Introduction
Lung cancer continues to be one of the most
prevalent malignancies and a leading cause of cancer-related
mortality worldwide. Non-small cell lung cancer (NSCLC)
constituting nearly 85% of all cases of lung cancer has been
treated by drugs targeting receptor tyrosine kinases (RTKs)
(1–4). Of these RTKs, the epidermal growth
factor receptor (EGFR) family, whose members include EGFR, ErbB2,
ErbB3 and ErbB4 has been widely recognized. Therefore, it has been
accepted that EGFR signaling may be crucial in cellular oncogenic
transformation, and dysregulation of EGFR has been implicated in
pathogenesis of various types of human cancers (5). EGFR-TKIs such as erlotinib (Tarceva)
and gefitinib (Iressa) have been evaluated in patients with NSCLCs
or other human cancers (6,7). Of note, activating mutations within
the EGFR tyrosine kinase domain including an amino acid
substitution at exon 21 (L858R) and in-frame deletions in exon 19
were found to be predictors of clinical response to EGFR-TKIs
(8). Initially, these competitive
and reversible EGFR-TKIs have been effective in patients with
NSCLCs. However, acquired resistance universally develops in many
patients responding to these drugs (9,10). In
the past several years, studies have revealed the two molecular
mechanisms for acquired resistance: a secondary T790M mutation in
EGFR and Met amplification (11–13).
To overcome the acquired resistance to EGFR-TKIs, conventional
approaches have focused on inhibiting the kinase activity of EGFR
with mutated T790M through selective inhibitors (14,15).
However, these approaches have shown that tumor cells may escape
such solitary inhibition through bypassing pathways that renew drug
resistance.
In this regard, a highly promising rational
multi-target drug may be an agent that targets heat shock protein
90 (Hsp90). Hsp90 is an ATP-dependent molecular chaperone which
modulates the stability of a large number of oncogenic proteins
including EGFR, c-Met, ErbB2, Raf1 and Akt (16–19).
Therefore, Hsp90 inhibition appears to be beneficial for anticancer
treatment as disruption of Hsp90 activity can induce the
degradation of oncogenic client proteins including mutant
T790M-EGFR in NSCLCs. Practically, geldanamycin ansamycin (GA) and
its derivatives have been reported to possess strong antitumor
effect on the growth of NSCLCs with T790M-EGFR (17,20,21).
However, the clinical use of Hsp90-targeting drugs including GA and
its derivatives are challenged by their intrinsic toxicity in spite
of their effective preclinical antitumor activities (22,23).
Therefore, these drawbacks have prompted the development of new GA
derivatives with less toxic outcomes. Consistent with this, we
previously reported the development of non-benzoquinone GA
derivatives, produced by a mutasynthetic approach and directed
biosynthetic method using genetically engineered Streptomyces
hygroscopicus (24,25). Here, the present study analyzes the
antitumor effects of WK88-1, a non-benzoquinone GA derivative, in
the gefitinib-resistant NSCLC H1975 cell line, with a secondary
T790M mutation in EGFR.
Materials and methods
Materials
Antibodies for phospho-EGFR (Tyr1068), ErbB2, ErbB3,
phospho-ErbB3, Met, phospho-Met (Tyr1234/1235), Akt, phospho-Akt
(Ser473), Hsp90, Hsp70, Erk1/2, phospho-Erk1/2 (Thr202/Tyr204),
caspase-3, cleaved caspase-3, cleaved PARP and β-actin were
purchased from Cell Signaling Technology (Beverly, MA, USA).
Antibodies specific for EGFR, Bcl-2 and p53 were purchased from
Santa Cruz Biotechnology (Santa Cruz, CA, USA). Gefitinib was
purchased from LC Laboratories (Woburn, MA, USA). Fetal bovine
serum (FBS), streptomycin and penicillin were obtained from Thermo
Scientific (South Logan, UT, USA). Halt™ Protease and Phosphatase
Inhibitor Cocktail (100X) and EDTA (100X) were purchased from
Thermo Fisher Scientific (Rockford, IL, USA). WK88-1 was purified
from a culture of S. hygroscopicus AC2, in which the AHBA
synthase gene was disrupted by the kanamycin-resistance gene,
supplemented with 3-aminobenzoic acid (24).
Cell culture
The human NSCLC cell line H1975 (EGFR L858R/T790M)
was maintained in RPMI-1640 with L-glutamine containing 10% fetal
bovine serum (FBS) and 1% penicillin/streptomycin. The cells were
cultured as a monolayer at 37°C with 5% CO2 in a
humidified incubator.
Cell proliferation assay
Cell proliferation was determined by MTS assay with
CellTiter96® Aqueous One Solution Reagent (Promega,
Madison, WI, USA). H1975 cells were plated in a 96-well plate at
2.5×103 cells/well. Following 24 h of incubation, the
cells were treated with the indicated concentration of the
compounds or DMSO and subsequently incubated for 24, 48 and 72 h at
37°C. After being incubated with compounds, 20 μl of
CellTiter96® Aqueous One Solution reagent was added to
the wells, and the plate was incubated at 37°C for an additional 1
h. Absorbance at 490 nm was then read on a Tecan Infinite F200 Pro
plate reader (Promega, Madison, WI, USA) and values were expressed
as percent of absorbance from cells incubated in DMSO alone.
Western blot analysis
H1975 cells were seeded in a 100-mm culture dish at
2×106 cells and allowed to attach. The cells were
treated with various concentrations of compound or DMSO and
incubated for 24 h. Cells were harvested in ice-cold lysis buffer
(50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% NP-40) with Halt
Protease-phosphatase inhibitor cocktail and EDTA. The cells were
incubated on ice for 30 min and centrifuged at 16,000 rcf for 20
min at 4°C. Subsequently, 20–30 μg of lysate/lane was separated by
SDS-PAGE followed by transfer to a PVDF membrane (Bio-Rad,
Hercules, NJ, USA). The membrane was blocked with 5% skim milk in
0.1% TBS-T for 2 h and then probed with the corresponding primary
antibodies overnight at 4°C. The membranes were washed with 0.1%
TBS-T and incubated with secondary antibodies for 1 h. The
membranes were developed by horseradish peroxidase-conjugated
secondary antibody, and proteins were visualized by SuperSignal
West Dura Extended Duration Substrate (Thermo Scientific, Waltham,
MA, USA). The membranes were imaged with LAS-3000 (Fuji, Japan)
according to the instructions of the manufacturer.
Migration and invasion assay
Assessment of cell migration and Matrigel invasion
capacity in H1975 cells was performed using BD cell-culture inserts
and/or BD BioCoat Matrigel invasion chamber (8-μm pore
size). The cells (5×104) were added into the upper
chambers, followed by a 24- (for migration) or 48-h (for invasion)
incubation at 37°C in 5% CO2. Culture media in inserts
were carefully removed, and the membrane containing the cells on
the lower surface of the inserts was fixed and stained with
hematoxylin and eosin staining (H&E). The cells that migrated
to the lower surface were quantified under a light microscope.
Flow cytometry
For determination of apoptotic cells, H1975 cells
were seeded at 5×105 cells/60-mm dish. Cell apoptosis
was measured using FITC Annexin V Apoptosis Detection kit I (BD
Biosciences Pharmingen) in accordance with the supplied protocols.
After exposure to the indicated compounds for 24 h, cells were
detached. The cells were washed twice with cold PBS, suspended in
1× binding buffer at a concentration of 5×105 cells/ml.
And then FITC Annexin V and propidium iodide (5 μl stock/100 μl
buffer) were added. After incubation for 15 min at RT in the dark,
400 μl of 1× binding buffer was added to each tube. The
cells were analyzed with a BD FACSVerse flow cytometer and BD
FACSuite software. The fraction of cell population in different
quadrants was analyzed using quadrant statistics.
Animals
For the in vivo xenograft assay, male athymic
nude mice (5 weeks of age) were obtained from Orient (Seoul, Korea)
and maintained under specific pathogen-free conditions based on the
guidelines established by the Seoul National University. Animals
were acclimated for 1 week before the study and housed in
climate-controlled quarters with a 12-h light/dark cycle.
Xenograft mouse model
Mice were divided into 2 groups for each cell line:
i) vehicle group (n=10); ii) 1 mg/kg of WK88-1 (n=10). H1975 cells
(1×106/100 μl) were suspended in RPMI-1640 medium and
inoculated with 100 μl Matrigel subcutaneously into the right flank
of each mouse. Vehicle or WK88-1 was injected 3 times/week. Tumor
volume was calculated from measurements of two diameters of the
individual tumor base using the following formula: Tumor volume
(mm3) = length × width × height × 0.52. Tumor volume was
measured every 3 or 4 days and the tumor weight was determined
after excision on the final day of the experiment. Mice were
monitored until tumors reached 1 cm3 total volume, at
which time mice were euthanized and tumors were extracted.
Statistical analysis
Quantitative data are presented as means value ± SD
unless indicated otherwise. The statistical significance of
compared measurements was measured using the Student’s t-test, and
P<0.05 was considered to indicate statistically significant
results.
Results
WK88-1, a non-benzoquinone GA derivative,
suppresses the proliferation of gefitinib-resistant H1975
cells
To date, one of the major obstacles to the
development of Hsp90 inhibitors concerns the issue of toxicity. Our
previous data revealed that a non-benzoquinone GA derivative,
WK88-1, exhibited little hepatotoxicity compared with GA (data not
shown), indicating that WK88-1 could be a potential alternative to
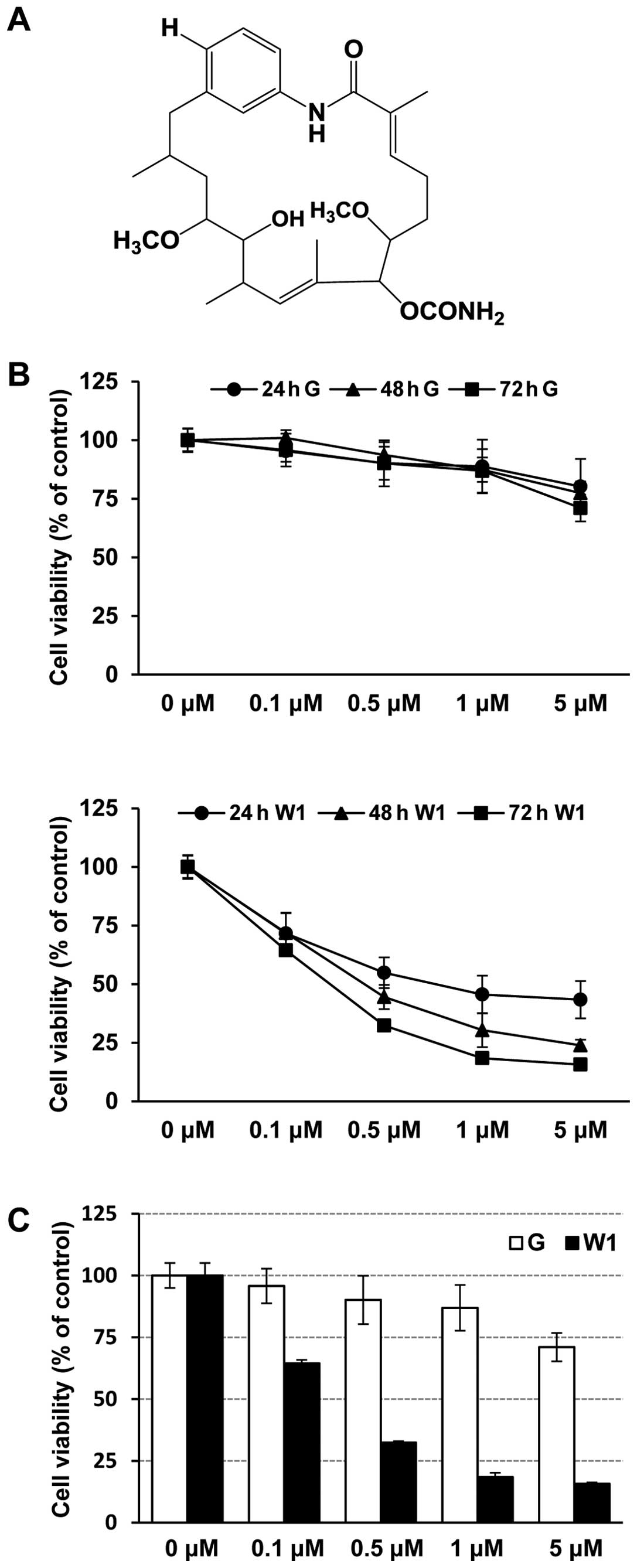
GA. WK88-1, a 18-dehydroxyl-17-demethoxyreblastatin (Fig. 1A) was synthesized through
mutasynthetic and directed biosynthetic approaches (24,25).
To investigate the effect of WK88-1, we used H1975, a human lung
adenocarcinoma cell line with the T790M/L858R mutation in EGFR. We
first assessed the anti-proliferative effects of gefitinib or
WK88-1 in H1975 cells. The cells were treated with the indicated
concentration of gefitinib or WK88-1 up to 72 h, and cell
proliferation was determined by MTS assay. Expectedly, our data
showed that H1975 cells were relatively resistant to gefitinib
treatment (Fig. 1B and C). However,
a potent growth-inhibitory effect was evidently observed in the
gefitinib-resistant H1975 cells which were treated with increasing
concentrations of WK88-1 (Fig. 1B and
C).
WK88-1 induces the degradation of RTKs
and inhibits downstream signaling in H1975 cells
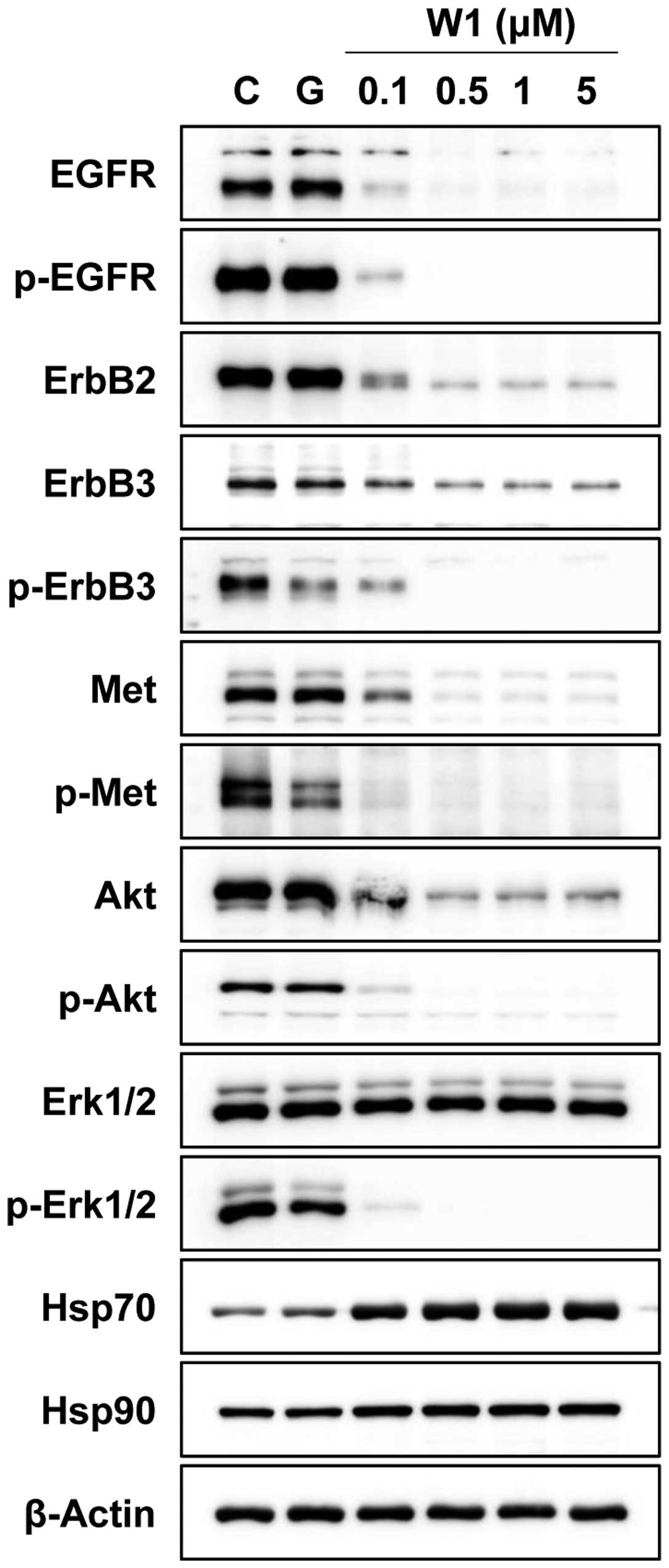
To investigate whether WK88-1 inhibits cancer cell
growth by Hsp90 inhibition, we examined the protein expression and
phosphorylation status of the client proteins of Hsp90 including
EGFR, Met and downstream signaling molecules in H1975 cells. The
cells were treated with 1 μM of gefitinib or various concentrations
of WK88-1 at 0.1, 0.5, 1 and 5 μM for 24 h. As expected,
treatment with 1 μM gefitinib did not show any inhibitory effect on
the phosphorylation of EGFR or activation of Akt and Erk1/2 in the
gefitinib-resistant H1975 cells (Fig.
2). In contrast, a robust decrease of total levels of EGFR,
ErbB2, ErbB3, Met and Akt proteins was observed in the
WK88-1-treated H1975 cells accompanied by upregulation of Hsp70
(Fig. 2). In addition, treatment
with WK88-1 inhibited the activity of downstream Akt and Erk1/2 by
inducing upstream protein degradation. Therefore, these findings
indicate that WK88-1 may circumvent gefitinib-resistance in H1975
cells through the inhibition of Hsp90 governing the stability of
ErbB3 as well as EGFR, ErbB2 and Met.
Effects of WK88-1 on the migration and
invasion in gefitinib-resistant H1975 cells
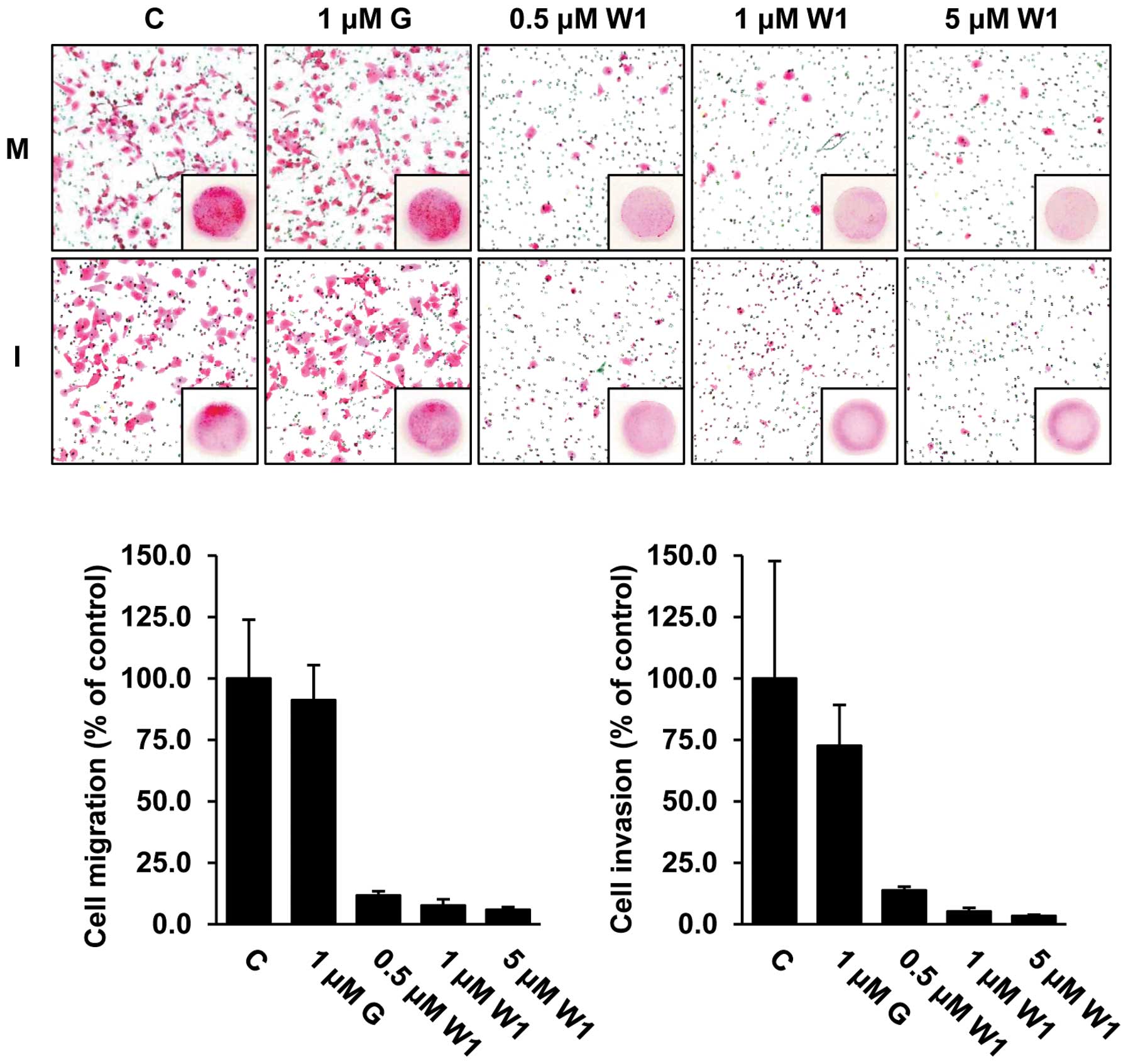
To assess whether WK88-1 has antitumor activity in
detail, we next performed in vitro migration and invasion
assays in the H1975 cells. As expected, our data revealed that
treatment with 1 μM gefitinib did not exhibit any significant
effect on the migratory and invasive capacities, whereas treatment
with WK88-1 strongly abrogated the migratory and invasive
capacities of the gefitinib-resistant H1975 cells (Fig. 3). Therefore, we conclude that a
significant inhibition of migration and invasion was observed in
the cells following treatment with WK88-1.
WK88-1 induces apoptosis in the
gefitinib-resistant H1975 cells
Notably, proliferation of tumor cells may be
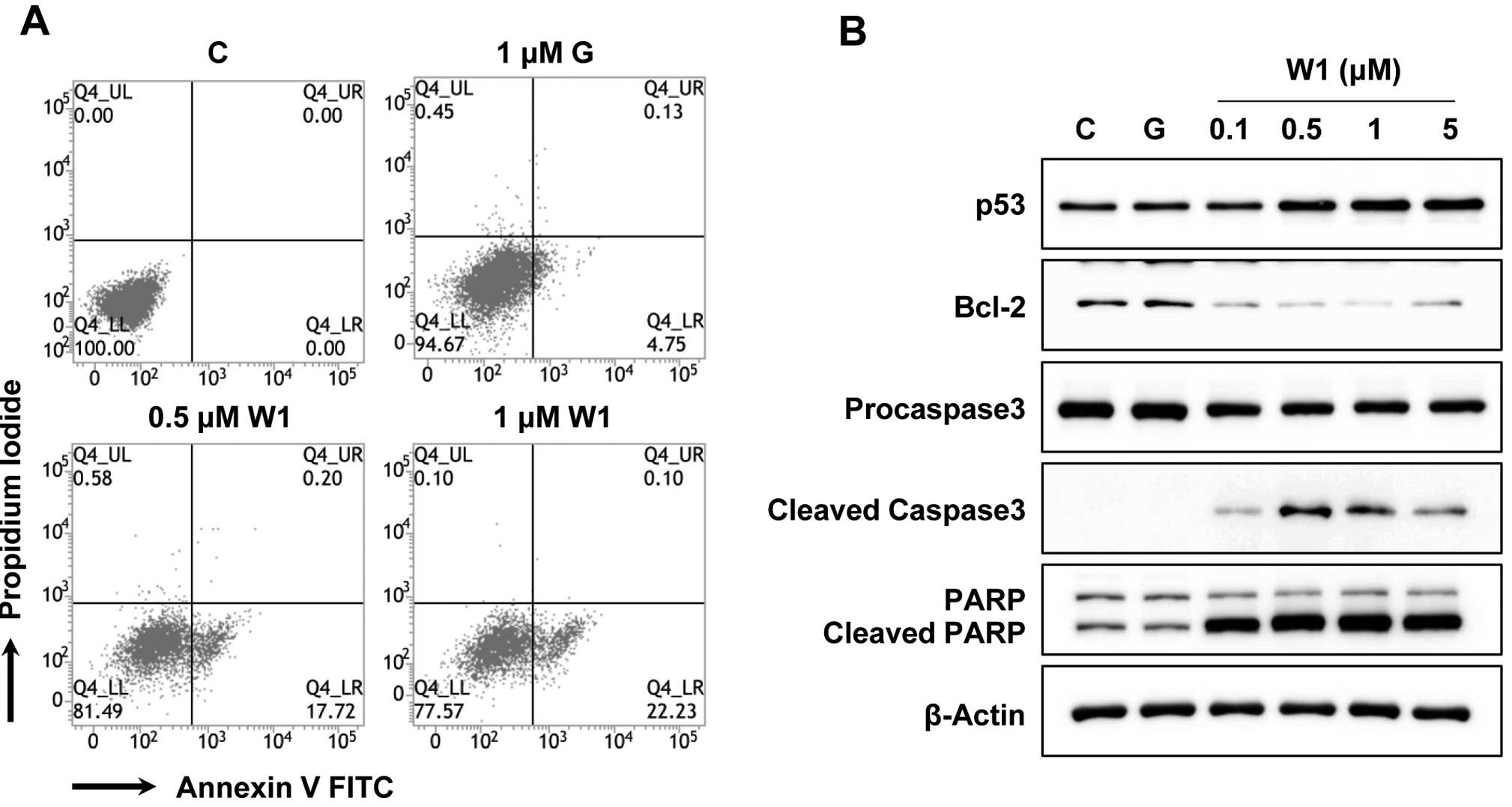
suppressed by the induction of apoptosis. To ascertain whether the
anti-proliferative activity of WK88-1 in the H1975 cells may be
related to the induction of apoptosis, flow-cytometric analyses
with Annexin V were performed. As a result, our data showed that
treatment with WK88-1 facilitated early apoptosis in the
gefitinib-resistant H1975 cells. As shown in Fig. 4A, dose-dependent treatment with
0.5–1 μM WK88-1 in the gefitinib-resistant H1975 cells markedly
induced apoptosis at a level of 17.72 and 22.23%, respectively, but
not in the cells treated with 1 μM gefitinib (4.75%). We next
investigated the effects of WK88-1 on apoptotic markers under the
same culture conditions. The p53 protein level was slightly
increased in the WK88-1-treated cells, whereas an anti-apoptotic
protein, Bcl-2, was markedly downregulated under the same
conditions (Fig. 4B). Notably,
treatment with WK88-1 resulted in the induction of cleaved
caspase-3 which mediates PARP cleavage (Fig. 4B). These findings clearly suggest
that treatment with WK88-1 is sufficient for inducing apoptosis
through the extrinsic apoptotic pathway in gefitinib-resistant
NSCLC with T790M mutation in EGFR.
WK88-1 inhibits gefitinib-resistant NSCLC
tumor growth in a xenograft model
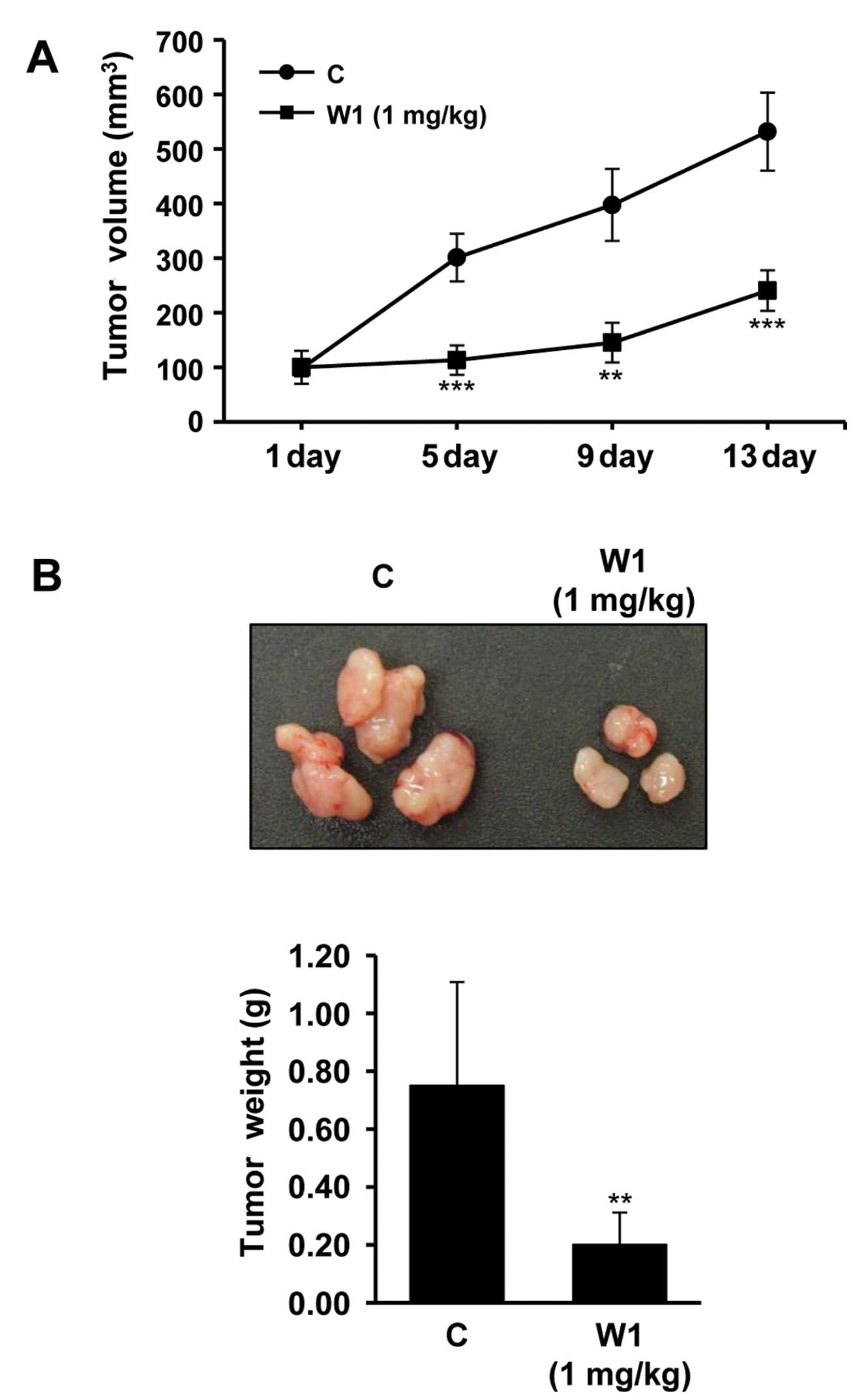
We next examined whether treatment with WK88-1
suppresses gefitinib-resistant tumor growth in vivo. Effects
of WK88-1 were assessed in xenograft models. To accomplish this,
gefitinib-resistant H1975 cells were injected subcutaneously in
nude mice. As expected, the H1975 cell line which harbors a mutant
T790M-EGFR showed rapid growth rate. Our data revealed that a
significant reduction in tumor growth rate (Fig. 5A) and tumor weight (Fig. 5B) was observed following 1 mg/kg
WK88-1 treatment. Therefore, we reasoned that WK88-1 has
significant antitumor efficacy in nude mice bearing H1975 tumor
xenografts, suggesting that WK88-1 could overcome gefitinib
resistance in NSCLCs.
Discussion
It has been suggested that Hsp90 contributes to the
six basic hallmarks of cancers such as escape from apoptosis,
self-sufficiency in growth signals, insensitivity to anti-growth
signals, sustained angiogenesis, limitless replicative potential
and tissue invasion and metastasis (26,27).
Consistently, pharmacological inhibition of Hsp90 activity has
emerged as a promising therapeutic strategy for antitumor
treatment. Hsp90 comprising ~2% of the total cellular protein is
routinely expressed in malignant cells, and mutated oncogenic
proteins are more reliant on Hsp90 function (28). Notably, tumor cells have been
reported to exhibit greater dependence on Hsp90’s chaperoning
function to restructure numerous unfolded and mutated proteins
found in tumors which are surrounded by a hypoxic, acidotic and
nutrient-starved microenvironment (29,30). A
previous report suggested that increased expression of Hsp90 is
linked to worse prognosis in NSCLC patients (31). Therefore, targeting Hsp90 has been
regarded as an attractive strategy for the treatment of NSCLCs. For
this reason, diverse naturally occurring Hsp90 inhibitors including
GA, 17-AAG and 17-DMAG have been extensively evaluated; however,
many of these drugs still have toxicity issues in clinical trials
despite their preclinical efficacy. Therefore, our research has
focused on developing novel GA derivatives targeting Hsp90 in
tumors with less toxicity. Previously, we reported diverse
non-benzoquinone GA derivatives produced by a mutasynthetic
approach (24,25).
In the present study, we evaluated the antitumor
activity of a non-benzoquinone derivative, WK88-1, in a
gefitinib-resistant H1975 cell line harboring the T790M mutation in
EGFR. Our data revealed that treatment with WK88-1 in the H1975
cells induced simultaneous degradation of oncogenic RTKs including
EGFR, ErbB2 and ErbB3, and subsequently inhibited downstream
signaling such as Akt phosphorylation. Treatment with WK88-1
inhibited ATP-binding activity and dissociated mutant T790M-EGFR in
the H1975 cells, resulting in destabilization of multiple proteins.
Of note, destabilization of mutant T790M-EGFR by WK88-1 may disrupt
EGFR-ErbB3 heterodimers, resulting in rapid depletion of phspho-Akt
considering that ErbB3 is used to couple EGFR to the PI3K-Akt
pathway (11,32). As many Hsp90 client proteins
including EGFR have been shown to be implicated in cancer cell
growth and survival (33), these
findings provide insight as to how treatment with WK88-1 abrogates
gefitinib-resistance in NSCLCs harboring mutant T790M-EGFR.
In conclusion, our findings revealed that WK88-1 has
significant antitumor efficacy, as confirmed by the significant
reduction in in vivo tumor growth in a mouse xenograft model
as well as decreased proliferation, migration and invasion in
gefitinib-resistant H1975 cells. Therefore, our data suggest that
WK88-1 may be a potential Hsp90 inhibitor for overcoming acquired
resistance to gefitinib in NSCLCs with T790M-EGFR.
Acknowledgements
This research was conducted by the Settlement
Research Grant of Keimyung University in 2011 (to C-H. Jeong).
References
|
1
|
Fukuoka M, Yano S, Giaccone G, et al:
Multi-institutional randomized phase II trial of gefitinib for
previously treated patients with advanced non-small-cell lung
cancer (The IDEAL 1 Trial) (corrected). J Clin Oncol. 21:2237–2246.
2003.Erratum in: J Clin Oncol 22: 4863, 2004.
|
|
2
|
Lynch TJ, Adjei AA, Bunn PA Jr, et al:
Summary statement: novel agents in the treatment of lung cancer:
advances in epidermal growth factor receptor-targeted agents. Clin
Cancer Res. 12:4365s–4371s. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Perez-Soler R, Chachoua A, Hammond LA, et
al: Determinants of tumor response and survival with erlotinib in
patients with non-small-cell lung cancer. J Clin Oncol.
22:3238–3247. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shepherd FA, Rodrigues Pereira J, Ciuleanu
T, et al: Erlotinib in previously treated non-small-cell lung
cancer. N Engl J Med. 353:123–132. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Laskin JJ and Sandler AB: Epidermal growth
factor receptor: a promising target in solid tumours. Cancer Treat
Rev. 30:1–17. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mitsudomi T, Morita S, Yatabe Y, et al:
Gefitinib versus cisplatin plus docetaxel in patients with
non-small-cell lung cancer harbouring mutations of the epidermal
growth factor receptor (WJTOG3405): an open label, randomised phase
3 trial. Lancet Oncol. 11:121–128. 2010. View Article : Google Scholar
|
|
7
|
Mok TS, Wu YL, Thongprasert S, et al:
Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N
Engl J Med. 361:947–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lynch TJ, Bell DW, Sordella R, et al:
Activating mutations in the epidermal growth factor receptor
underlying responsiveness of non-small-cell lung cancer to
gefitinib. N Engl J Med. 350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jackman D, Pao W, Riely GJ, et al:
Clinical definition of acquired resistance to epidermal growth
factor receptor tyrosine kinase inhibitors in non-small-cell lung
cancer. J Clin Oncol. 28:357–360. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pao W, Wang TY, Riely GJ, et al: KRAS
mutations and primary resistance of lung adenocarcinomas to
gefitinib or erlotinib. PLoS Med. 2:e172005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Engelman JA, Zejnullahu K, Mitsudomi T, et
al: MET amplification leads to gefitinib resistance in lung cancer
by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kobayashi S, Boggon TJ, Dayaram T, et al:
EGFR mutation and resistance of non-small-cell lung cancer to
gefitinib. N Engl J Med. 352:786–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pao W, Miller VA, Politi KA, et al:
Acquired resistance of lung adenocarcinomas to gefitinib or
erlotinib is associated with a second mutation in the EGFR kinase
domain. PLoS Med. 2:e732005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li D, Shimamura T, Ji H, et al: Bronchial
and peripheral murine lung carcinomas induced by T790M-L858R mutant
EGFR respond to HKI-272 and rapamycin combination therapy. Cancer
Cell. 12:81–93. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhou W, Ercan D, Chen L, et al: Novel
mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature.
462:1070–1074. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
da Rocha Dias S, Friedlos F, Light Y,
Springer C, Workman P and Marais R: Activated B-RAF is an Hsp90
client protein that is targeted by the anticancer drug
17-allylamino-17-demethoxygeldanamycin. Cancer Res. 65:10686–10691.
2005.PubMed/NCBI
|
|
17
|
Shimamura T, Lowell AM, Engelman JA and
Shapiro GI: Epidermal growth factor receptors harboring kinase
domain mutations associate with the heat shock protein 90 chaperone
and are destabilized following exposure to geldanamycins. Cancer
Res. 65:6401–6408. 2005. View Article : Google Scholar
|
|
18
|
Webb CP, Hose CD, Koochekpour S, et al:
The geldanamycins are potent inhibitors of the hepatocyte growth
factor/scatter factor-met-urokinase plasminogen activator-plasmin
proteolytic network. Cancer Res. 60:342–349. 2000.PubMed/NCBI
|
|
19
|
Yang S, Qu S, Perez-Tores M, et al:
Association with HSP90 inhibits Cbl-mediated down-regulation of
mutant epidermal growth factor receptors. Cancer Res. 66:6990–6997.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pratilas CA, Hanrahan AJ, Halilovic E, et
al: Genetic predictors of MEK dependence in non-small cell lung
cancer. Cancer Res. 68:9375–9383. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shimamura T, Li D, Ji H, et al: Hsp90
inhibition suppresses mutant EGFR-T790M signaling and overcomes
kinase inhibitor resistance. Cancer Res. 68:5827–5838. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sausville EA, Tomaszewski JE and Ivy P:
Clinical development of 17-allylamino, 17-demethoxygeldanamycin.
Curr Cancer Drug Targets. 3:377–383. 2003. View Article : Google Scholar
|
|
23
|
Supko JG, Hickman RL, Grever MR and
Malspeis L: Preclinical pharmacologic evaluation of geldanamycin as
an antitumor agent. Cancer Chemother Pharmacol. 36:305–315. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim W, Lee D, Hong SS, et al: Rational
biosynthetic engineering for optimization of geldanamycin
analogues. Chembiochem. 10:1243–1251. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim W, Lee JS, Lee D, et al: Mutasynthesis
of geldanamycin by the disruption of a gene producing starter unit:
generation of structural diversity at the benzoquinone ring.
Chembiochem. 8:1491–1494. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar
|
|
27
|
Koga F, Kihara K and Neckers L: Inhibition
of cancer invasion and metastasis by targeting the molecular
chaperone heat-shock protein 90. Anticancer Res. 29:797–807.
2009.PubMed/NCBI
|
|
28
|
Whitesell L and Lindquist SL: HSP90 and
the chaperoning of cancer. Nat Rev Cancer. 5:761–772. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jolly C and Morimoto RI: Role of the heat
shock response and molecular chaperones in oncogenesis and cell
death. J Natl Cancer Inst. 92:1564–1572. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Workman P: Altered states: selectively
drugging the Hsp90 cancer chaperone. Trends Mol Med. 10:47–51.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gallegos Ruiz MI, Floor K, Roepman P, et
al: Integration of gene dosage and gene expression in non-small
cell lung cancer, identification of HSP90 as potential target. PloS
One. 3:e00017222008.PubMed/NCBI
|
|
32
|
Engelman JA, Janne PA, Mermel C, et al:
ErbB-3 mediates phosphoinositide 3-kinase activity in
gefitinib-sensitive non-small cell lung cancer cell lines. Proc
Natl Acad Sci USA. 102:3788–3793. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shimamura T and Shapiro GI: Heat shock
protein 90 inhibition in lung cancer. J Thorac Oncol. 3:S152–S159.
2008. View Article : Google Scholar : PubMed/NCBI
|