Introduction
Epithelial ovarian cancer (EOC) is one of the most
common tumors of the female reproductive system, and the mortality
rate of this disease ranks first among gynecological malignant
tumors. In spite of continuous efforts to improve the therapeutic
response, the prognosis for patients with EOC is still very poor,
with an overall 5-year survival of ~50%. Moreover, over 70% of
patients with EOC suffer from relapse, although most advanced-stage
patients undergo cytoreductive surgery and platinum-based
chemotherapy (1).
Recently, several studies have revealed that long
non-coding RNAs (lncRNAs), which have lengths of >200
nucleotides (nt), play important roles in tumor development
(2). The maternally expressed 3
gene (MEG3) is an imprinted gene located on chromosome 14q32
that encodes an lncRNA-MEG3 RNA (3). MEG3 is expressed in many human
tissues, such as ovary, testes and brain (4). However, recent studies have shown that
expression of MEG3 is lost in multiple types of tumors including
neuroblastomas, hepatocellular cancers and gliomas and in many
cancer cell lines (5). Promoter
methylation may account for the loss of expression of MEG3 in
cancer. Studies have shown that promoter methylation plays a major
role in silencing of the MEG3 gene in clinically
non-functioning adenomas (NFAs) (6), meningiomas (7) and neuroblastoma cell lines (8). Moreover, the results of Zhou et
al (5) indicate that
inactivation of the MEG3 gene in these tumors can be
attributed in part to promoter silencing by hypermethylation.
MEG3 may act as a tumor suppressor during
tumorigenesis. Overexpression of MEG3 inhibits the growth of cancer
cell lines such as MCF7, HeLa (4),
HCT116 and IOMM-Lee (9) and
promotes apoptosis in HepG2, PLC/PRF/5, U251 and U87 MG cells
(10,11). Zhou et al determined that the
antiproliferative activity of MEG3 was through regulation of tumor
suppressor p53 (9).
However, the relationship between MEG3 and EOC has
not been studied, and the role of MEG3 in EOC is still unknown.
Therefore, on the basis of previous studies, we hypothesized that
MEG3 may be a novel tumor-suppressor gene in EOC. In the
present study, we determined whether or not MEG3 RNA is
downregulated in human EOC and inhibits EOC proliferation via
targeting of p53.
Materials and methods
Tumor tissues and cell lines
Tissues from 20 normal ovaries and 20 EOCs were
obtained from the Third Affiliated Hospital of Guangzhou Medical
University and the General Hospital of Guangzhou Military Command
of the People’s Liberation Army (Table
I) and frozen at −80°C. The histopathologic diagnoses were
determined by the hospital’s pathologist using International
Federation of Gynecology and Obstetrics (FIGO) and morphologic
criteria. Human ovarian cancer cell lines OVCAR3, SKOV3, HP8910 and
ES-2 were purchased from the American Type Culture Collection
(ATCC; Manassas, VA, USA), maintained in Dulbecco’s modified
Eagle’s medium (DMEM) containing 10% FBS (Gibco, Carlsbad, CA, USA)
and cultured at 37°C with 5% CO2. The present study was
approved by the Institutional Review Board of Guangzhou Medical
University.
 | Table IRelationship between MEG3 RNA
expression and promoter methylation status. |
Table I
Relationship between MEG3 RNA
expression and promoter methylation status.
| Samples | MEG3 RNA level | Percent | Promoter
status | Percent |
|---|
| Normal |
| N01a | High | 85% High | U | 0% M |
| N02 | High | 15% Low | U | 15% MU |
| N03 | High | | U | 85% U |
| N04 | High | | U | |
| N05 | High | | U | |
| N06 | High | | U | |
| N07 | High | | U | |
| N08 | High | | U | |
| N09 | High | | U | |
| N10 | Low | | MU | |
| N11 | High | | U | |
| N12 | High | | U | |
| N13 | High | | U | |
| N14 | Low | | MU | |
| N15 | High | | U | |
| N16 | High | | U | |
| N17 | High | | U | |
| N18 | Low | | MU | |
| N19 | High | | U | |
| N20 | High | | U | |
| Cancer |
| Grade I |
| C03 | Lost | 50% Lost | M | 50% M |
| C11 | High | 50% High | U | 50% U |
| Grade II |
| C04 | Lost | 75% Lost | M | 75% M |
| C06 | Low | 25% Low | U | 25% U |
| C10 | Lost | | M | |
| C16 | Lost | | M | |
| Grade III |
| C01 | Low | 71.4% Lost | U | 57.2% M |
| C02 | Lost | 28.6% Low | M | 35.7% MU |
| C05 | Low | | MU | 7.1% U |
| C07 | Lost | | M | |
| C08 | Lost | | MU | |
| C09 | Lost | | M | |
| C12 | Lost | | M | |
| C13 | Lost | | M | |
| C14 | Lost | | MU | |
| C15 | Low | | MU | |
| C17 | Lost | | M | |
| C18 | Lost | | M | |
| C19 | Lost | | M | |
| C20 | Low | | MU | |
RNA extraction and RT-PCR
Total RNA was extracted from 20 EOC and 20 normal
ovarian tissue samples using RNAiso reagent (Takara, Japan). The
reverse-transcription reactions were performed with PrimeScript RT
Master Mix (Takara). PCR was performed using a Premix Taq version
2.0 (Takara) in accordance with the instructions from the
respective manufacturer. The primers used to amplify MEG3
were as follows: 5′-GCC CTA GGG GAG TGA CTA CA (forward) and 5′-ACT
CGG GAC ATA CCT GCT CT (reverse). The primers used to detect
expression of p53, GDF15 and RB1 were as
follows: p53, 5′-CCT CAG CAT CTT ATC CGA GTG (forward) and
5′-TCA TAG GGC ACC ACC ACA C (reverse); GDF15, 5′-AAG AAC
TCA GGA CGG TGA ATG (forward) and 5′-CCG CAA CTC TCG GAA TCT G
(reverse); RB1, 5′-GAA CTG TGG GGA ATC TGT ATC TTT (forward)
and 5′-AAC TGC TGG GTT GTG TCA AAT A (reverse). β-actin was used as
an internal reference. The reaction conditions were as follows: 30
cycles of 94°C for 5 min, 94°C for 30 sec, 58–60°C for 30 sec and
72°C for 30 sec, and 1 cycle of 72°C for 10 min. The PCR products
were resolved by electrophoresis through 2–3% agarose gels and
visualized by ethidium bromide staining and the use of
Semi-quantitative software (ImageJ). Each sample was analyzed in
triplicate.
Methylation-specific PCR (MSP)
DNA from human tissues was extracted from 20 EOC and
20 normal ovarian tissue samples using the QIAmp DNA Mini kit
(Qiagen). Bisulfite treatment was performed with the EpiTect
Bisulfite kit (Qiagen) in accordance with the manufacturer’s
instructions. The methylation status of the MEG3 gene was
determined using conventional MSP. For the MSP, we used the EpiTect
MSP kit (Qiagen). The sequences of the methylation-specific primers
were as follows: the methylated pair (M), 5′-GTT AGT AAT CGG GTT
TGT CGG C (forward) and 5′-AAT CAT AAC TCC GAA CAC CCG CG
(reverse); the unmethylated pair (U), 5′-GAG GAT GGT TAG TTA TTG
GGG T (forward) and 5′-CCA CCA TAA CCA ACA CCC TAT AAT CAC A
(reverse). MEG3 DNA was amplified in a DNA Engine Peltier thermal
cycler (Bio-Rad) by 1 cycle of 95°C for 15 min followed by 5 cycles
of 94°C for 30 sec, 70°C for 30 sec and 72°C for 30 sec; 5 cycles
of 94°C for 30 sec, 65°C for 30 sec and 72°C for 30 sec; 30 cycles
of 94°C for 30 sec, 60°C for 30 sec and 72°C for 30 sec; and 1
cycle of 72°C for 7 min. The PCR products were identified by
electrophoresis through a 2.5% agarose gel and ethidium bromide
staining (12).
Treatment with 5-aza-2-deoxycytidine
To explore the functional role of DNA methylation in
the silencing of MEG3 transcription in ovarian cancer cells,
OVCAR3, SKOV3, HP8910 and ES-2 cells were seeded onto 6-well plates
at 1.0×105 cells/well and cultured in DMEM containing 1,
5, 10 or 20 μM 5-aza-2-deoxycytidine (5-aza-CdR) (Sigma-Aldrich,
USA) for 6 days (6–8,10).
Cells cultured in the absence of 5-aza-CdR were used as a control.
The media, which contained different concentrations of 5-aza-CdR,
were changed every day. RT-PCR and MSP were performed to measure
the expression of MEG3 RNA and the methylation status of the
MEG3 promoter, respectively, as described above.
Cell proliferation assay (MTT assay)
To express MEG3, the MEG3 cDNA was cloned
into the pcDNA3.1 vector (referred to as pcDNA3.1-MEG3). The empty
pcDNA3.1 vector (referred to as pcDNA3.1-empty) was used in the
negative control (NC) group, and phosphate-buffered saline (PBS)
was used in the blank group. Cellular proliferation was measured by
the MTT assay. OVCAR3 cells were plated in 96-well plates at
~5×103 cells/well in quintuplicate. After ~12 h in
culture, the cells were transfected with pcDNA3.1-MEG3,
pcDNA3.1-empty or PBS using Lipofectamine 2000 (Life Technologies,
USA). The number of cells/well was determined by measuring the
absorbance (490 nm) of MTT at 12, 24 and 48 h after transfection.
Each experiment was repeated at least 3 times.
5-Ethynyl-2′-deoxyuridine (EdU)
incorporation assay
OVCAR3 cells were plated in 24-well plates at
~5.0×104 cells/well. The cells were transfected with
pcDNA3.1-MEG3, pcDNA3.1-empty or PBS when they reached 80%
confluency. Forty-eight hours after transfection, EdU (50 mM) (Cell
Light EdU DNA Imaging kit; Guangzhou RiboBio, China) was added, and
the cells were cultured for an additional 2 h. The cells were then
stained with 100 mM Apollo 567 fluorescent azide and Hoechst (5
mg/ml) for 30 min. Images were acquired and analyzed using the High
Content Imaging Pathway 855 (BD, USA), and the number of
EdU-positive cells was calculated as follows: Number of
EdU-positive cells/number of Hoechst-stained cells × 100% (13). Each experiment was repeated at least
3 times.
Flow cytometric analysis
For the cell cycle assay, 1.0×105 OVCAR3
cells were plated in 6-well plates and then transfected with
pcDNA3.1-MEG3, pcDNA3.1-empty or PBS when they reached 80%
confluencey. The cells were collected after 48 h and stained with
propidium iodide (PI) and RNase (Cell Cycle Detection kit; KeyGene
Biotech, China) to analyze the cell cycle with FACS (BD).
To assay apoptosis, OVCAR3 cells
(1.0×105/well) in 6-well plates were transfected with
pcDNA3.1-MEG3, pcDNA3.1-empty or PBS. After 48 h, the cells were
harvested, stained with Annexin V and PI (Annexin V-FITC Apoptosis
Detection kit; KeyGene Biotech) in accordance with the
manufacturer’s protocol, and subjected to FACS analysis (BD
Biosciences, USA).
Luciferase reporter assays
Since MEG3 has been reported to activate p53 by
binding to the p53 promoter, we tested whether it can regulate the
activity of p53 in EOC cells. OVCAR3 cells were plated in 6-well
plates and transfected with pcDNA3.1-MEG3 and pGL3-p53 using
Lipofectamine 2000. Forty-eight hours later, the cells were
harvested, and luciferase activity was determined using the
Dual-Luciferase kit (Promega, Madison, WI, USA) in accordance with
the manufacturer’s instructions. Luciferase activity was normalized
to that of pRL-SV40 (Promega).
Western blotting
After being transfected with pcDNA3.1-MEG3 or
pcDNA3.1-empty, OVCAR3 cells were lysed with RIPA lysis buffer.
Proteins were harvested and the concentrations of the samples were
determined with the BCA protein assay kit (Beyotime, China). Then,
the proteins were resolved on a 10% SDS denatured polyacrylamide
gel and transferred to a PVDF membrane. After the membranes were
blocked in 5% skim milk overnight at 4°C, they were incubated with
rabbit anti-human antibodies at the recommended dilution [p53,
1:500; GDF15, 1:700; RB1, 1:700 (all from BioWorld, USA)] and then
incubated with the appropriate secondary antibody for ~1 h. Lab
Works™ Image Acquisition and Analysis Software (UVP, USA) were used
to quantify the intensities of the bands. GAPDH was used as a
loading control.
Statistical analysis
All data are expressed as the means ± SD from at
least 3 separate experiments. Statistical differences were
calculated using one-way ANOVA. The relationship between the extent
of CpG methylation and expression of MEG3 RNA was assessed by
association analysis. All calculations were performed with the
software package GraphPad Prism 5.0. A probability (P)-value
<0.05 was considered to indicate a statistically significant
result.
Results
Downregulation of the lncRNA MEG3 in
ovarian cancer
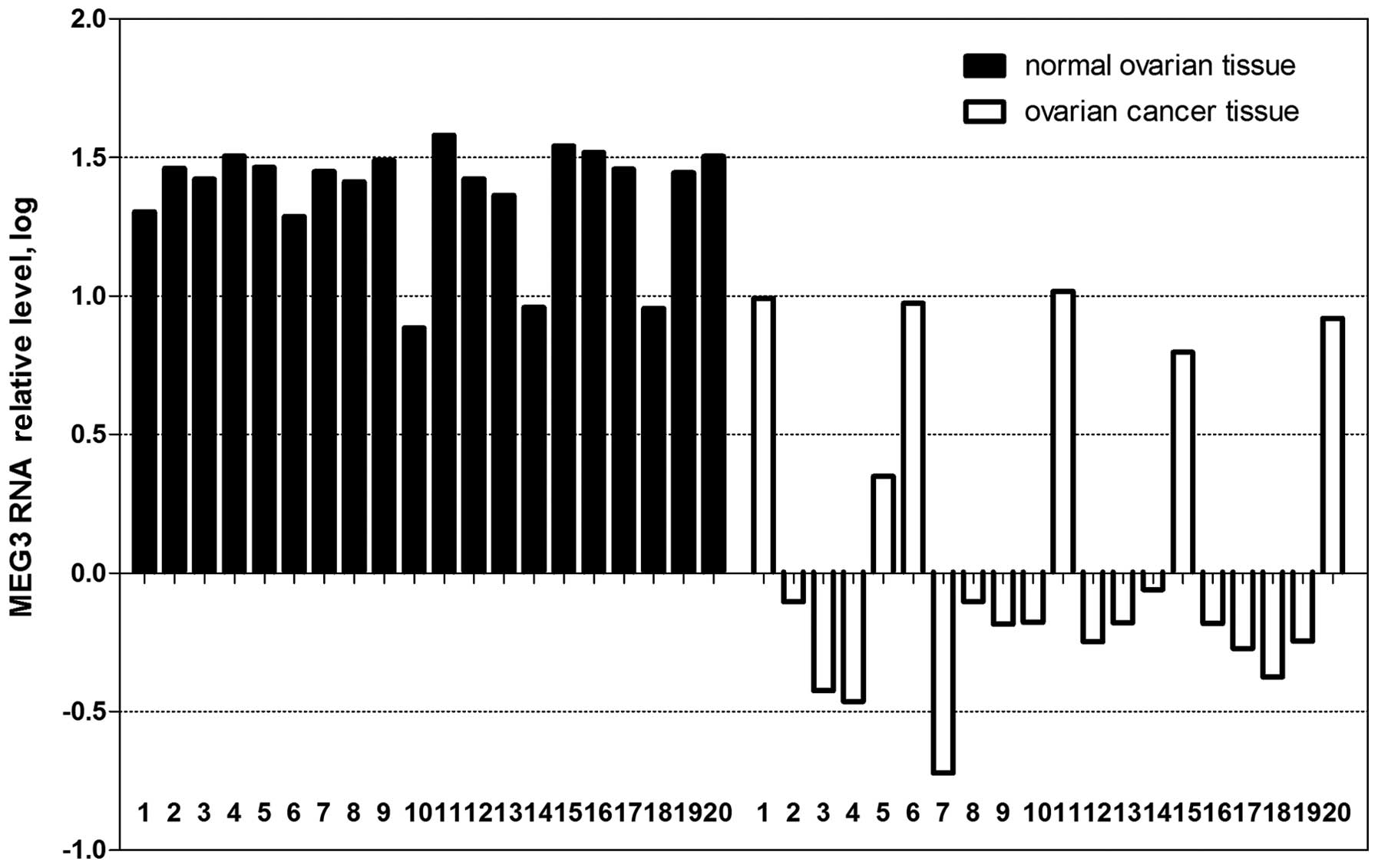
As shown in Fig. 1,
the expression of MEG3 was downregulated in 25% (5 of 20) or absent
in 70% (14 of 20) of the ovarian cancer tissues. In comparison, 85%
(17 of 20) of the normal ovarian tissues expressed MEG3 at high
levels. The difference in expression of MEG3 RNA between normal
samples and cancer samples was significant (P<0.01). Moreover,
as shown in Table I, MEG3 RNA was
lost in 50% (1 of 2) of grade I tumors, 75% (3 of 4) of grade II
tumors and 71% (10 of 14) of grade III tumors. Furthermore, MEG3
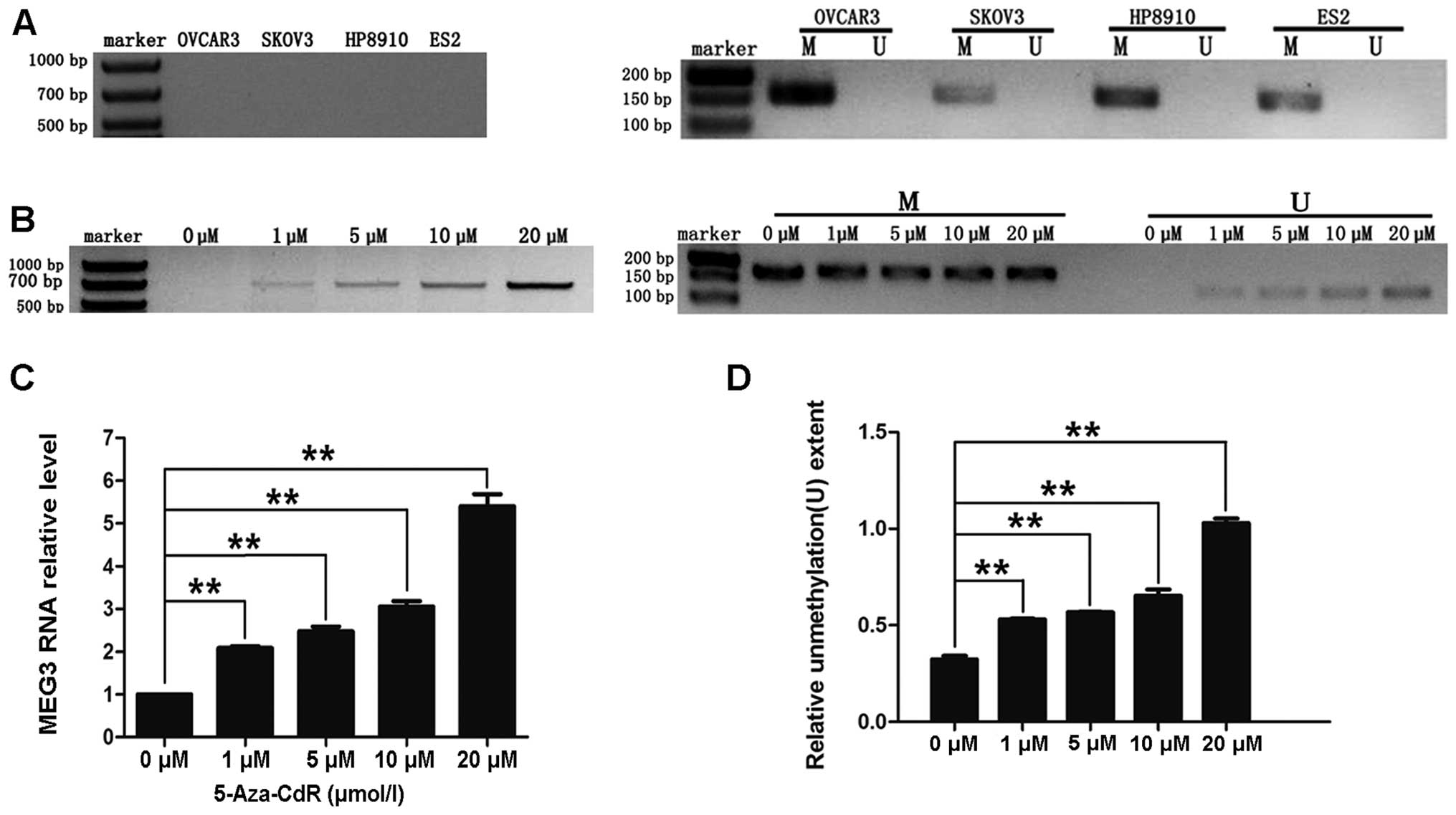
RNA was not detected in any of the ovarian cancer cell lines
(OVCAR3, SKOV3, HP8910 or ES-2; Fig.
3A).
Partial downregulation of MEG3 due to
promoter methylation
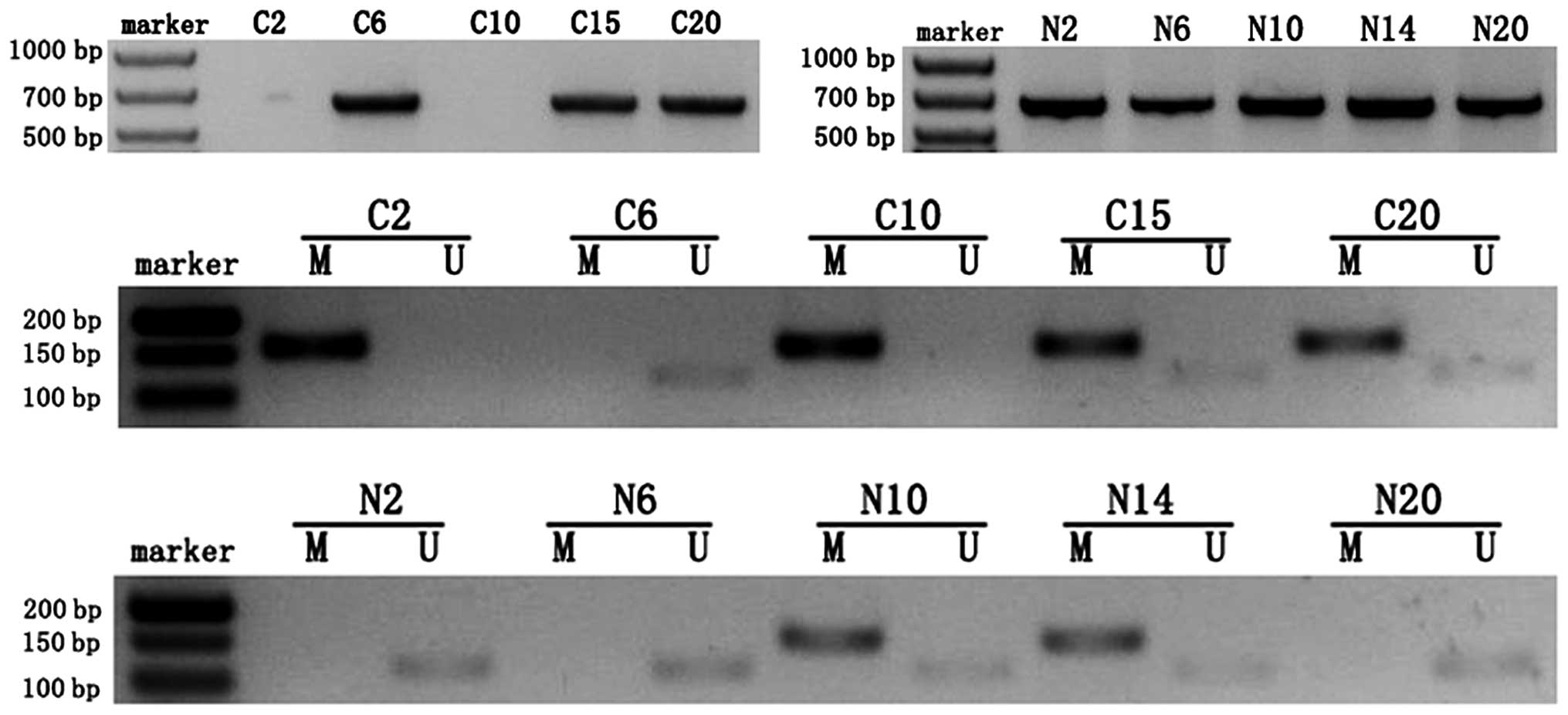
An abnormal methylation pattern (M) of the MEG3
differentially methylated region (DMR) was observed in 60% (12 of
20) of patients with ovarian cancer who were evaluated. An
unmethylated pattern (U) and a partially methylated pattern (M and
U) were observed in 15 and 25%, respectively, of the 20 cancer
samples. In comparison, most of the normal ovarian tissue samples
(85%, 17 of 20) displayed an unmethylated pattern (U). The
difference in MEG3 expression between normal and cancer samples was
significant (P<0.01) after using the Chi-square test to correct
for continuity. As shown in Fig. 2
and Table I, all of the ovarian
cancer samples that were categorized as ‘lost expression’ displayed
a hypermethylated pattern (M) while all of the normal samples that
were categorized as ‘high-expression’ displayed an unmethylated
pattern (U). Furthermore, the MEG3 promoter was
hypermethylated in 50% (1 of 2) of grade I tumors, 75% (3 of 4) of
grade II tumors and 57% (8 of 14) of grade III tumors. Association
analysis indicated that a significant negative correlation existed
between the extent of CpG methylation and the expression of MEG3
RNA (χ2=32.73, r=−0.905, P<0.01). Methylation in the
4 ovarian cancer cell lines (OVCAR3, SKOV3, HP8910 and ES-2) was
detected by MSP. This analysis revealed that all 4 of the cell
lines were abnormally methylated (M; Fig. 3A). Collectively, the data illustrate
that partial downregulation of MEG3 was due to hypermethylation of
the MEG3 promoter and that the methylation status correlates
with EOC grade.
Expression of MEG3 RNA resumes after
treatment with 5-aza-CdR
We treated human ovarian cancer OVCAR3 cells with
different concentrations of 5-aza-CdR. As shown in Fig. 3B–D, treatment with 1, 5, 10 or 20 μM
of 5-aza-CdR resulted in re-expression of MEG3 and partially
reversed the abnormal methylation pattern of the MEG3
promoter. These results indicate that CpG methylation may lead to
downregulation of MEG3 RNA in ovarian cancer cell lines.
MEG3 inhibits proliferation and promotes
apoptosis of ovarian cancer cells
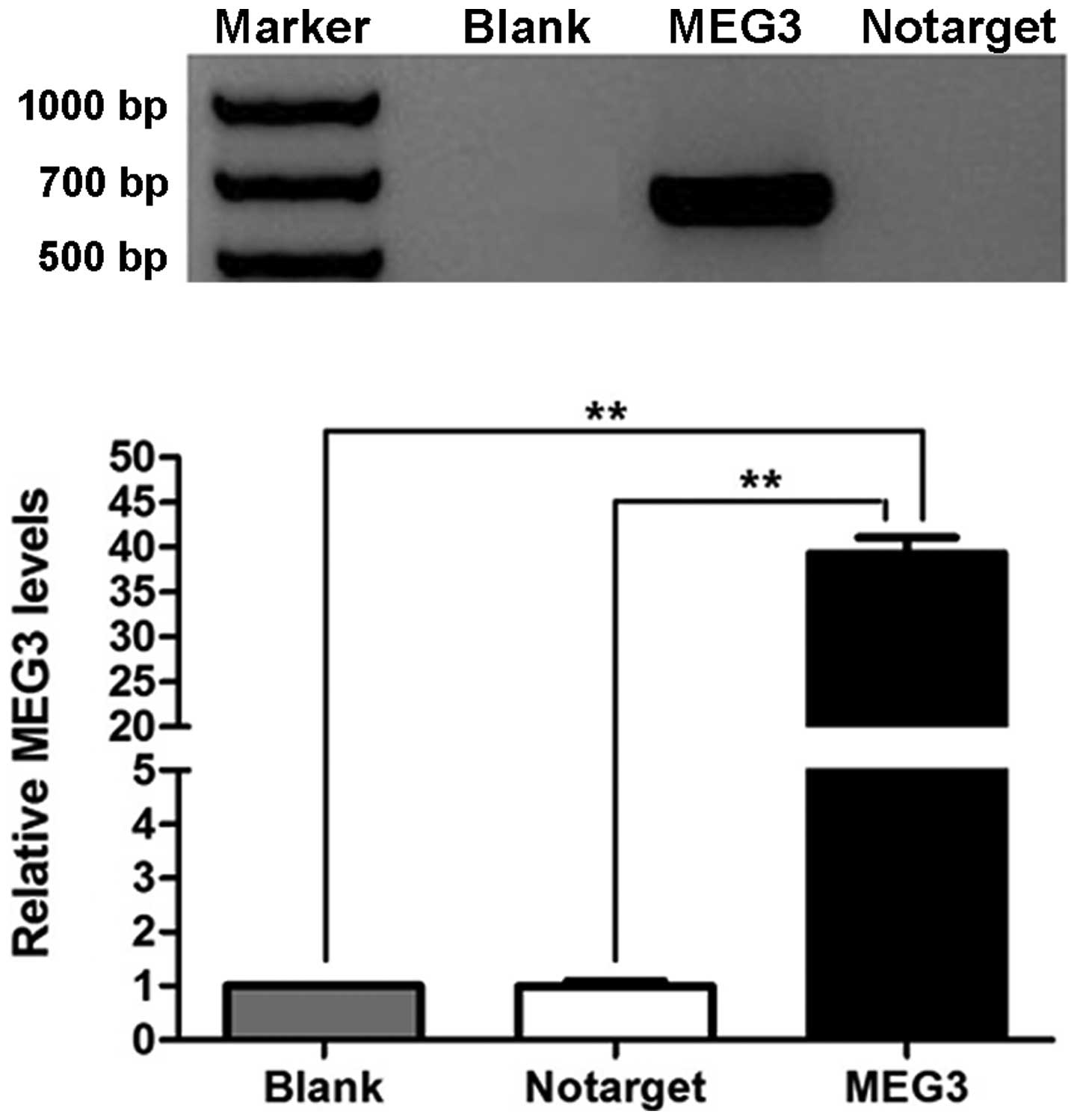
We used RT-PCR to determine that MEG3 RNA was
re-expressed in OVCAR3 cells after they were transfected with
pcDNA3.1-MEG3. MEG3 RNA was not detected in cells of the NC and
blank groups (Fig. 4A). The
difference in the expression of MEG3 between the MEG3 group and the
NC and blank groups was significant (P<0.01).
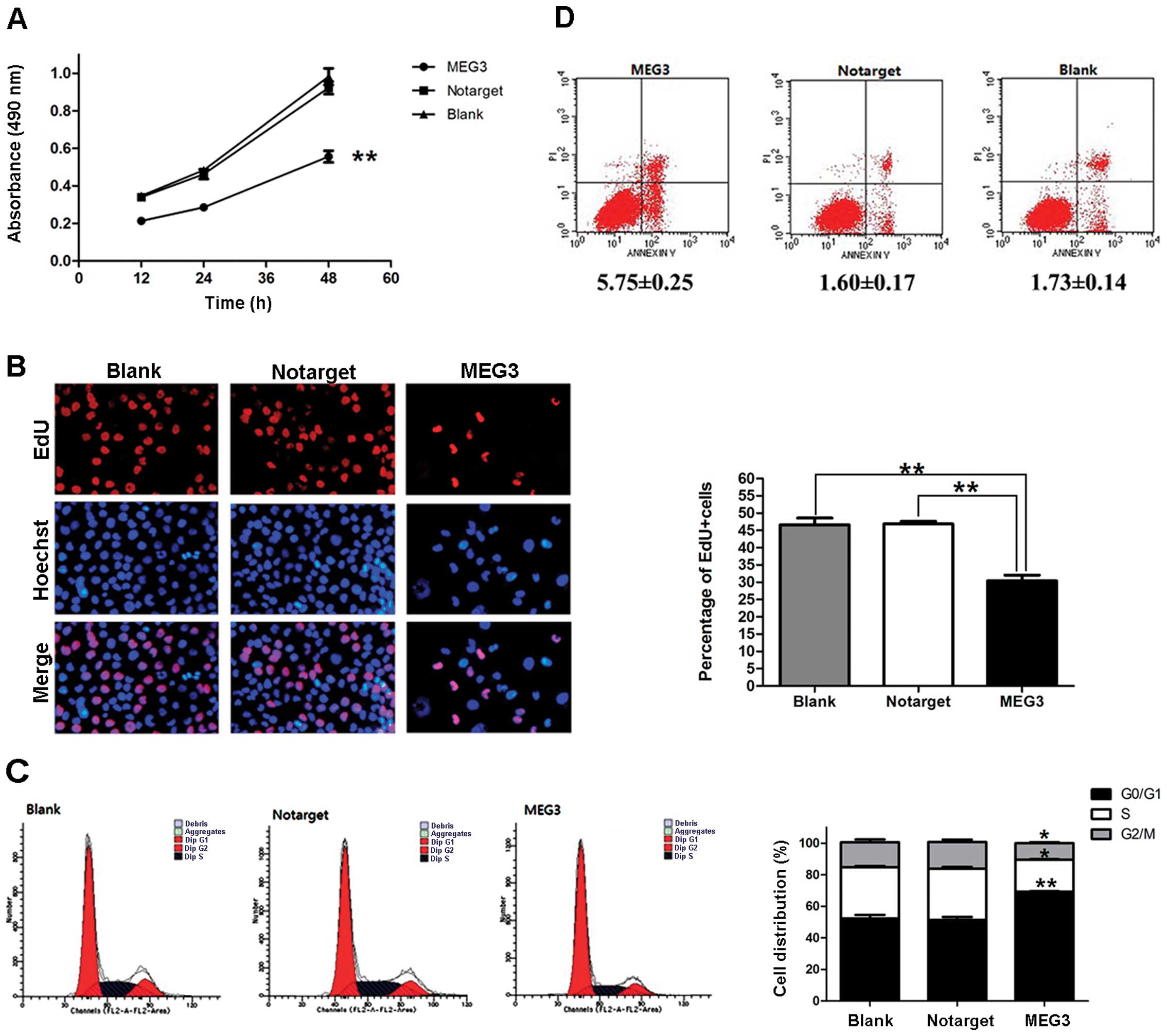
To investigate the role of MEG3 in cell growth,
OVCAR3 cells were transfected with pcDNA3.1-MEG3 and then subjected
to proliferation and apoptosis assays. Results of the MTT assay
showed that proliferation was significantly inhibited in cells that
overexpressed MEG3. In contrast to the NC and blank groups, the
inhibition ratio of the MEG3 group at 12, 24 and 48 h after
transfection was significantly different (37.26, 38.19 and 39.73%,
respectively; P<0.01; Fig. 5A).
An EdU assay showed that the percentage of cells in the S phase was
reduced by 35.01±2.68% in OVCAR3 cells that overexpress MEG3
(P<0.01; Fig. 5B). The outcome
of analysis of the cell cycle indicated that OVCAR3 cells were
arrested at the G0/G1 phase and that the percentages of cells in
the S and G2/M phases were also decreased (Fig. 5C). An apoptosis assay showed that
overexpression of MEG3 caused OVCAR3 cells to undergo apoptosis
(Fig. 5D). All of the data
presented above support the hypothesis that MEG3 negatively
regulates the growth of ovarian cancer cells.
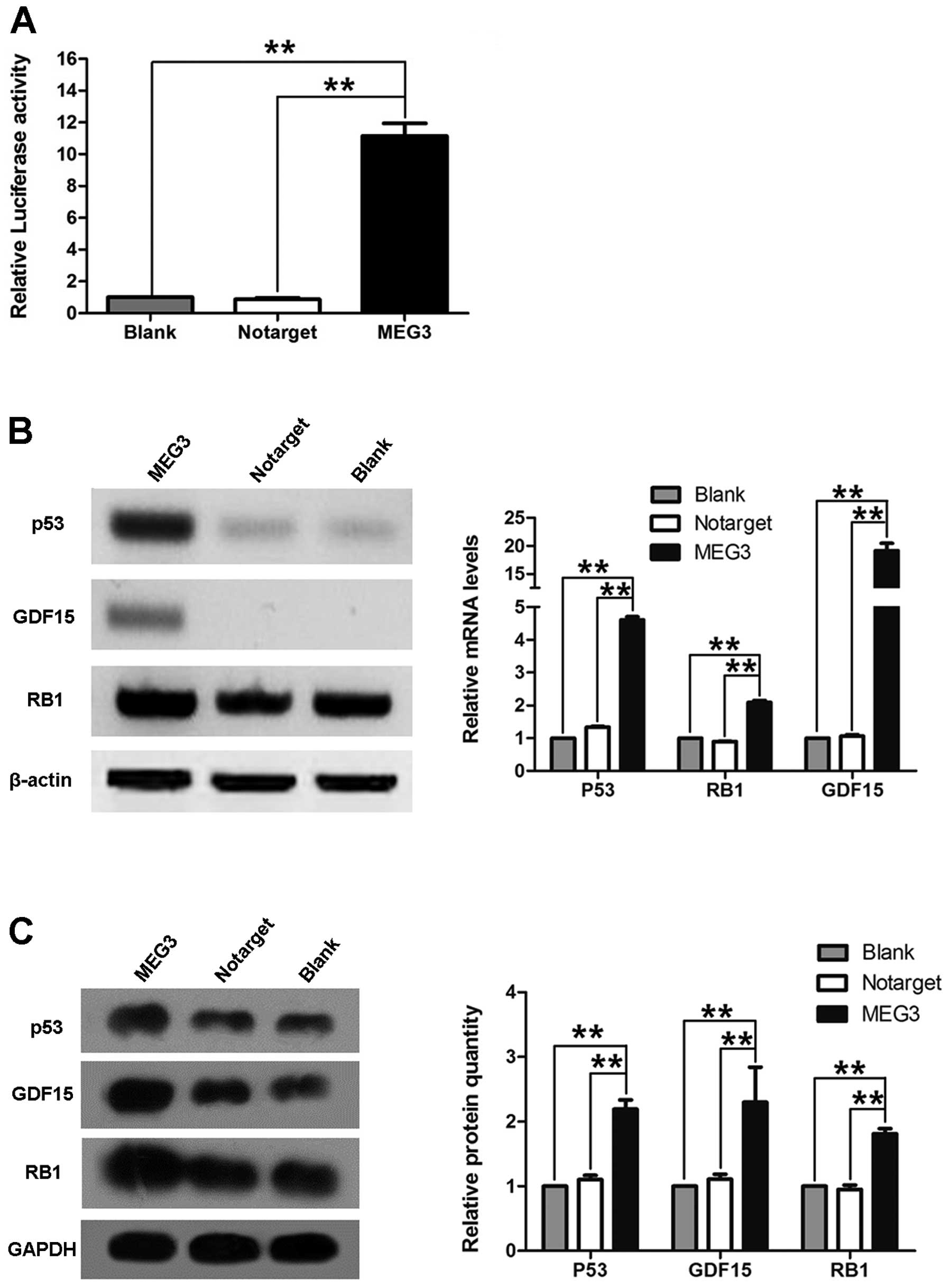
Association of MEG3 and p53
To understand the molecular mechanism by which MEG3
suppresses the growth of ovarian cancer cells, we investigated
whether MEG3 regulates the activation of p53. OVCAR3 cells were
cotransfected with pGL3-p53 and pcDNA3.1-MEG3. Fig. 6A shows that cells transfected with
pcDNA3.1-MEG3 had significantly increased levels of p53 activity.
Additionally, we also found that overexpression of MEG3 in OVCAR3
cells increased p53, GDF15 and RB1 mRNA and
protein levels (Fig. 6B and C).
Discussion
We determined that MEG3 RNA is highly expressed in
most normal human ovarian tissues. However, expression of MEG3 in
epithelial ovarian cancers (EOCs) was sharply decreased and
sometimes even lost. Expression of MEG3 RNA was not detected in
>70% of the EOC samples, and this loss of expression was
associated with the grade of the tumor. We also discovered that
MEG3 RNA was not expressed in the OVCAR3, SKOV3, HP8910 and ES-2
cell lines. On the other hand, the MEG3 promoter was
hypermethylated in >60% of the EOC samples and in 100% of the
ovarian cancer cell lines. As was determined in previous reports,
the demethylating agent 5-aza-CdR reversed the hypermethylation of
the MEG3 promoter in OVCAR3 cells, with the effectiveness
increasing with increasing concentrations of the drug.
Is there a relationship between downregulation or
loss of expression of MEG3 RNA and hypermethylation of the
MEG3 promoter?
MEG3 is a long non-coding RNA (lncRNA). The
MEG3 gene, also known as gene trap locus 2 (gtl2), is
located on human chromosome 14q32 (Meg3 is located on mouse
chromosome 12) and is reciprocally imprinted with the paternally
expressed gene DLK1 (15). Two DMRs
are located upstream of the MEG3 gene, IG-DMR and MEG3-DMR,
which controls expression in the placenta and the body,
respectively (16). Zhao et
al used bisulfite sequencing to determine that the methylation
in regions 1 and 4 of the MEG3 promoter is significantly higher in
NFAs than in normal pituitary and that the MEG3 promoter
overlaps with the MEG3-DMR (6).
Hypermethylation of the MEG3 promoter is also observed in
meningiomas (7) and neuroblastoma
cell lines (8). Downregulation or
loss of expression of MEG3 RNA is consistent with abnormal
methylation of the MEG3 promoter in these types of cancers.
In contrast, MEG3 is expressed in many normal human tissues whose
promoters do not display CpG hypermethylation. These studies
indicate that inactivation of the MEG3 gene and
downregulation of MEG3 RNA in these tumors partially result from
silencing of the promoter by hypermethylation (5). Moreover, treating cells with 5-aza-CdR
triggers re-expression of MEG3 in multiple human breast cancer
tumor cell lines (6), meningiomas
(7), neuroblastomas (8) and hepatocellular carcinomas (10).
Epigenetic silencing of tumor-suppressor genes by
methylation is now arousing more and more interest in cancer
research. DNA methylation is an epigenetic modification that is
important in transcriptional control (17). Hypermethylation of the promoters of
these genes is associated with gene silencing. Similar to gene
mutation, epigenetic silencing is another mechanism by which
tumor-suppressor genes are inactivated (18). Epigenetic changes have been
implicated in malignant transformation and progression of different
types of cancers, including ovarian cancer (18). For example, the RASSF1A gene
is methylated in ~40–50% of ovarian cancers (19,20).
Moreover, hypermethylation of the BRCA1 promoter is highly
correlated with decreased expression of BRCA1 and
BRCA2 mRNA and is significantly correlated with tumor stage
(21).
Our findings are consistent with those of the
studies mentioned above and strongly indicate that hypermethylation
of the MEG3 promoter plays a significant role in
inactivation of the MEG3 gene in EOC.
Functionally, in a colony-formation assay,
overexpression of MEG3 in MCF7 and HeLa cells was found to result
in a significantly lower number of colonies in these cells than in
cells transfected with the controls (4). In addition, ectopic expression of MEG3
significantly inhibited the incorporation of BrdU in HCT116 and
IOMM-Lee cells (15). Braconi et
al (10) and Wang et al
(11) reported that ectopic
expression of MEG3 caused apoptosis in PLC/PRF/5 and U251 and U87
MG cells, respectively. We proposed that MEG3 may play the same
role in EOC. The results of the present study indicated that
overexpression of MEG3 inhibited proliferation and promoted
apoptosis in ovarian cancer cells. Downregulation of MEG3 in
ovarian cancer cells appears to enhance the growth of the
neoplasm.
In general, the tumor suppressor p53 potently
inhibits cell growth by blocking proliferation or by activating
cell death programs (22). Recent
studies have demonstrated that lncRNAs physically associate with
p53, suggesting that they may play an important role in regulating
cell growth via the p53 pathway (23). Normally, the level of p53 is very
low as the ubiquitin-proteasome system induces its rapid
degradation. MDM2 is a member of this system, which inhibits p53
function and promotes p53 protein degradation (24). MEG3 targets p53 by either directly
interacting with p53 or indirectly suppressing the negative
regulator MDM2 (9). These effects
may result in selective activation of downstream targets of p53
such as GDF15 and other yet-to-be-identified proteins with
antiproliferative and tumor-suppressive functions (5). The RB pathway may be the other
mechanism by which MEG3, independently of p53, functions as a tumor
suppressor (9). Transfecting HCT116
cells with an MEG3 expression vector induces a significant increase
in p53 (9). In the present study,
p53 mRNA and protein levels and the activity of the
p53 promoter were increased in OVCAR3 cells transfected with
pcDNA3.1-MEG3. Moreover, in the present study, expression of
GDF15 and RB1 was 2-fold greater than that in control
cells. These data indicate that MEG3 targets p53 to exert an
antiproliferative function.
Although Braconi et al (10) determined that microRNA-29 can
regulate the expression of MEG3 in hepatocellular cancer by
modulating DNA methyltransferase (DNMT) 1 and 3b, the mechanisms
upstream of DNMT regulation of MEG3 need to be identified. Staub
et al (25) found that
HSULF1 is epigenetically silenced in ovarian cancer and that
epigenetic therapy targeting HSULF1 rendered ovarian tumors
sensitive to conventional first-line therapies. Our future studies
will confirm whether or not targeting MEG3 with
5-aza-2-deoxycytidine treatment may sensitize EOC to
chemotherapy.
In conclusion, our data suggest that MEG3 may play
an important role as a tumor suppressor in ovarian cancer cells
since: i) MEG3 is located at the chromosome 14q32 locus; ii)
MEG3 is highly expressed in normal ovarian tissues, but its
expression is decreased or absent in EOC tissues and ovarian cancer
cells; iii) hypermethylation of the MEG3 promoter is
negatively correlated with the expression of MEG3 RNA in EOCs; iv)
re-expression of MEG3 in ovarian cancer cells strongly suppresses
growth and promotes apoptosis; and finally v) MEG3 indirectly or
directly targets p53 and/or RB1 to control proliferation of ovarian
cancer cells (15). Further
investigation of the mechanism of action of MEG3 could therefore
possibly provide new therapeutic strategies for EOC.
Acknowledgements
We thank Qicai Liu and Jiachun Lu for the helpful
advice with experiments and careful reading of this manuscript.
This study was supported by grants from the Science and Information
Technology Agency of Guangzhou (no. 2010GN-E00221).
References
|
1
|
Itamochi H: Targeted therapies in
epithelial ovarian cancer: molecular mechanisms of action. World J
Biol Chem. 1:209–220. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gibb EA, Brown CJ and Lam WL: The
functional role of long non-coding RNA in human carcinomas. Mol
Cancer. 10:382011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Miyoshi N, Wagatsuma H, Wakana S, et al:
Identification of an imprinted gene, Meg3/Gtl2 and its human
homologue MEG3, first mapped on mouse distal chromosome 12
and human chromosome 14q. Genes Cell. 5:211–220. 2000.
|
|
4
|
Zhang X, Zhou Y, Mehta KR, et al: A
pituitary-derived MEG3 isoform functions as a growth suppressor in
tumor cells. J Clin Endocrinol Metab. 88:5119–5126. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhou Y, Zhang X and Klibanski A:
MEG3 noncoding RNA: a tumor suppressor. J Mol Endocrinol.
48:R45–R53. 2012. View Article : Google Scholar
|
|
6
|
Zhao J, Dahle D, Zhou Y, Zhang X and
Klibanski A: Hypermethylation of the promoter region is associated
with the loss of MEG3 gene expression in human pituitary
tumors. J Clin Endocrinol Metab. 90:2179–2186. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang X, Gejman R, Mahta A, et al:
Maternally expressed gene 3, an imprinted noncoding RNA
gene, is associated with meningioma pathogenesis and progression.
Cancer Res. 70:2350–2358. 2010. View Article : Google Scholar
|
|
8
|
Astuti D, Latif F, Wagner K, et al:
Epigenetic alteration at the DLK1-GTL2 imprinted domain in
human neoplasia: analysis of neuroblastoma, phaeochromocytoma and
Wilms’ tumour. Br J Cancer. 92:1574–1580. 2005.PubMed/NCBI
|
|
9
|
Zhou Y, Zhong Y, Wang Y, et al: Activation
of p53 by MEG3 non-coding RNA. J Biol Chem. 282:24731–24742. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Braconi C, Kogure T, Valeri N, et al:
microRNA-29 can regulate expression of the long non-coding RNA gene
MEG3 in hepatocellular cancer. Oncogene. 30:4750–4756. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang P, Ren Z and Sun P: Overexpression of
the long non-coding RNA MEG3 impairs in vitro glioma cell
proliferation. J Cell Biochem. 113:1868–1874. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Benetatos L, Hatzimichael E, Dasoula A, et
al: CpG methylation analysis of the MEG3 and SNRPN imprinted genes
in acute myeloid leukemia and myelodysplastic syndromes. Leuk Res.
34:148–153. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Feng S, Cong S, Zhang X, et al:
MicroRNA-192 targeting retinoblastoma 1 inhibits cell proliferation
and induces cell apoptosis in lung cancer cells. Nucleic Acids Res.
39:6669–6678. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Anwar SL, Krech T, Hasemeier B, et al:
Loss of imprinting and allelic switching at the DLK1-MEG3
locus in human hepatocellular carcinoma. PLoS One. 7:e494622012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Benetatos L, Vartholomatos G and
Hatzimichael E: MEG3 imprinted gene contribution in tumorigenesis.
Int J Cancer. 129:773–779. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kagami M, O’Sullivan MJ, Green AJ, et al:
The IG-DMR and the MEG3-DMR at human chromosome 14q32.2:
hierarchical interaction and distinct functional properties as
imprinting control centers. PLoS Genet. 6:e10009922010.PubMed/NCBI
|
|
17
|
Jones PA and Baylin SB: The fundamental
role of epigenetic events in cancer. Nat Rev Genet. 3:415–428.
2002.PubMed/NCBI
|
|
18
|
Baylin SB and Ohm JE: Epigenetic gene
silencing in cancer - a mechanism for early oncogenic pathway
addiction? Nat Rev Cancer. 6:107–116. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yoon JH, Dammann R and Pfeifer GP:
Hypermethylation of the CpG island of the RASSF1A gene in
ovarian and renal cell carcinomas. Int J Cancer. 94:212–217.
2001.PubMed/NCBI
|
|
20
|
Ibanez de Caceres I, Battagli C, Esteller
M, et al: Tumor cell-specific BRCA1 and RASSF1A
hypermethylation in serum, plasma, and peritoneal fluid from
ovarian cancer patients. Cancer Res. 64:6476–6481. 2004.
|
|
21
|
Chan KY, Ozçelik H, Cheung AN, Ngan HY and
Khoo US: Epigenetic factors controlling the BRCA1 and
BRCA2 genes in sporadic ovarian cancer. Cancer Res.
62:4151–4156. 2002.PubMed/NCBI
|
|
22
|
Kim T, Veronese A, Pichiorri F, et al: p53
regulates epithelial-mesenchymal transition through microRNAs
targeting ZEB1 and ZEB2. J Exp Med. 208:875–883. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Huarte M, Guttman M, Feldser D, et al: A
large intergenic noncoding RNA induced by p53 mediates global gene
repression in the p53 response. Cell. 142:409–419. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Brooks CL and Gu W: p53 regulation by
ubiquitin. FEBS Lett. 585:2803–2809. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Staub J, Chien J, Pan Y, et al: Epigenetic
silencing of HSulf-1 in ovarian cancer:implications in
chemoresistance. Oncogene. 26:4969–4978. 2007. View Article : Google Scholar : PubMed/NCBI
|