of total flavonoids and dihydromyricetin in
Ampelopsis grossedentala(Hand-Mazz) (W.T. Wang). Zhongguo
Zhong Yao Za Zhi. 25:423–425. 2000.(In Chinese).
|
|
6
|
Du Q, Cai W, Xia M and Ito Y: Purification
of (+)-dihydromyricetin from leaves extract of Ampelopsis
grossedentata using high-speed countercurrent chromatograph
with scale-up triple columns. J Chromatogr A. 973:217–220.
2002.
|
|
7
|
Wang Y, Zhou L, Li R and Wang Y:
Determination of ampelopsin in the different parts of Ampelopsis
grossedentata in different seasons by RP-HPLC. Zhong Yao Cai.
25:23–24. 2002.(In Chinese).
|
|
8
|
Murakami T, Miyakoshi M, Araho D, et al:
Hepatoprotective activity of tocha, the stems and leaves of
Ampelopsis grossedentata, and ampelopsin. Biofactors.
21:175–178. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shen Y, Lindemeyer AK, Gonzalez C, Shao
XM, Spigelman I, OIsen RW and Liang J: Dihydromyricetin as a novel
anti-alcohol intoxication medication. J Neurosci. 32:390–401. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang YS, Ning ZX, Yang SZ and Wu H:
Antioxidation properties and mechanism of action of
dihydromyricetin from Ampelopsis grossedentata. Yao Xue Xue
Bao. 38:241–244. 2003.PubMed/NCBI
|
|
11
|
He G, Du F, Yang W, Pei G and Zhu Y:
Effects of tengcha flavonoids on scavenging oxygen free radicals
and inhibiting lipid-peroxidation. Zhong Yao Cai. 26:338–340.
2003.(In Chinese).
|
|
12
|
Zhong ZX, Qin JP, Zhou GF and Chen XF:
Experimental studies of hypoglycemic action on total flavone of
Ampelopsis grossedentata from Guangxi. Zhongguo Zhong Yao Za
Zhi. 27:687–689. 2002.(In Chinese).
|
|
13
|
Qi S, Xin Y, Guo Y, Diao Y, Kou X, Luo L
and Yin Z: Ampelopsin reduces endotoxic inflammation via repressing
ROS-mediated activation of PI3K/Akt/NF-κB signaling pathways. Int
Immunopharmacol. 12:278–287. 2012.PubMed/NCBI
|
|
14
|
Ni F, Gong Y, Li L, Abdolmaleky HM and
Zhou JR: Flavonoid Ampelopsin inhibits the growth and metastasis of
prostate cancer in vitro and in mice. PloS One. 7:e388022012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang B, Dong S, Cen X, et al: Ampelopsin
sodium exhibits antitumor effects against bladder carcinoma in
orthotopic xenograft models. Anticancer Drugs. 23:590–596. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zeng S, Liu D, Ye Y, Wang L and Wang W:
Anti-tumor effects of ampelopsin on human lung cancer GLC-82
implanted in nude mice. Zhong Yao Cai. 27:842–845. 2004.(In
Chinese).
|
|
17
|
Gao Q, Yang X and Ou M: Effect of serum
containing tengcha total flavonoid and dihydromyricetin on
proliferation and apoptosis of HepG2 cells. Zhongguo Zhong Yao Za
Zhi. 36:500–503. 2011.(In Chinese).
|
|
18
|
Luo GQ, Zeng S and Liu DY: Inhibitory
effects of ampelopsin on angiogenesis. Zhong Yao Cai. 29:146–150.
2006.(In Chinese).
|
|
19
|
Liu D and Luo M: Study on inhibitory
effect of ampelopsin on melanoma by serologic pharmacological
method. Zhong Yao Cai. 24:348–350. 2001.(In Chinese).
|
|
20
|
Zheng HQ and Liu DY: Anti-invasive and
anti-metastatic effect of ampelopsin on melanoma. Ai Zheng.
22:363–367. 2003.(In Chinese).
|
|
21
|
Liu DY, Zheng HQ and Luo GQ: Effects of
ampelopsin on invasion and metastasis of B16 mouse melanoma in vivo
and in vitro. Zhongguo Zhong Yao Za Zhi. 28:957–961. 2003.(In
Chinese).
|
|
22
|
Massaoka MH, Matsuo AL, Figueiredo CR, et
al: Jacaranone induces apoptosis in melanoma cells via ROS-mediated
downregulation of Akt and p38 MAPK Activation and displays
antitumor activity in vivo. PLoS One. 7:e386982012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Crighton D, Wilkinson S, O’Prey J, et al:
DRAM, a p53-induced modulator of autophagy, is critical for
apoptosis. Cell. 126:121–134. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ferlay J, Parkin DM and
Steliarova-Founcher E: Estimates of cancer incidence and mortatlity
in Europe in 2008. Eur J Cancer. 46:765–781. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sondak VK, Sabel MS and Mulé JJ:
Allogeneic and autologous melanoma vaccines: where have we been and
where are we going? Clin Cancer Res. 12:2337s–2341s. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Garbe C, Peris K, Hauschild A, et al:
Diagnosis and treatment of melanoma: European Consensus-based
interdisciplinary guideline. Eur J Cancer. 46:270–283. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kruse JP and Gu W: Modes of p53
regulation. Cell. 137:609–622. 2009. View Article : Google Scholar
|
|
28
|
Tang Y, Luo J, Zhang W and Gu W:
Tip60-dependent acetylation of p53 modulates the decision between
cell-cycle arrest and apoptosis. Mol Cell. 24:827–839. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sykes SM, Mellert HS, Holbert MA, Li K,
Marmorstein R, Lane WS and McMahon SB: Acetylation of the p53
DNA-binding domain regulates apoptosis induction. Mol Cell.
24:841–851. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Armstrong MJ, Stang MT, Liu Y, et al:
Interferon regulatory factor 1 (IRF-1) induces
p21WAF1/CIP1 dependent cell cycle arrest and
p21WAF1/CIP1 independent modulation of suivivin in
cancer cells. Cancer Lett. 319:56–65. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tu Ys, Kang XL, Zhou JG, Lv XF, Tang YB
and Guan YY: Involvement of Chk1-Cdc25A-cyclin A/CDK2 pathway in
simvastatin induced S-phase cell cycle arrest and apoptosis in
multiple myeloma cells. Eur J Pharmacol. 670:356–364. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bonnefont J, Laforge T, Plastre O, Beck B,
Sorce S, Dehay C and Krause KH: Primate-specific RFPL1 gene
controls cell-cycle progression through cyclin B1/Cdc2 degradation.
Cell Death Differ. 18:293–303. 2011.
|
|
33
|
Watanabe G, Behrns KE, Kim JS and Kim RD:
Heat shock protein 90 inhibition abrogates hepatocellular cancer
growth through cdc2-mediated G2/M cell cycle arrest and
apoptosis. Cancer Chemother Pharmacol. 64:433–443. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yonish-Rouach E, Resnitzky D, Lotem J,
Sachs L, Kimchi A and Oren M: Wild-type p53 induces apoptosis of
myeloid leukaemic cells that is inhibited by interleukin-6. Nature.
352:345–357. 1991. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Donauer J, Schreck I, Liebel U and Weiss
C: Role and interaction of p53, BAX and the stress-activated
protein kinases p38 and JNK in benzo(a)pyrene-diolepoxide induced
apoptosis in human colon carcinoma cells. Arch Toxical. 86:329–337.
2012. View Article : Google Scholar
|
|
36
|
Tomita Y, Marchenko N, Erster S, et al: WT
p53, but not tumor-derived mutants, bind to Bcl2 via the DNA
binding domain and induce mitochondrial permeabilization. J Biol
Chem. 281:8600–8606. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Qin JZ, Ziffra J, Stennett L, Bodner B,
Bonish BK, Chaturvedi V, et al: Proteasome inhibitors trigger
NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res.
65:6282–6293. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Escárcega RO, Fuentes-Alexandro S,
Garcı́a-Carrasco M, Gatica A and Zamora A: The transcription factor
nuclear factor-kappa B and cancer. Clin Oncol. 19:154–161.
2007.
|
|
39
|
Uchida S, Yoshioka K, Kizu R, et al:
Stress-activated mitogen-activated protein kinase c-Jun
NH2-terminal kinase and p38 target Cdc25B for degradation. Cancer
Res. 69:6438–6444. 2009. View Article : Google Scholar : PubMed/NCBI
|