Introduction
Epidermal growth factor receptor (EGFR) tyrosine
kinase inhibitors (TKI) have shown marked therapeutic effects
against non-small cell lung cancer (NSCLC) with EGFR activating
mutations, such as exon 19 deletions and L858R point mutations
(1). However, acquired resistance
to EGFR-TKIs develops in almost all patients, usually within 1
year, thus, limiting improvement in patient outcomes (2,3).
Approximately 50% of patients resistant to the first generation
EGFR-TKIs gefitinib or erlotinib (also known as reversible
EGFR-TKIs) present tumors with a secondary mutation in exon 20 of
EGFR, which involves the substitution of threonine at position 790
by methionine (T790M) in the tyrosine kinase functional domain;
~20% have tumors with bypass signaling caused by proto-oncogene Met
amplification or overexpression which activates downstream
pathways, including phosphatidylinositol 3-kinase/protein kinase B
(PI3K/AKT) and mitogen-activated protein kinase
kinase/extracellular signal-regulated kinase (MEK/ERK) (4,5).
The T790M mutation was shown to confer resistance by
increasing EGFR affinity for ATP relative to that for first
generation EGFR-TKIs, resulting in continuous activation of
downstream pro-survival signaling pathways, such as PI3K/AKT and
MEK/ERK (6). The second generation
EGFR-TKIs (also known as irreversible EGFR-TKIs), such as BIBW2992,
were designed to covalently bind the kinase domain and are,
therefore, less affected by the increase in ATP-binding affinity
compared with the reversible inhibitors (7). However, IC50 values of
irreversible EGFR-TKIs are >400 times higher in NSCLC cell lines
with the T790M mutation than in NSCLC cells without the T790M
mutation, markedly diminishing the clinical value of irreversible
EGFR-TKIs (8). Several studies have
demonstrated that although MET-TKI shows marginal efficacy in NSCLC
cell lines with the T790M mutation when administered alone, its
combinations with gefitinib/erlotinib are effective in mutated
cells (9–11). These observations might be explained
by the fact that many growth factor signaling pathways overlap and
interact with each other, suggesting a redundancy in cell
signaling. For instance, activation of one tyrosine kinase receptor
may co-activate the downstream signaling pathway of other tyrosine
kinase receptors. Therefore, strategies to interrupt receptor
cross-signaling or to target more than one pathway may result in
increased effects on tumor inhibition.
To the best of our knowledge, no previous study has
reported the effects of a combination of BIBW2992 and ARQ 197 (MET
inhibitor) on proliferation, apoptosis and downstream signaling
pathways of EGFR/MET in NSCLC cell lines. Therefore, we
investigated the effects of such a combination (BIBW2992 and ARQ
197) on the NSCLC cell line H1975 harboring an EGFR T790M mutation,
aimed at the two molecular mechanisms of acquired resistance to
reversible EGFR-TKIs as mentioned above. Our results showed that
the BIBW2992 and ARQ 197 combination inhibited cell growth, induced
cell apoptosis and cell cycle arrest at the
G0/G1 phase, and reduced the phosphorylation
of AKT and ERK1/2, primary downstream effectors of the EGFR and
c-MET signaling pathways.
Materials and methods
Cell culture, chemicals and
antibodies
Human lung adenocarcinoma cell line H1975 was
purchased from the Cell Biology Institute of the Chinese Academy of
Sciences, Beijing, China. Cells were cultured in RPMI-1640
(Invitrogen, Carlsbad, CA, USA) supplemented with 10% FBS,
penicillin (100 units/ml) and streptomycin (100 mg/ml) at 37°C in a
humidified environment containing 5% CO2.
BIBW2992 and ARQ 197 were purchased from Sigma
Chemical (St. Louis, MO, USA). Erlotinib hydrochloride was obtained
from Roche China. These drugs were dissolved in DMSO and used at
the indicated concentrations. The Annexin V-PI double staining
apoptosis detection kit was purchased from Jingmei Biotech Co.,
Ltd. (Shanghai, China).
Genomic studies of the EGFR and MET
genes
Genomic DNA was purified from H1975 cells using the
Qiagen DNAeasy Kit (Qiagen, Shanghai, China) according to the
manufacturer’s instructions. Mutation analysis of the EGFR, MET and
KRAS genes was carried out by direct sequencing after OneStep
reverse transcriptase-PCR (RT-PCR) using the Qiagen OneStep Reverse
Transcription-PCR kit (Qiagen).
MET gene copy number detection
Genomic copy number variation of the MET gene in
H1975 cells was assessed using real-time PCR with Power SYBR-Green
PCR Master Mix (Applied Biosystems, Shanghai, China) on an ABI
PRISM 7900-HT System. PCR reactions were set following the standard
ΔCT method according to the manual. MET q-PCR primers
were purchased from ABI (ABI assay no. Hs01565582_g1). RNaseP was
used as a reference gene.
Cytotoxicity assays
Cell viability was assessed using the MTT
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)
colorimetric assay (12). Briefly,
cells were seeded in 96-well plates at a density of
1×104 cells/well for 16–20 h. After treatment for 48 h
with erlotinib (2 μM), BIBW2992 (1 μM), ARQ 197 (2 μM), and the 2
μM erlotinib/2 μM ARQ 197 and 1 μM BIBW2992/2 μM ARQ 197
combinations, the medium was removed and the cells were incubated
with 0.5 mg/ml MTT in complete medium for 4 h. The absorbance was
measured at 565 nm using an OptiMax plate reader (GE Healthcare,
Shanghai, China), and the viability was expressed as a percentage
relative to the control cells.
Apoptosis assay
Apoptosis rates were determined by staining cells
using an Annexin V-fluorescein isothiocyanate (FITC) and propidium
iodide (PI) kit (Jingmei Biotechnology Co., Ltd.) according to the
manufacturer’s instructions. Briefly, 1×106 H1975 cells
were incubated in the presence of erlotinib (2 μM), BIBW2992 (1
μM), ARQ 197 (2 μM), and the 2 μM erlotinib/2 μM ARQ 197 and 1 μM
BIBW2992/2 μM ARQ 197 combinations. After 48 h of incubation, cells
were washed twice with phosphate-buffered solution (PBS; pH 7.4),
resuspended in 500 μl binding buffer before addition of 5 μl
Annexin V and 1 mg/ml PI, and analyzed on an LSR flow cytometer (BD
Biosciences, San Jose, CA, USA) using CellQuest software (BD
Biosciences). Early apoptotic cells were positive for Annexin V and
negative for PI while late apoptotic cells were positive for both
Annexin V and PI.
Analysis of cell cycle distribution
H1975 cells (1×106) were seeded in p60
Petri dishes in complete medium and incubated for 16–20 h. After
treatment with erlotinib (2 μM), BIBW2992 (1 μM), ARQ 197 (2 μM),
and 2 μM erlotinib/2 μM ARQ 197 and 1 μM BIBW2992/2 μM ARQ 197
combinations for 48 h, cells were collected and fixed with ice-cold
70% ethanol overnight at −20°C. Upon centrifugation, cell pellets
were treated with 4 mg/ml PI solution containing 1% Triton X-100
and 100 mg/ml RNase for 30 min. To avoid cell aggregation, cell
suspensions were filtered with nylon membranes (BD Biosciences),
and the samples were analyzed on an LSR flow cytometer using
CellQuest software. A minimum of 1×104 cells were
analyzed for DNA content, and the percentages of cells in various
cell cycle phases were quantified using the ModFit LT ver. 3.0 (BD
Biosciences).
Western blot analysis
H1975 cells were incubated in the presence of
erlotinib (2 μM), BIBW2992 (1 μM), ARQ 197 (2 μM), and 2 μM
erlotinib/2 μM ARQ 197 and 1 μM BIBW2992/2 μM ARQ 197 combinations
for 48 h, collected and lysed in ice-cold cell extraction buffer
(Life Technologies, Shanghai, China). Protein concentrations were
determined using the BCA protein assay kit (Pierce, Rockford, IL,
USA), and equal protein amounts (50–100 μg/well) were subjected to
electrophoresis on 12% SDS-polyacrylamide gels. After
electrophoretic transfer of proteins onto nitrocellulose membranes,
samples were sequentially incubated with primary antibodies and
goat anti-rabbit secondary antibodies conjugated to horseradish
peroxidase (Cell Signaling Technology Inc., Beverly, MA, USA).
Primary antibodies raised in rabbit against human EGFR, p-EGFR
(Y1068), MET and p-MET (Y1234/1235) were purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA); rabbit anti-human AKT, p-AKT
(S473), ERK1/2, p-ERK1/2 (T202/Y204) and β-actin antibodies were
purchased from Abcam (Cambridgeshire, UK).
Finally, protein bands were detected using the
enhanced chemiluminescence kit (ECL; Pierce), and the membranes
were exposed to X-ray film and imaged.
RNA interference
Duplexed Stealth RNA interference (Invitrogen)
against MET and Stealth RNA Interference Negative Control Low GC
Duplex 3 (Invitrogen) were used for RNA interference assays.
Briefly, 1×105 H1975 cells suspended in 2 ml
antibiotic-free medium were seeded in 6-well plates and incubated
at 37°C for 24 h. Then, cells were transfected with small
interfering RNA (siRNA; 250 pmol) or scramble RNA (siSCR) using
Lipofectamine 2000 (5 μl) (Invitrogen) following the manufacturer’s
instructions. After 48 h of incubation, cells were used in
proliferation and apoptosis assays as described above. MET and EGFR
knockdown were confirmed by western blot analysis, and the siRNA
sequences were as follows: MET forward, 5′-UCCAGAAGAUCAGUUUCCUA
AUUCA-3′ and reverse, 5′-UGAAUUAGGAAACUGAUCU UCUGGA-5′; EGFR
forward, 5′-UUUAAAUUCACCAAUA CCUAUUCCG-3′ and reverse,
5′-CGGAAUAGGUAUUGG UGAAUUUAAA-5′.
Statistical analysis
Data are expressed as means ± SD and were nalyzed by
the Student’s t-test or one way analysis of variance (ANOVA) for
comparison between multiple groups. P<0.05 was considered to
indicate a statistically significant result.
Results
EGFR and MET genotypes in the H1975
cells
Direct DNA sequencing showed L858R and T790M
mutations in the EGFR gene of the H1975 cells, whereas no mutations
were detected in the MET and KRAS genes in this cell line. Using
qPCR analysis, we found that MET had 1.1 copies in H1975 cells.
Effects of the BIBW2992/ARQ 197
combination on H1975 cell growth
H1975 cells were incubated with erlotinib, BIBW2992,
ARQ 197 and the combinations of erlotinib/ARQ 197 and BIBW2992/ARQ
197 for 48 h, respectively, and cell proliferation was evaluated by
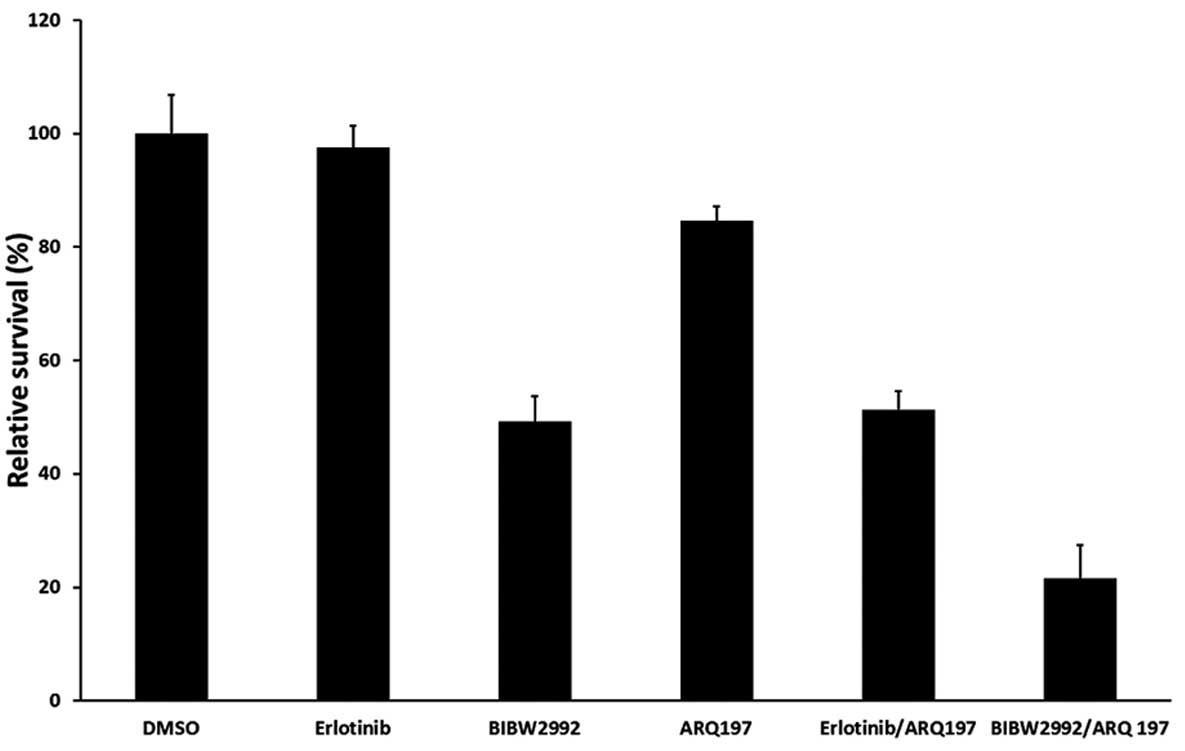
MTT assays. Erlotinib and ARQ 197 used individually exhibited
little cytotoxicity on H1975 cells, with 97.5 and 84.6% viable
cells, respectively, while BIBW2992 alone and the erlotinib and ARQ
197 combination showed moderate cytotoxicity (with 49.2 and 57.3%
viable cells, respectively; P<0.05 compared with DMSO). Notably,
treatment with the BIBW2992/ARQ 197 combination resulted in
pronounced growth inhibition of H1975 cells, with only 21.5% viable
cells detected (P<0.01 compared with DMSO and the erlotinib/ARQ
197 combination) (Fig. 1).
Effects of the BIBW2992/ARQ 197
combination on apoptosis in the H1975 cells
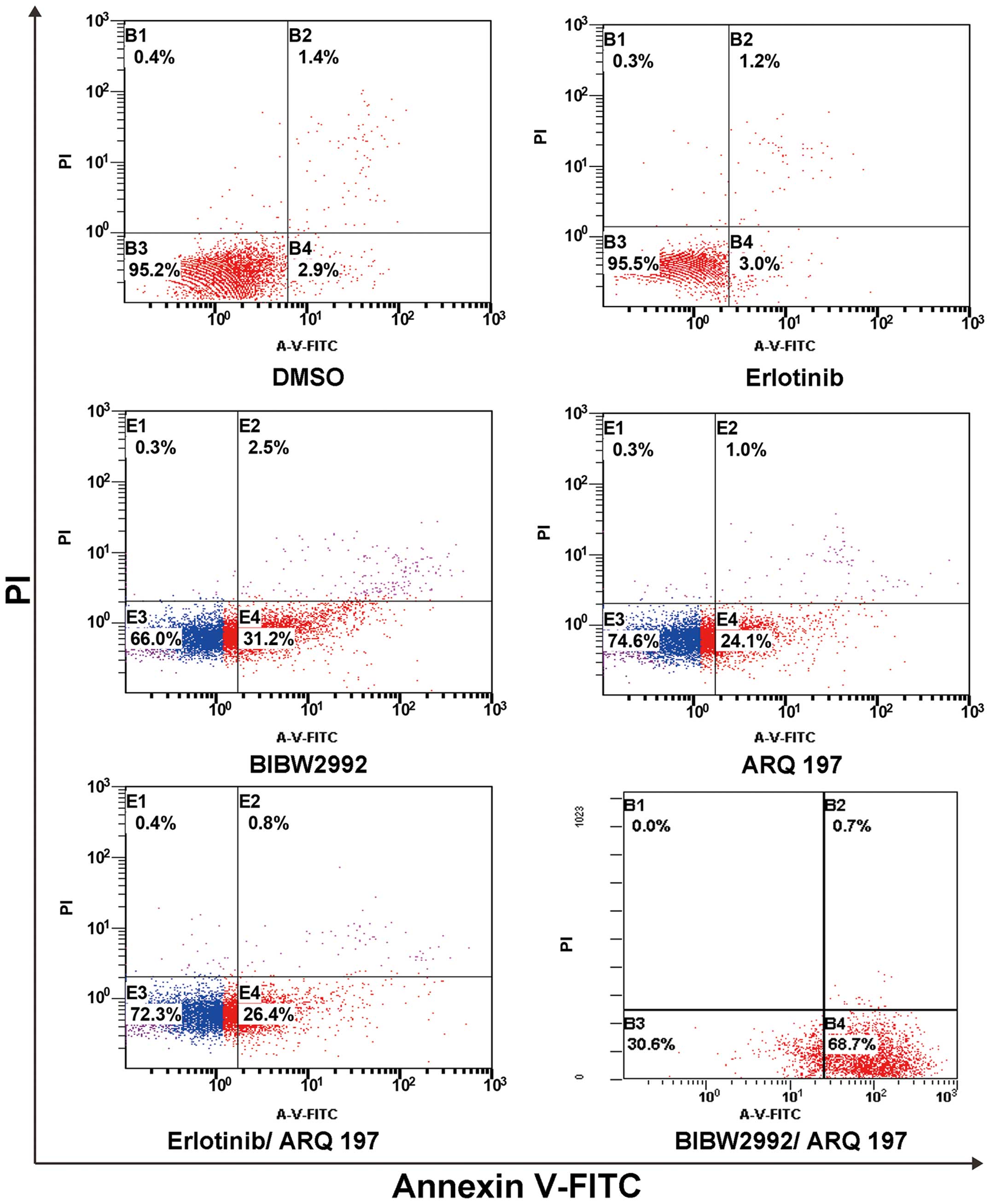
H1975 cells were treated as described for the cell
viability assays, and apoptosis was assessed by Annexin V-PI double
staining. Annexin V can be used to detect externalization of
phosphatidylserine during the progression of apoptosis, therefore
identifying cells in early apoptosis. H1975 cells treated with
erlotinib displayed an early apoptosis rate of 3.0%, similar to the
controls, indicating H1975 cell resistance to erlotinib. Treatment
with BIBW2992, ARQ 197 and the erlotinib/ARQ 197 combination
resulted in 31.2, 24.1 and 26.4% early apoptosis, respectively
(P<0.001 compared with the control cells). Importantly, the
early apoptosis rate of H1975 cells after treatment with the
combination of BIBW2992/ARQ 197 was highest at 68.4% as shown in
Fig. 2 (P<0.01 compared with the
control, erlotinib, BIBW2992, ARQ 197 and erlotinib/ARQ 197
combination). These data indicate that the BIBW2992/ARQ 197
combination was much stronger in inducing apoptosis in H1975 cells
in comparison with BIBW2992, ARQ 197 and the erlotinib/ARQ 197
combination.
Effects of BIBW2992/ARQ 197 combination
on the H1975 cell cycle
H1975 cells were treated as described above, and the
percentages of cells in the cell cycle phases were assessed by flow
cytometry. After treatment with erlotinib, BIBW2992, ARQ 197 and
the combination of erlotinib/ARQ 197, slightly more H1975 cells
were detected in the G0/G1 phase in
comparison with the control samples, but with no statistical
significance. However, 72.9% of the H1975 cells were found in the
G0/G1 phase after treatment with the
BIBW2992/ARQ 197 combination; a rate much higher than that in any
other treatment group (Table I;
P<0.01 compared with the controls, erlotinib, BIBW2992, ARQ 197
and the erlotinib/ARQ 197 combination).
 | Table IEffects of the BIBW2992/ARQ 197
combination on the cell cycle distribution of H1975 cells. |
Table I
Effects of the BIBW2992/ARQ 197
combination on the cell cycle distribution of H1975 cells.
| Cell cycle phase
(%) |
|---|
|
|
|---|
| Drugs |
G0/G1 | S | G2/M |
|---|
| DMSO | 38.6±3.1 | 42.1±2.6 | 19.3±3.3 |
| Erlotinib | 38.9±2.8 | 43.8±3.7 | 17.3±1.8 |
| BIBW2992 | 40.5±3.4 | 33.8±2.2 | 25.7±4.6 |
| ARQ 197 | 43.1±0.8 | 32.2±1.6 | 24.7±3.5 |
| Erlotinib/ARQ
197 | 46.4±2.3 | 30.8±4.4 | 22.8±1.7 |
| BIBW2992/ARQ 197 | 72.9±3.7a | 15.8±3.1 | 11.3±0.6 |
Effects of the BIBW2992/ARQ 197
combination on the expression of EGFR, MET and downstream
effectors
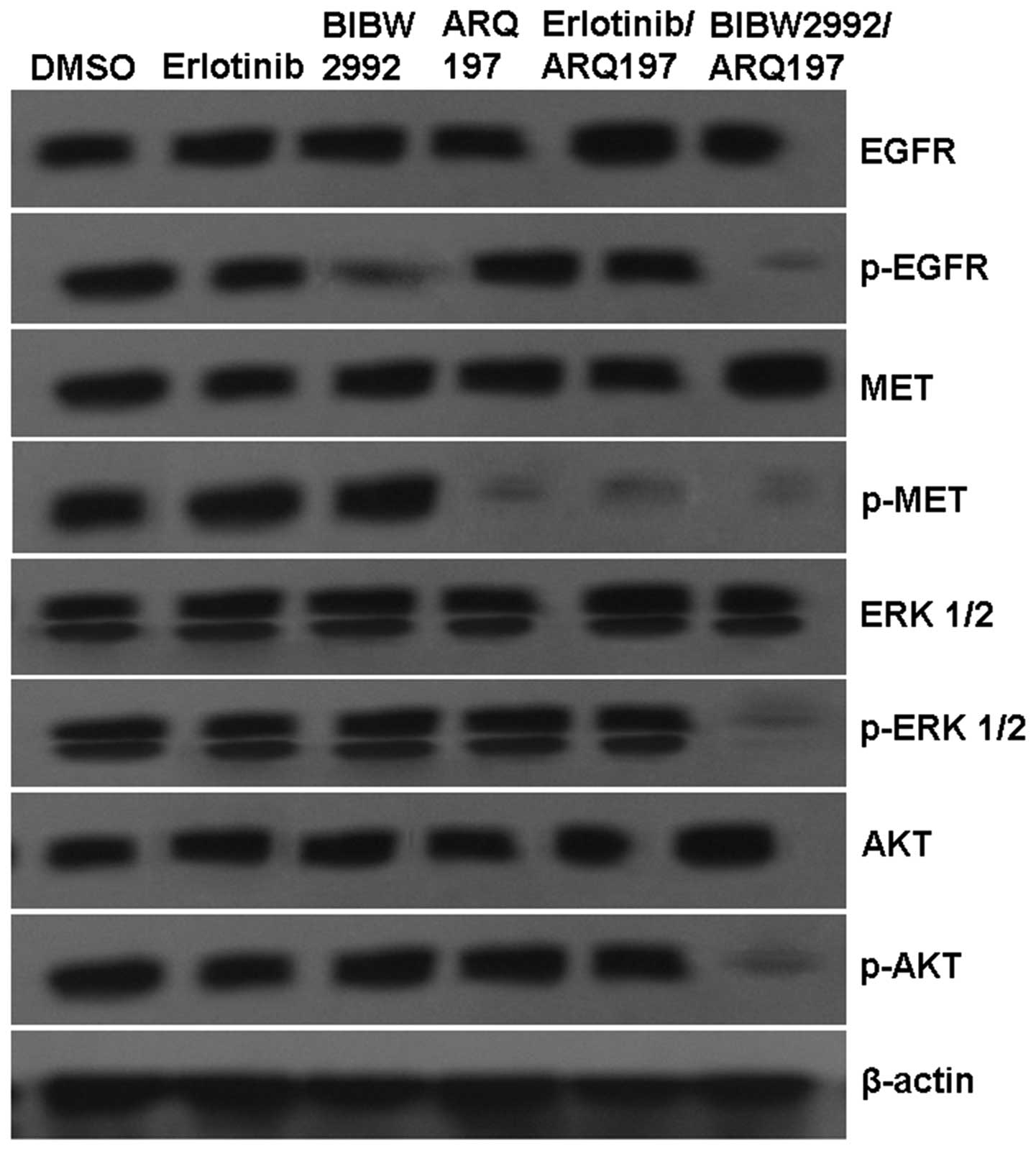
H1975 cells were treated as described above, and the
proteins were detected by western blot analysis. As shown in
Fig. 3, control cells robustly
expressed total MET and phosphorylated MET. Combined with our gene
analysis results, these data confirmed MET activation with no
genomic amplification or mutation in H1975 cells. In addition,
erlotinib did not significantly affect EGFR, MET, AKT or ERK1/2.
Our data showed decreased levels of phosphorylated EGFR after
treatment with BIBW2992 and reduced levels of phosphorylated MET in
the presence of the erlotinib/ARQ 197 combination and ARQ 197
alone. Of note, expression levels of total and phosphorylated AKT
or ERK1/2 were not markedly altered in these groups. In contrast to
the control and other treatments, expression levels of
phosphorylated EGFR, MET, AKT and ERK1/2 were significantly
decreased after treatment with the BIBW2992/ARQ 197 combination
(Fig. 3).
Effects of specific EGFR and MET
downregulation on growth and apoptosis in H1975 cells
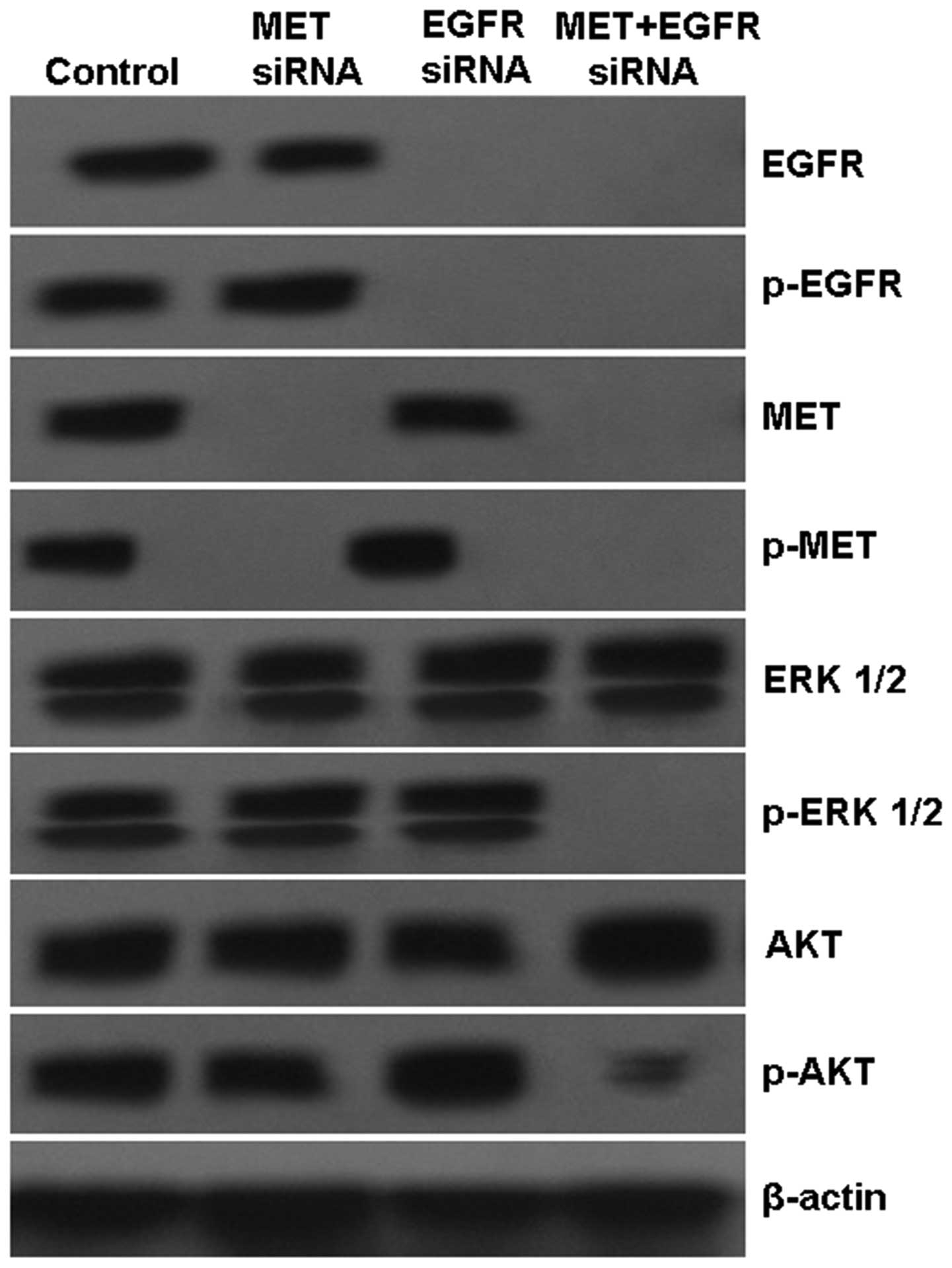
To further validate this combination dual TKI
strategy, H1975 cells were subjected to pathway-specific siRNA
knockdown of MET and EGFR kinase signaling pathways, either alone
or in combination. After 48 h of culture, optimal inhibition of
downstream signaling molecules (phosphorylated-AKT and ERK1/2) was
achieved, as observed with siRNA knockdown of both MET and EGFR
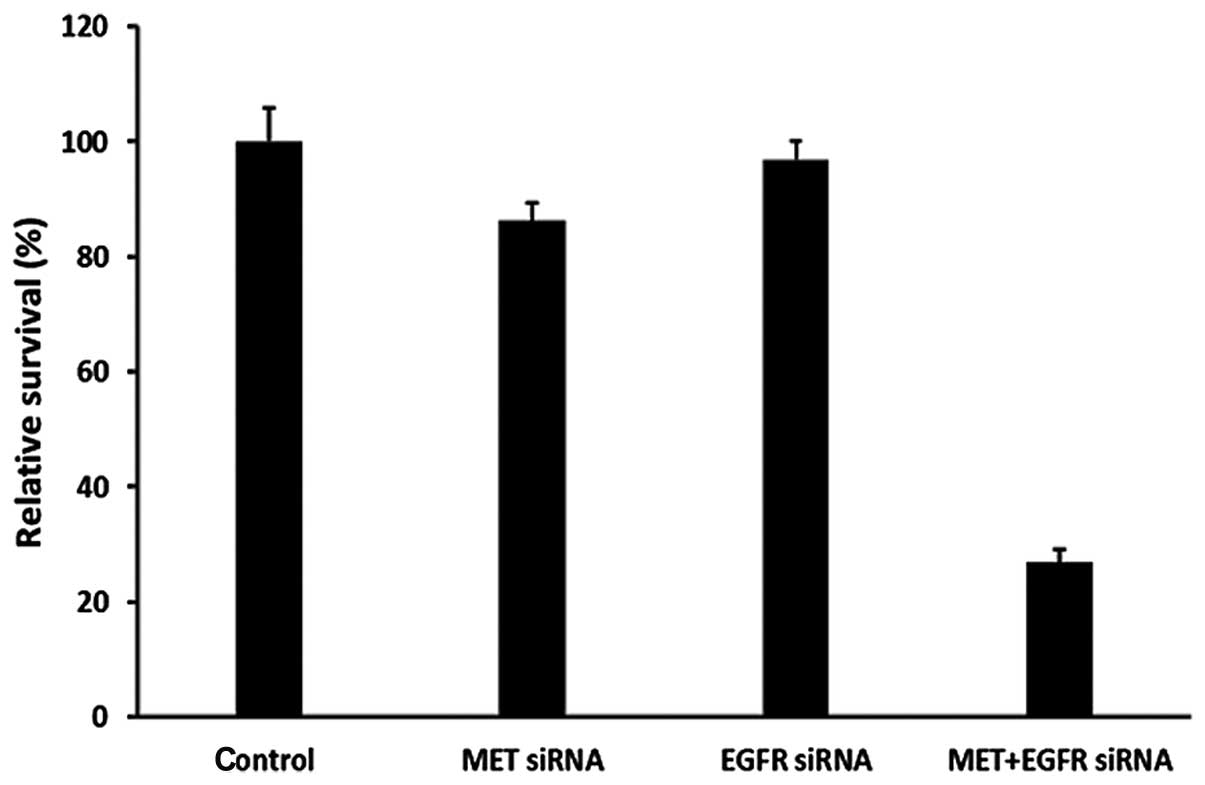
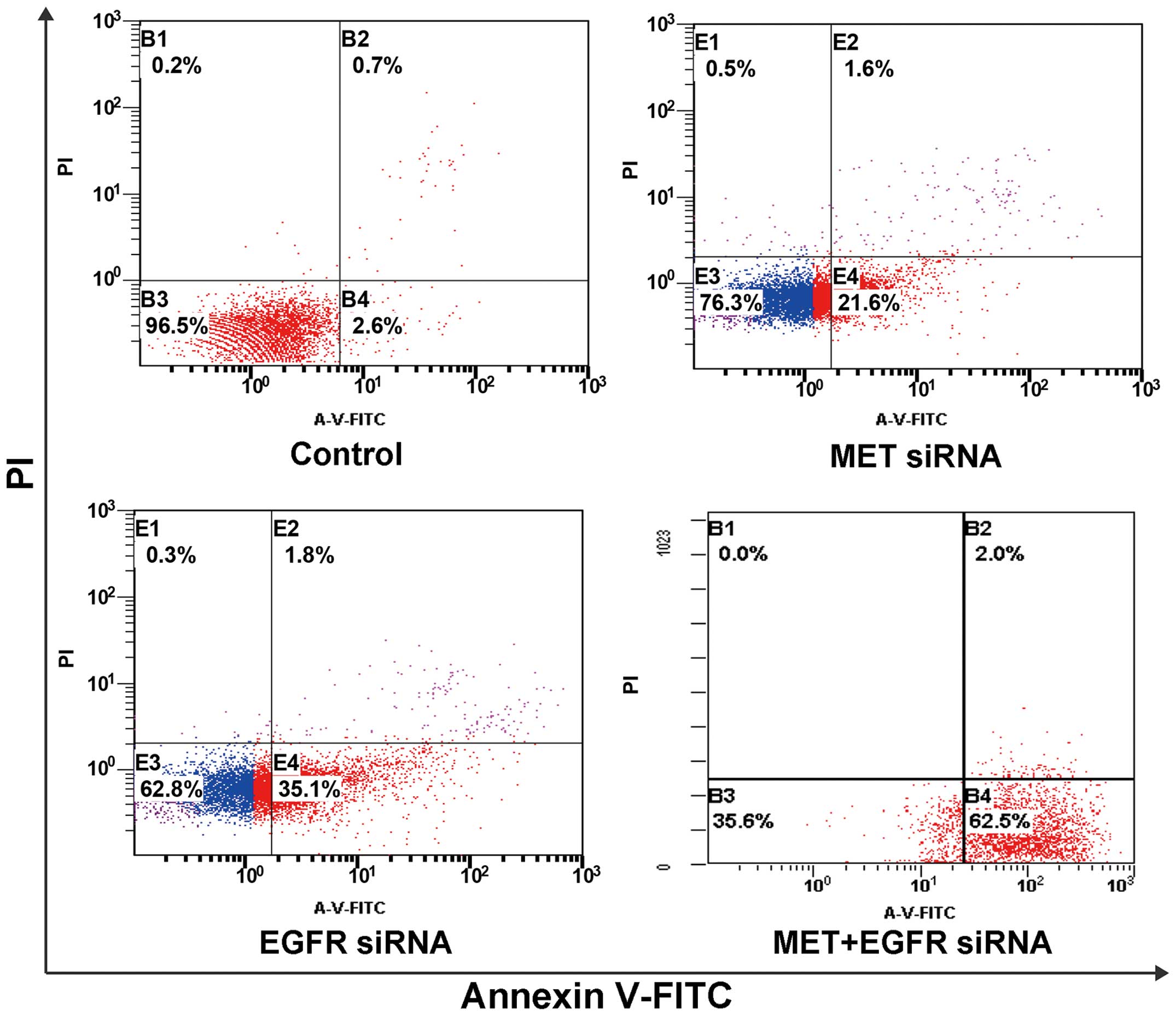
targets, compared with single target knockdown (Fig. 4). We found a much higher cell growth
inhibition and increased early cell apoptosis rates after knockdown
of both MET and EGFR targets compared with single target knockdown
(Figs. 5 and 6). Taken together, these data indicate
that cooperative enhanced inhibition using dual TKIs against the
MET and EGFR pathways may constitute an effective treatment
strategy to inhibit H1975 cells with acquired T790M-EGFR-mediated
TKI resistance.
Discussion
In the early days of anticancer therapy, the
observation that tumors are hyperproliferative lesions prompted the
development of antimitotic and cytostatic compounds with the hope
that they would function indiscriminately on each cancer type. As
it became evident that cancers have multiple etiologies, and that
neoplastic progression is associated with a combination of genetic
and epigenetic alterations, the task of developing therapies
suitable for treatment of the full spectrum of cancers appeared
almost impossible (13).
To date, it is well established that inactivation of
individual oncogenes can block tumor growth and even lead to tumor
regression; despite the multitude of genetic alterations harbored
by transformed cells (14). These
findings have given an unprecedented clinical value to the concept
that considers cancer a disease of genes, allowing a novel
classification of tumors based on the presence of defined genetic
lesions. Classical histopathological diagnosis is still important
to evaluate the extent of phenotypic aggressiveness, but
personalized molecular diagnosis is needed to understand whether a
tumor in a given patient carries a particular genetic alteration
that could be targeted by therapy (15).
Recent prospective studies have demonstrated that
the EGFR-TKIs gefitinib and erlotinib are associated with a high
response rate and prolong progression-free survival in patients
with EGFR activating mutant lung cancer. Responders to these
agents, however, later relapse after acquiring EGFR-TKI resistance,
making it urgent to develop novel therapeutic agents to overcome
acquired resistance to EGFR-TKIs (1–3,5).
Gatekeeper mutations, including the T315I mutation
in ABL associated with resistance to imatinib (16), the L1196M mutation in ALK associated
with resistance to crizotinib (17), and the T790M mutation in EGFR
associated with resistance to gefitinib and erlotinib, are common
mechanisms by which tumor cells acquire resistance to molecularly
targeted drugs. Although irreversible EGFR-TKIs have been developed
to overcome T790M-mediated resistance to gefitinib and erlotinib,
recent clinical trials have demonstrated that monotherapy with
irreversible EGFR-TKIs have failed to provide benefits in patients
with NSCLC refractory to gefitinib or erlotinib (18), at least in part, due to the
relatively low efficacy of this class of compounds to T790M EGFR in
clinically relevant concentrations.
Met amplification and/or overexpression is also
associated with acquired resistance to gefitinib and erlotinib in
mutated EGFR lung cancer. MET is known as the only specific
receptor for hepatocyte growth factor (HGF) (19). Upon activation, MET forms a
heterodimer and transduces strong signals to various pathways,
including PI3K/Akt and MEK/ERK. Engelman et al (20) reported that overexpressed Met
protein utilizes ErbB3 as an adaptor protein to mediate survival
signals through activation of the PI3K-Akt pathway. Various
targeted inhibitory strategies are being evaluated to antagonize
MET/HGF signaling in human cancers, including small-molecule kinase
inhibitors, antibodies to the ligand HGF, and receptor MET itself
(11). Owing to the unique
intrinsic properties of MET regulating cellular ‘invasive
signaling’, MET has been proposed as playing a role in ‘oncogene
addiction’ in a small subset of human cancers as well as in
‘oncogene expedience’ by inducing an enhanced transformed tumor
malignant ‘fitness’ in a much larger range of cancers, leading to
promotion of tumor progression (21). In the latter case, activated MET can
intercept with various other oncogenic signals, including
mutant-EGFR, in maintaining and enhancing the tumor
invasive-progressive phenotype, thereby also creating the
opportunity for MET to be a therapeutic target even in late
advanced metastatic disease (22,23).
Several studies have demonstrated that the EGFR
T790M mutation and MET activation have complementary roles in
acquired resistance to reversible EGFR-TKI, through mediation of
collaborative signaling with receptor cross-activation (3,22). For
example, Tang et al (21)
found a direct association between MET and EGFR harboring the T790M
mutation in H1975 cells, interaction enhanced by HGF resulting in
augmented phosphorylation of Akt and ERK1/2. These observations
indicate that EGFR and MET have joint downstream pro-survival
signaling pathways, and activation of EGFR or MET could
reciprocally trigger their downstream signaling pathways.
Therefore, monotherapy with an irreversible EGFR-TKI alone not only
requires high concentrations resulting in adverse effects, but also
induces acquired resistance through MET activation. Accordingly,
monotherapy with MET-TKI alone or MET-TKI combined with a
reversible EGFR-TKI has little therapeutic effects on T790M-EGFR
NSCLC cells, since pro-survival signals mediated by T790M-EGFR are
continuously activated in these conditions (12). Therefore, neither monotherapy is
sufficient to overcome the acquired resistance. However, afatinib,
a novel irreversible inhibitor of the ErbB family member EGFR, was
recently shown to display preclinical efficacy in NSCLC with common
EGFR-activating mutations and the T790M mutation typically
associated with EGFR TKI resistance (24–26).
In the present study, we found that MET was neither
genomically amplified nor mutated in the erlotinib-resistant H1975
cell line (L858R/T7980M-EGFR). Our data showed that MET had 1.1
copies in H1975 cells, in agreement with previous studies (12,27,28).
However, we did find that MET was activated in these cells,
possessing constitutive (ligand-independent) receptor activation.
The double activation of MET and EGFR not only conferred resistance
to erlotinib but also resulted in markedly enhanced catalytic
kinases, demonstrated in cell cytotoxicity assays and western blot
analysis of EGFR, MET and their downstream signaling pathways in
agreement with previous reports (19). When treating H1975 cells with
different inhibitors, BIBW2992 and ARQ 197 alone and the
erlotinib/ARQ 197 combination showed only little or moderate
effects on cell growth inhibition, apoptosis induction and cell
cycle arrest. BIBW2992 and ARQ 197 decreased EGFR and MET
phosphorylation, respectively. However, BIBW2992 or ARQ 197 alone
had no impact on AKT and ERK1/2 phosphorylation. In contrast, the
combination of BIBW2992 and ARQ 197 showed pronounced effects on
cell growth inhibition, apoptosis induction and cell cycle arrest,
and markedly decreased phosphorylation of AKT and ERK1/2. The
therapeutic effects of BIBW2992 combined with ARQ 197 were more
evident than that of the erlotinib/ARQ 197 combination as shown
above. This might result from stronger cytotoxic effects of
BIBW2992 compared with reversible EGFR-TKIs in T790M-EGFR NSCLC
cells. Finally, dual MET and EGFR siRNA knockdown experiments in
H1975 cells provided further validation of the therapeutic value of
an irreversible EGFR-TKI combined with MET-TKI. Dual inhibition
appears to be superior to single target inhibition. Our present
report demonstrated the efficacy of dual receptor tyrosine
kinase-targeted inhibition against EGFR (BIBW2992) and MET (ARQ
197) as a strategy to achieve optimized inhibition of cell
viability, cell cycle progression and cellular signaling in
T790M-EGFR-mediated erlotinib resistance in H1975 cells.
Overall, our findings suggest that the combination
of MET-TKI and irreversible EGFR-TKI may be effective in
controlling H1975 cells with acquired resistance to erlotinib.
Downregulation of the PI3K/AKT and MEK/ERK signaling pathways were
related to the cytotoxic effects of this combinational therapeutic
approach. Further studies should focus on the exact mechanisms of
apoptosis induction and cross-talk between EGFR and MET downstream
pro-survival signaling pathways.
References
|
1
|
Vincent MD, Kuruvilla MS, Leighl NB and
Kamel-Reid S: Biomarkers that currently affect clinical practice:
EGFR, ALK, MET, KRAS. Curr Oncol. 19:S33–S44. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Capelletto E and Novello S: Emerging new
agents for the management of patients with non-small cell lung
cancer. Drugs. 72(Suppl 1): 37–52. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Suda K, Mizuuchi H, Maehara Y and
Mitsudomi T: Acquired resistance mechanisms to tyrosine kinase
inhibitors in lung cancer with activating epidermal growth factor
receptor mutation - diversity, ductility, and destiny. Cancer
Metastasis Rev. 31:807–814. 2012. View Article : Google Scholar
|
|
4
|
Liu Y, Shi QF, Ye YC, Tashiro S, Onodera S
and Ikejima T: Activated O2•− and
H2O2 mediated cell survival in
SU11274-treated non-small-cell lung cancer A549 cells via
c-Met-PI3K-Akt and c-Met-Grb2/SOS-Ras-p38 pathways. J Pharmacol
Sci. 119:150–159. 2012.
|
|
5
|
Ayoola A, Barochia A, Belani K and Belani
CP: Primary and acquired resistance to epidermal growth factor
receptor tyrosine kinase inhibitors in non-small cell lung cancer:
an update. Cancer Invest. 30:433–446. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu L, Kikuchi E, Xu C, et al: Combined
EGFR/MET or EGFR/HSP90 inhibition is effective in the treatment of
lung cancers codriven by mutant EGFR containing T790M and MET.
Cancer Res. 72:3302–3311. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li D, Ambrogio L, Shimamura T, et al:
BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in
preclinical lung cancer models. Oncogene. 27:4702–4711. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Takezawa K, Okamoto I, Tanizaki J, et al:
Enhanced anticancer effect of the combination of BIBW2992 and
thymidylate synthase-targeted agents in non-small cell lung cancer
with the T790M mutation of epidermal growth factor receptor. Mol
Cancer Ther. 9:1647–1656. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dulak AM, Gubish CT, Stabile LP, Henry C
and Siegfried JM: HGF-independent potentiation of EGFR action by
c-Met. Oncogene. 30:3625–3635. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nakachi I, Naoki K, Soejima K, et al: The
combination of multiple receptor tyrosine kinase inhibitor and
mammalian target of rapamycin inhibitor overcomes erlotinib
resistance in lung cancer cell lines through c-Met inhibition. Mol
Cancer Res. 8:1142–1151. 2010. View Article : Google Scholar
|
|
11
|
Liu X, Newton RC and Scherle PA:
Development of c-MET pathway inhibitors. Expert Opin Investig
Drugs. 20:1225–1241. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nakagawa T, Takeuchi S, Yamada T, et al:
Combined therapy with mutant-selective EGFR inhibitor and Met
kinase inhibitor for overcoming erlotinib resistance in EGFR-mutant
lung cancer. Mol Cancer Ther. 11:2149–2157. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Comoglio PM, Giordano S and Trusolino L:
Drug development of MET inhibitors: targeting oncogene addiction
and expedience. Nat Rev Drug Discov. 7:504–516. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Brambilla E and Gazdar A: Pathogenesis of
lung cancer signalling pathways: roadmap for therapies. Eur Respir
J. 33:1485–1497. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Suda K, Tomizawa K, Osada H, et al:
Conversion from the ‘oncogene addiction’ to ‘drug addiction’ by
intensive inhibition of the EGFR and MET in lung cancer with
activating EGFR mutation. Lung Cancer. 76:292–299. 2012.
|
|
16
|
Branford S, Rudzki Z, Walsh S, et al: High
frequency of point mutations clustered within the adenosine
triphosphate-binding region of BCR/ABL in patients with chronic
myeloid leukemia or Ph-positive acute lymphoblastic leukemia who
develop imatinib (STI571) resistance. Blood. 99:3472–3475. 2002.
View Article : Google Scholar
|
|
17
|
Choi YL, Soda M, Yamashita Y, et al:
EML4-ALK mutations in lung cancer that confer resistance to ALK
inhibitors. N Engl J Med. 363:1734–1739. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wong KK, Fracasso PM, Bukowski RM, et al:
A phase I study with neratinib (HKI-272), an irreversible pan ErbB
receptor tyrosine kinase inhibitor, in patients with solid tumors.
Clin Cancer Res. 15:2552–2558. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guo A, Villen J, Kornhauser J, et al:
Signaling networks assembled by oncogenic EGFR and c-Met. Proc Natl
Acad Sci USA. 105:692–697. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Engelman JA, Zejnullahu K, Mitsudomi T, et
al: MET amplification leads to gefitinib resistance in lung
cancer by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar
|
|
21
|
Tang Z, Du R, Jiang S, et al: Dual
MET-EGFR combinatorial inhibition against T790M-EGFR-mediated
erlotinib-resistant lung cancer. Br J Cancer. 99:911–922. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Suda K, Murakami I, Katayama T, et al:
Reciprocal and complementary role of MET amplification and
EGFR T790M mutation in acquired resistance to kinase
inhibitors in lung cancer. Clin Cancer Res. 16:5489–5498.
2010.PubMed/NCBI
|
|
23
|
Bonanno L, Jirillo A and Favaretto A:
Mechanisms of acquired resistance to epidermal growth factor
receptor tyrosine kinase inhibitors and new therapeutic
perspectives in non small cell lung cancer. Curr Drug Targets.
12:922–933. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bowles DW, Weickhardt A and Jimeno A:
Afatinib for the treatment of patients with EGFR-positive non-small
cell lung cancer. Drugs Today. 49:523–535. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nelson V, Ziehr J, Agulnik M and Johnson
M: Afatinib: emerging next-generation tyrosine kinase inhibitor for
NSCLC. Onco Targets Ther. 6:135–143. 2013.PubMed/NCBI
|
|
26
|
Chen X, Zhu Q, Zhu L, et al: Clinical
perspective of afatinib in non-small cell lung cancer. Lung Cancer.
81:155–161. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang W, Li Q, Takeuchi S, et al: Met
kinase inhibitor E7050 reverses three different mechanisms of
hepatocyte growth factor-induced tyrosine kinase inhibitor
resistance in EGFR mutant lung cancer. Clin Cancer Res.
18:1663–1671. 2012. View Article : Google Scholar
|
|
28
|
Koizumi H, Yamada T, Takeuchi S, et al:
Hsp90 inhibition overcomes HGF-triggering resistance to EGFR-TKIs
in EGFR-mutant lung cancer by decreasing client protein expression
and angiogenesis. J Thorac Oncol. 7:1078–1085. 2012. View Article : Google Scholar : PubMed/NCBI
|