Introduction
Hypoxia is an important micro-environmental factor
in promoting tumor progression. In hypoxia, several transcription
factors are induced to respond to the decreased oxygen level. Among
these transcription factors, hypoxia-inducible factor-1 (HIF-1) is
one of the most important factors that play a critical role in
controlling oxygen delivery and metabolic adaptation to hypoxic
conditions. HIF-1 is a heterodimeric protein consisting of a
constitutively expressed HIF-1β subunit, known as the aryl
hydrocarbon receptor nuclear translocator and HIF-1α subunit, which
are basic helix-loop-helix-PAS domain proteins; only HIF-1α is
regulated by the oxygen tension (1,2). The
regulation of HIF-1α occurs at the posttranslational level
involving modifications of hydroxylation, acetylation and
phosphorylation (3–7). Under normoxic conditions, prolyl
hydroxylases (PHDs) hydroxylate the cite-specific proline residues
of HIF-1α in a reaction that uses O2 as a substrate. The
modified HIF-1α interacts with Von Hippel-Lindau (VHL), which is
part of the E3 ubiquitin ligase complex targeting HIF-1α for 26S
proteasomal degradation. Under hypoxia, HIF-1α is stabilized due to
the lack of O2 and dimerizes with HIF-1β interacting
with the co-activator CBP/p300 to bind to the hypoxia response
element (HRE; 5′-GACGTG-3′) on the promoter region in various
target genes (1,2).
HIF-1 plays a central role in tumor progression and
angiogenesis in vivo. For instance, it can be activated by
oncogenic mutations of PTEN, VHL, the RAS/mitogen-activated protein
kinase (MAPK) pathway and the phosphorylation of
phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of
rapamycin (mTOR) pathway. Furthermore, HIF-1α is also stabilized by
reactive oxygen species (ROS), which block PHD activities (8). To date, HIF-1 is known to
transcriptionally upregulate >100 genes (9,10).
Exposure to a variety of growth factors has also been shown to
increase HIF-1 activity in normoxic and hypoxic conditions. HIF-1α
overexpression is associated with increased mortality in patients
with various tumors; this association is primarily based on the
HIF-1-mediated regulation of genes that play pivotal roles in the
central features of cancer pathogenesis such as angiogenesis,
invasion, metastasis and anti-apoptosis. All of these activities
make the HIF-1 transcription factor an attractive target for the
development of new anticancer therapeutics (11–13).
As part of our continuing search for HIF-1α
inhibitors from natural products, we identified celastrol, a
quinone methide triterpene, as a pharmacologically active compound
in Tripterygium wilfordii Hook F root extracts. Celastrol
has been widely used to treat autoimmune diseases, chronic
inflammation, neurodegenerative diseases as well as several types
of cancer (14). However, the
mechanism by which celastrol inhibits HIF-1-mediated tumor growth
is not fully understood. In present study, we found that celastrol
inhibited HIF-1 activation. This compound rapidly downregulates not
only HIF-1α, by decreasing its protein synthesis without affecting
mRNA levels or protein degradation, but also the expression of HIF
target genes such as vascular endothelial growth factor (VEGF) and
erythropoietin (EPO), which are essential for tumor growth. The
HIF-1 activation-inhibitory effects of this compound were
associated with the suppression of mTOR/ribosomal protein S6 kinase
(p70S6K)/eukaryotic initiation factor 4E (eIF4E) and extracellular
signal-regulated kinase (ERK) signaling pathways. We further
confirmed our in vitro observations by showing profound
antitumor activity of celastrol in a murine xenograft model with no
apparent toxicity to the animals.
Materials and methods
Cell culture and reagents
Hep3B, SK-Hep1, and HeLa cells were grown in DMEM
with penicillin (100 U/ml)-streptomycin (100 U/ml) (Invitrogen,
Carlsbad, CA, USA) and 10% heat-inactivated fetal bovine serum
(Hyclone, Logan, UT, USA). All cells were purchased from American
Type Culture Collection (ATCC, Manassas, VA, USA). MG-132 and
cycloheximide (CHX) were from Sigma (St. Louis, MO, USA). The
hypoxic culture was kept in a gas-controlled chamber (Thermo
Electron Corp., Marietta, OH, USA) maintained at 1% O2,
94% N2, and 5% CO2 at 37°C. In some
experiments, cobalt chloride was used to induce hypoxia mimicking
conditions. Cobalt chloride was reported as a widely used mimetic
of hypoxia in a large range of cells; the molecule is known to
inhibit prolyl hydroxylases leading to HIF-1α stabilization
(15). Celastrol was isolated from
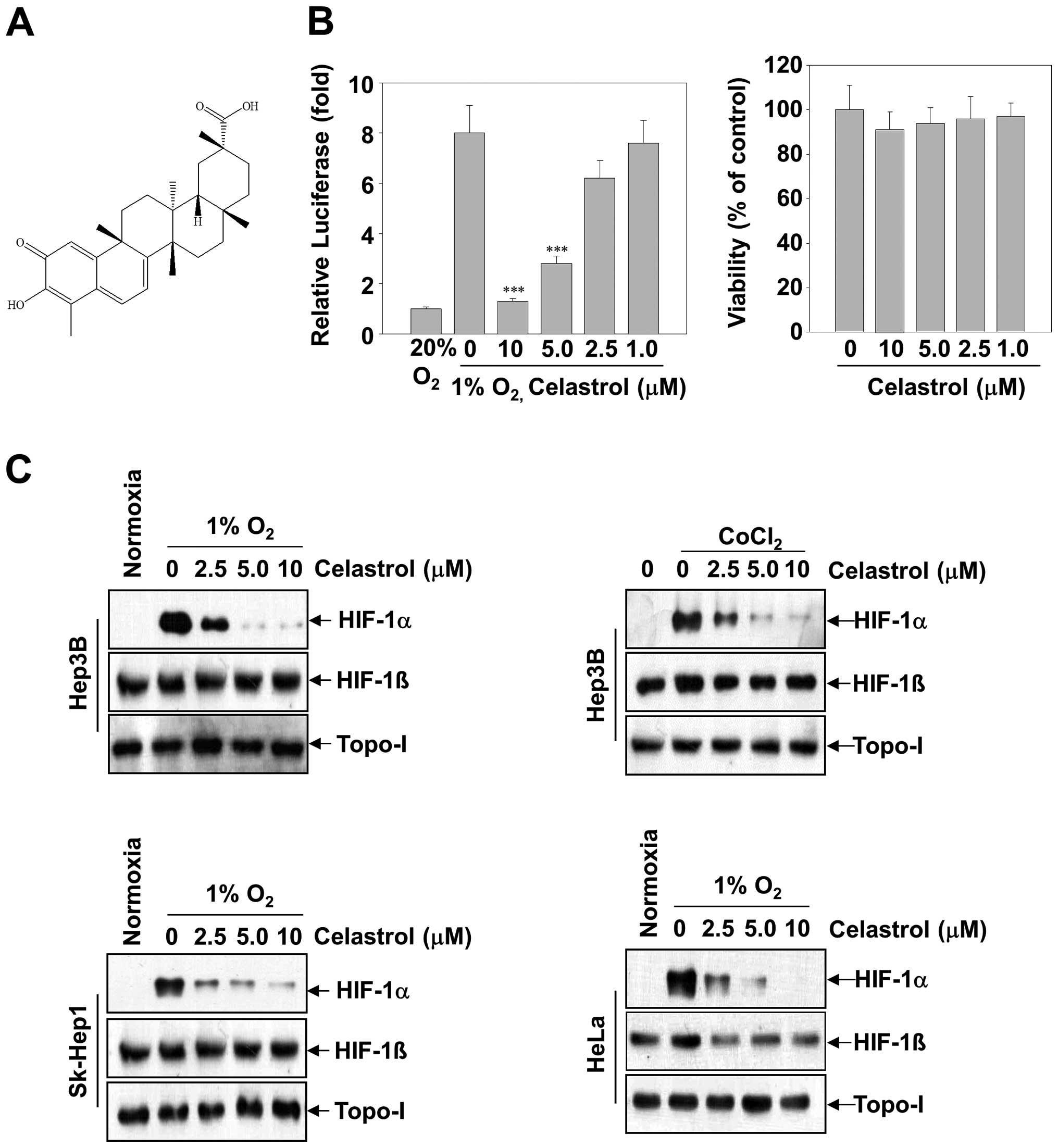
T. kirilowii and its structure is shown in Fig. 1A. The purity of celastrol was
>98% in the HPLC analysis.
Plasmids, transfections and luciferase
reporter assay
The ability of the compound to inhibit hypoxia
inducible factor was determined by HRE-dependent reporter assay as
previously described (16). In
brief, at 50–80% confluence, Hep3B cells, which were co-transfected
with the vectors for pGL3-HRE-Luciferase plasmid containing six
copies of HREs derived from the human VEGF gene and with pRL-CMV
(Promega, Madison, WI, USA) using Lipofectamine plus reagent
(Invitrogen, Carlsbad, CA, USA). Following 24 h incubation, the
cells were treated with various concentrations of celastrol and
incubated for 16 h in hypoxia. Luciferase assay was performed using
dual-luciferase reporter assay system according to the instructions
of the manufacturer (Promega). Luciferase activity was determined
in Microlumat plus luminometer (EG&G Berthold, Bad Wildbad,
Germany) by injecting 100 μl of assay buffer containing luciferin
and measuring light emission for 10 sec. The results were
normalized to the activity of renilla expressed by
cotransfected Rluc gene under the control of a constitutive
promoter.
Measurement of in vitro invasion and cell
viability
The ability of cells to invade through
Matrigel-coated filters (invasion) was determined using a modified
24-well Boyden chamber (Corning Costar, Cambridge, MA, USA; 8 μm
pore size) as previously described (17). Hep3B cells were seeded at a density
of 5×104 cells in 100 μl DMEM containing 10% FBS in the
upper compartment of transwell. To determine the effect of
celastrol, various concentrations of celastrol were added to the
lower or upper compartment of transwell. After incubation for 24 h
at 37°C in 5% CO2, the cells that did not penetrate the
filter were completely wiped out with a cotton swab, and the cells
that had migrated to the lower surface of the filter were fixed,
stained and counted in 5 randomly selected microscopic fields
(×100) per filter. Cell viability was measured by an MTT
[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide]
assay (Sigma). Briefly, untreated cells or treated cells with
celastrol in a 96-well plate were incubated for 24 h followed by
the addition of MTT to the cells. Optical densities were determined
on a microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Western blot analysis
Whole-cell extracts were obtained by lysing cells in
ice-cold lysis buffer (50 mM Tris-HCl, pH 7.5, 1% Nonidet P-40, 1
mM sulfonyl fluoride) supplemented with the protease inhibitor
cocktail (BD Biosciences, San Diego, CA, USA). HIF-1α protein was
analyzed in nuclear extracts prepared from cells using NE-PER
reagent (Pierce, Rockford, IL, USA), according to the instructions
of the manufacturer. An aliquot of protein extracts was used to
determine protein concentration by the Bradford method. Fifty
micrograms of whole-cell extracts or 30 μg of nuclear extract
protein per lane was separated by SDS-polyacrylamide gels and
followed by transferring to a polyvinylidene difluoride membrane
(Millipore, Bedford, MA, USA). The membrane was blocked with 5%
skim milk, and then incubated with the corresponding antibody.
Antibody for HIF-1α was obtained from BD Biosciences (1:250).
Antibodies for mTOR, phospho (Ser2448)-specific mTOR, p70S6K,
phospho (Thr389)-specific p70S6K, ERK1/2, phospho
(Thr202/Tyr204)-specific ERK1/2, eIF4E, phospho (Ser209)-specific
eIF4E, p38, phospho-specific p38, SAPK/JNK and phospho-specific
SAPK/JNK were purchased from Cell Signaling Technology (Beverly,
MA, USA). Antibodies for HIF-1β and topo-I were obtained from Santa
Cruz Biotechnology (Santa Cruz, CA, USA). Antibody for α-tubulin
was from Sigma. After binding of an appropriate secondary antibody
coupled to horseradish peroxidase, proteins were visualized by
enhanced chemiluminescence according to the instructions of the
manufacturer (Amersham Pharmacia Biotech, Buckinghamshire, UK).
VEGF ELISA
Hep3B cells were plated in 96-well plates at a
density of 1×105 cells/well and treated with various
concentrations of celastrol for 16 h under normoxic or hypoxic
conditions. The VEGF levels in the culture supernatant were
determined by ELISA using the Duo-Set ELISA development kit
(R&D Systems, Inc., Minneapolis, MN, USA), according to the
manufacturer’s instructions.
RT-PCR analysis
Total RNA from Hep3B cells was obtained using RNA
Mini kit (Qiagen, Valencia, CA, USA). Total RNA (2 μg) was used to
perform reverse transcription-PCR (RT-PCR) using RT-PCR kit
(Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s
protocol. The PCR primers were: VEGF, 5′-GCTCTACCTCCACCATGCCAA-3′
(sense) and 5′-TGGAAGATGTCCACCAGGGTC-3′ (antisense); EPO,
5′-CACTTTCCGCAAACTCTTCCG-3′ (sense) and 5′-GTCA CAGCTTGCCACCTAAG-3′
(antisense); HIF-1α, 5′-CTCA AAGTCCGACAGCCTCA-3′ (sense) and
5′-CCCTGCAGT AGGTTTCTGCT-3′ (antisense); GAPDH, 5′-ACCACAGTC
CATGCCATCAC-3′ (sense) and 5′-TCCACCACCCTGTT GCTGTA-3′ (antisense).
The oligonucleotide sequences of the reaction products were
confirmed by sequencing.
In vivo xenograft assay
All surgical procedures and careful handling of the
animals were in accordance with IACUC guidelines. Six-week-old
specific pathogen-free Crj:BALB/c nu/nu female athymic nude mice
(Vital River, China) were randomly assigned to three groups, each
of which consisted of five mice (n=5 per group), and were then
subcutaneously inoculated with 0.2 ml of Hep3B cells
(5×107 cells/ml) in the right flank region. Celastrol,
dissolved in DMSO, was administered orally three times a week for
35 days at a dose of 3 and 10 mg/kg body weight starting from the
ten days post cell implantation with mice. Tumor volume was
calculated every five days using the equation: (Length ×
(width)2)/2. Tumors were harvested 4 h after the last treatment,
followed by homogenizing in RIPA for western blotting analysis.
Statistical analysis
All values are expressed as mean ± SD. A comparison
of the results was performed with one-way ANOVA and Tukey’s
multiple comparison tests (Graphpad Software, Inc., San Diego, CA,
USA). Statistically significant differences between groups were
defined as P-values <0.01.
Results
Celastrol inhibits HIF-1α expression in
tumor-derived cells
To investigate whether celastrol inhibited HIF-1α
transcriptional activation, we transfected Hep3B cells with a
luciferase reporter gene driven by six specific HREs. A substantial
increase of luciferase activity was observed in cells cultured in
hypoxic conditions, whereas celastrol dose-dependently inhibited
hypoxia-induced luciferase activity (Fig. 1B, left panel). Given that the
inhibition of HIF-1α transcriptional activation might be correlated
with celastrol-induced cytotoxicity, parallel studies of cell
viability were performed (Fig. 1B,
right panel). After the Hep3B cells were treated with celastrol (up
to 10 μM) for 24 h, no significant alteration of cell viability was
observed relative to the untreated control group.
To explore the mechanism underlying celastrol
activity, we investigated its effect on HIF-1α protein levels. In
Hep3B cells, HIF-1α protein is undetectable under normoxia, whereas
it is stabilized under hypoxia or in the presence of
CoCl2 and becomes readily detectable by western
blotting. Following 12-h treatment, celastrol exerted
dose-dependent inhibition of HIF-1α protein levels induced by
hypoxia or CoCl2 in Hep3B cells. (Fig. 1C, top two panels). In contrast to
the decrease of HIF-1α levels, celastrol had almost no effect on
the levels of HIF-1β and topo-I proteins. Next, in order to address
whether the inhibition of HIF-1α by celastrol was cell
line-specific, we extended these studies to a diverse set of tumor
cell lines with tissues of various origins, including the hepatic
cancer cell lines SK-Hep1 and epithelial cervical cancer cell lines
HeLa cells. Fig. 1C shows that,
under hypoxic conditions, HIF-1α accumulation was strongly
suppressed by celastrol in all cell lines.
Celastrol inhibits the protein synthesis
of HIF-1α but not its degradation
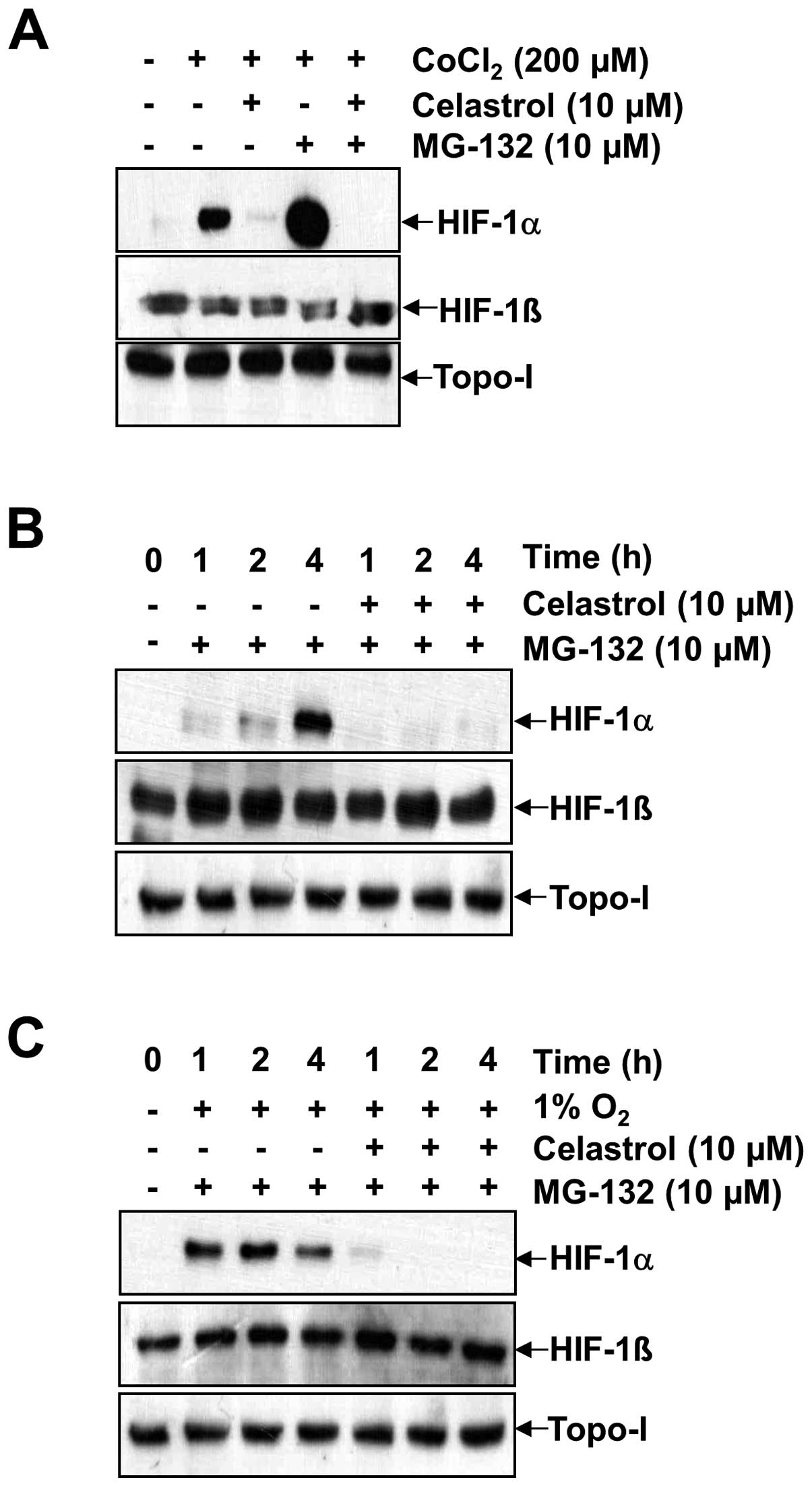
Generally, the accumulation of HIF-1α is dependent
on the balance between its protein synthesis and degradation. To
specifically investigate whether celastrol modulates HIF-1α protein
synthesis, we used the proteasome inhibitor MG-132 to prevent
HIF-1α degradation. This allowed us to observe HIF-1α stabilization
and accumulation following de novo protein synthesis. Due to
rapid proteosomal destruction and undetectable HIF-1α levels in
normoxic cells, the accumulation rate of HIF-1α via proteosomal
inhibition reflects the synthesis rate of the protein (18). As shown in Fig. 2A, B and C, HIF-1α rapidly
accumulated over a period of 4 h in the presence of MG-132 under
both normoxia and hypoxia, as well as in the presence of
CoCl2. In contrast, co-treatment with celastrol and
MG-132 resulted in a much slower rate of HIF-1α accumulation
(compare lane 4 with lane 5 for CoCl2 in Fig. 2A, lanes 2–4 with lanes 5–7 under
normoxia in Fig. 2B, and lanes 2–4
with lanes 5–7 under hypoxia in Fig.
2C). No effects were observed on HIF-1β and topo-I. These
results indicate that HIF-1α protein synthesis in Hep3B cells is
markedly impaired in the presence of celastrol. To address the
effect of celastrol on HIF-1α protein stability, the protein
translation inhibitor CHX was used to prevent de novo HIF-1α
protein synthesis. Accordingly, we determined whether celastrol
affects HIF-1α protein stability by the addition of 200 μM
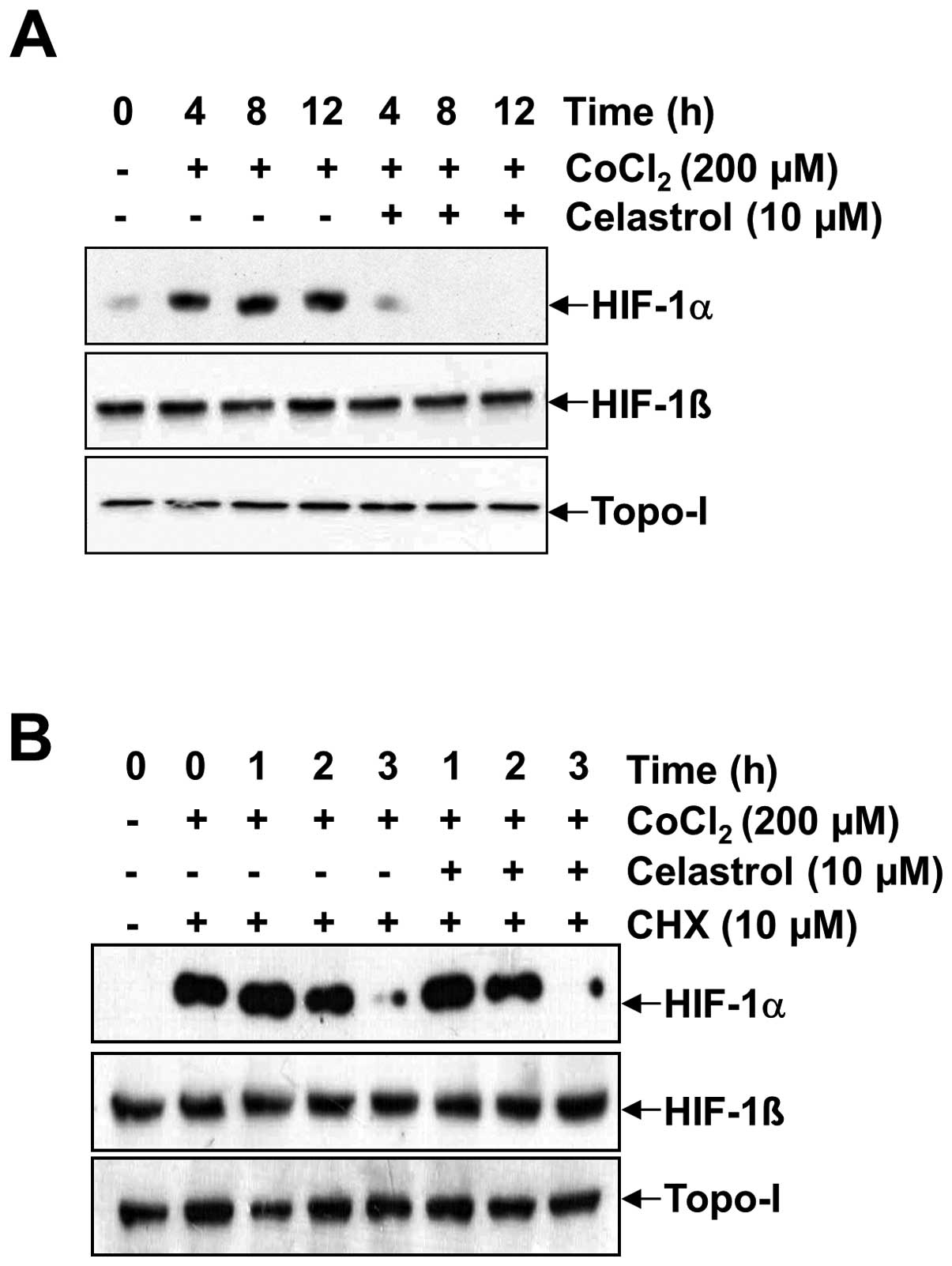
CoCl2 (Fig. 3). HIF-1α
accumulation increased in a time-dependent manner, but the addition
of celastrol resulted in the significant abrogation of HIF-1α
accumulation at every time point (Fig.
3A). To determine whether HIF-1α protein half-life was affected
by celastrol, we firstly induced HIF-1α accumulation in the
presence of 200 μM CoCl2 for 4 h, and then added CHX
alone or in combination with celastrol. In the presence of CHX,
HIF-1α levels rapidly declined in both the celastrol-untreated and
celastrol-treated cells, showing a half-life was ~1.5 h in Hep3B
cells. These results indicated that celastrol did not modify the
degradation rate of HIF-1α (Fig.
3B, lanes 3–5 with lanes 6–8).
To determine whether HIF-1α synthesis inhibition by
celastrol was a downstream effect from decreased HIF-1α gene
transcription or HIF-1α mRNA stability, we analyzed HIF-1α mRNA
levels by RT-PCR. Celastrol did not change HIF-1α mRNA levels under
either normoxia or hypoxia during 16 h of treatment (Fig. 5A). This suggests that
celastrol-mediated decrease of HIF-1α synthesis is due to
downregulation of HIF-1α mRNA translation.
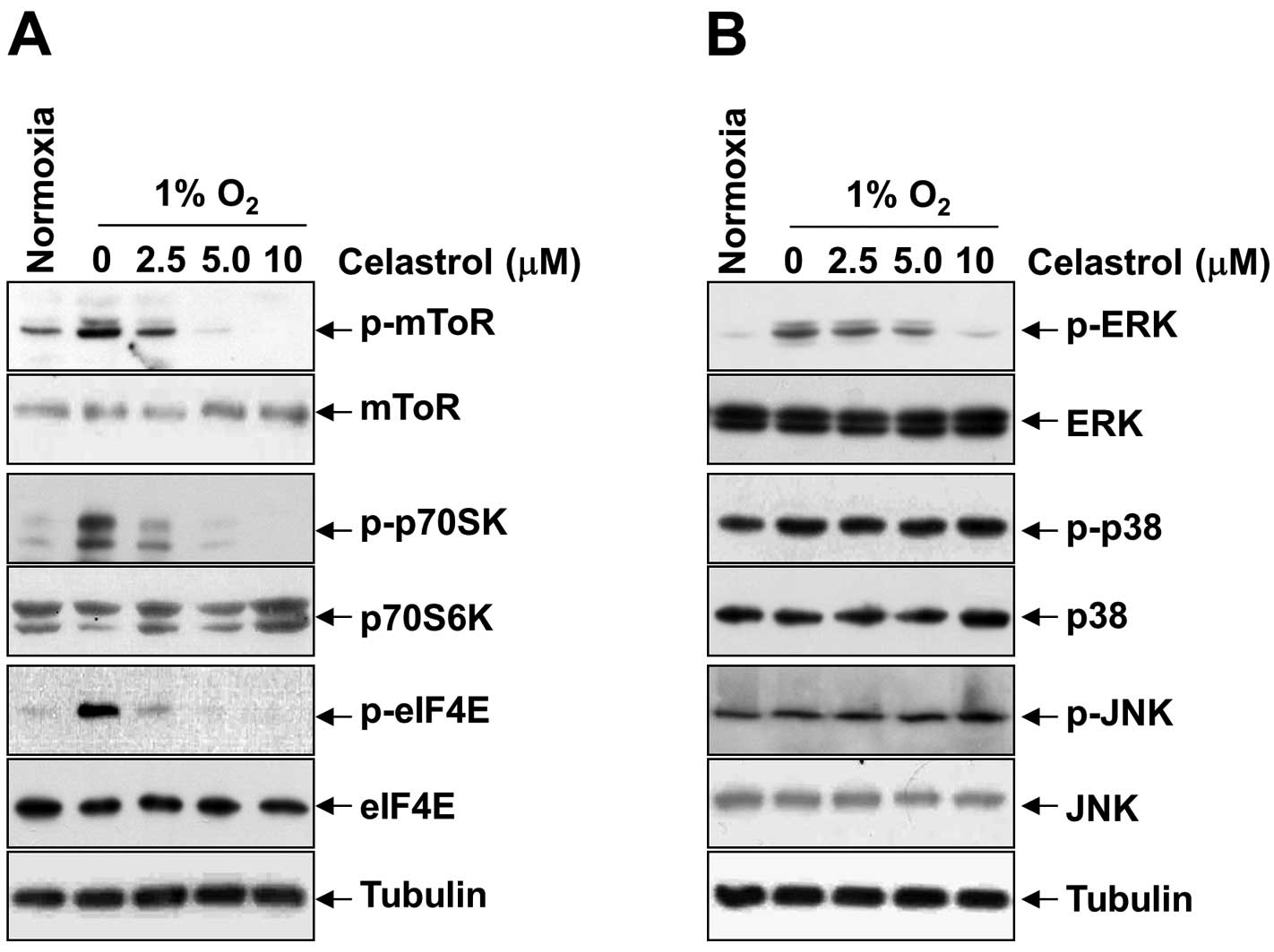
Downregulation of mTOR/p70S6K/eIF4E and
ERK1/2 phosphorylation by celastrol correlates with inhibition of
HIF-1α synthesis
To reveal the underlying mechanism by which
celastrol inhibits hypoxia-induced activation and translation of
HIF-1α, we first examined the phosphorylation status of translation
initiation factors such as mTOR, p70S6K, eIF4E, and ERK1/2 under
hypoxia. As shown in Fig. 4A and B,
in parallel with the alteration of HIF-1α protein, celastrol
dose-dependently inhibited the expression of phospho-mTOR,
phospho-p70S6K, phospho-eIF4E, and phospho-ERK1/2, induced by
hypoxia; however, it had no inhibitory effect on phospho-p38 and
phospho-JNK, and total protein levels for mTOR, p70S6K, eIF4E,
ERK1/2, p38, and JNK. These findings were partially in line with a
report showing that mTOR/p70S6K and ERK pathways are involved in
HIF-1α protein synthesis via functional activation of the
translational regulatory protein eIF4E in various cells (19). All of the results consistently
support the idea that celastrol inhibits hypoxia-induced activation
of HIF-1α through inhibition of mTOR/p70S6K/eIF4E and ERK
signaling.
Celastrol decreases expression of HIF-1α
target genes and suppresses the invasiveness of tumor cells
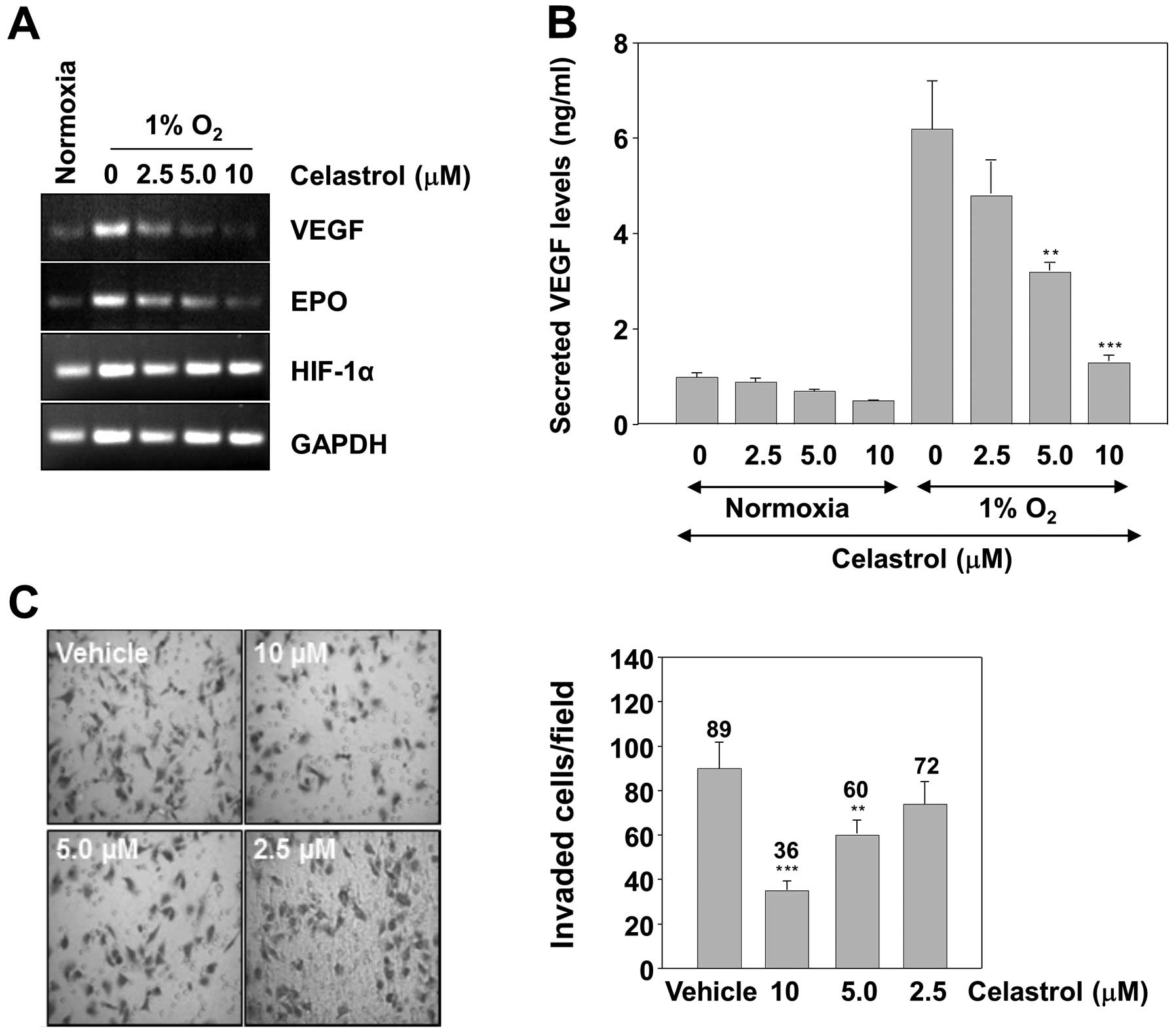
The expression of VEGF and EPO, which are involved
in tumor cell proliferation, angiogenesis, invasion and metastasis,
is known to be regulated by HIF-1α (11). We therefore examined whether
celastrol can suppress the expression of these genes. VEGF and EPO
mRNA levels were measured by RT-PCR analysis in Hep3B cells.
Treatment of the cells with celastrol resulted in a dose-dependent
inhibition of VEGF and EPO mRNA expression (Fig. 5A). The concentrations to inhibit the
expression of HIF-1α target genes were comparable with those of
HIF-1α protein accumulation. This result led us to measure the VEGF
protein concentration in the culture supernatant by ELISA.
Consistently, the hypoxic induction of secreted VEGF protein was
dose-dependently inhibited by celastrol (Fig. 5B). Reduced expression of VEGF and
EPO might be responsible for diminished invasion of tumor cells in
celastrol treatment. Therefore, whether celastrol modulates
invasion activity was examined in vitro with a Matrigel
invasion assay. Hep3B cells were seeded in the top chamber of a
Matrigel invasion chamber and were incubated with various
concentrations of celastrol for 16 h. The result showed that
celastrol significantly decreased invasiveness compared to the
vehicle control, accounting for the anti-invasive activity of
celastrol (Fig. 5C).
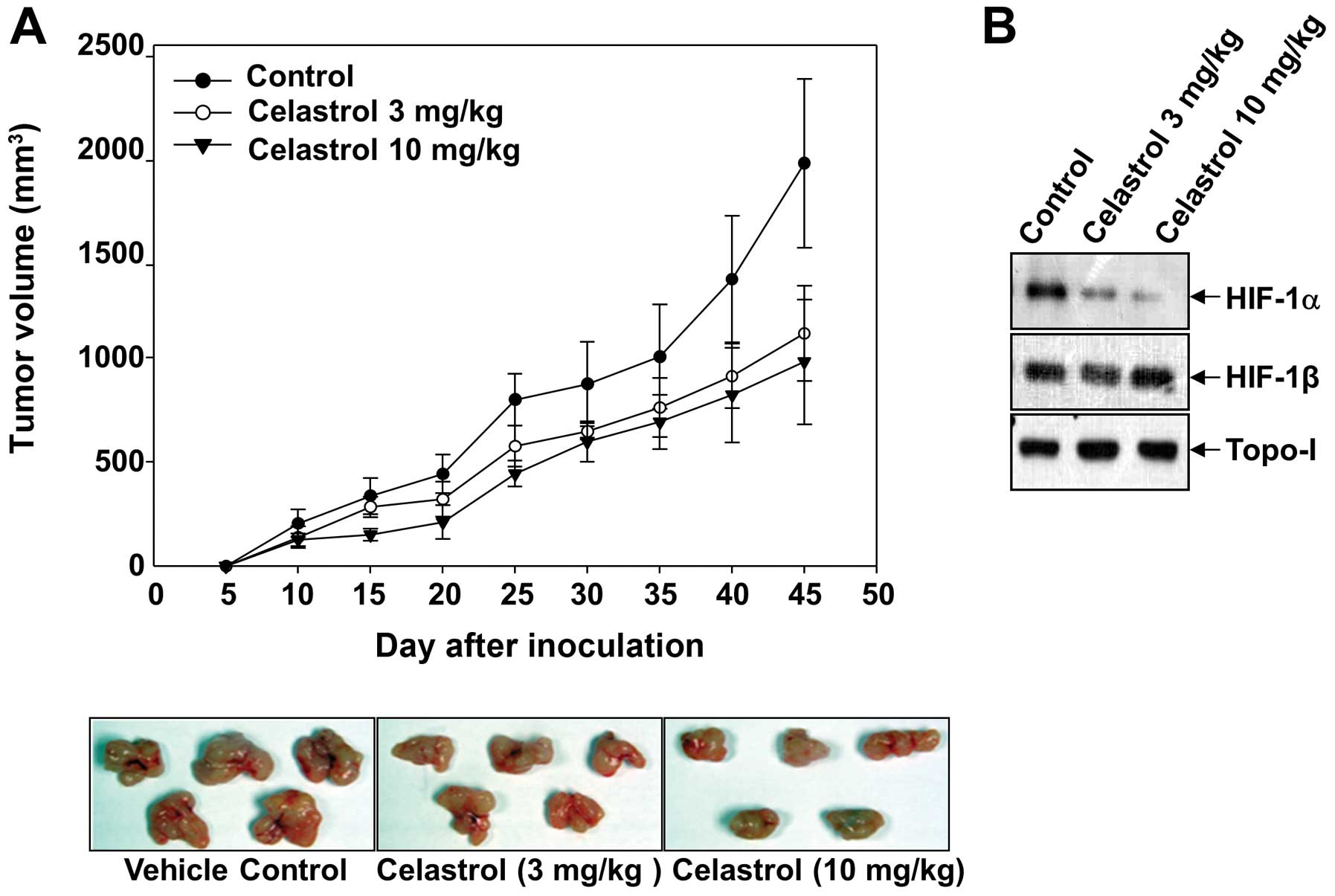
Celastrol inhibits growth of Hep3B cells
in a xenograft tumor model
Since celastrol suppresses both the expression of
HIF-1α target genes and the invasiveness of tumor cells, we next
determined whether these results could be translated into an in
vivo xenograft model. Hep3B cells were subcutaneously implanted
in athymic nude mice, and the experimental mice were treated with
celastrol (3 and 10 mg/kg) 3 days a week until the end of the
study. As expected, the administration of celastrol (10 mg/kg)
significantly inhibited Hep3B tumor growth up to 52.9%, compared to
that of the vehicle-treated control group (Fig. 6A). Due to the key roles of HIF-1α in
tumor angiogenesis, we studied its expression in the tumors by
western blotting. Consistent with the finding in cultured cells,
celastrol significantly decreased the protein levels of HIF-1α in
the tumors, whereas no differences were observed in topo-I
(Fig. 6B).
Discussion
Celastrol, a pharmacologically active compound in
Tripterygium wilfordii Hook F, has been used traditionally
for the treatment of arthritis and other diseases (14). It has been reported that celastrol
exerts potent anti-inflammatory activities in various experimental
models and strong cytotoxicity against various cancer cell-lines;
celastrol is also known to inhibit NF-κB activation by directly
targeting cysteine 179 in the IKK and this feature was supposed to
contribute to its anti-inflammatory and antitumor activities
through mechanisms associated with the inhibition of NF-κB
(20). Previous studies have also
shown that celastrol has antitumor and antiangiogenic effects in
several tumor cells, and the underlying mechanisms were shown to be
associated with their inhibitory effect on the proteasome, the
AKT/mTOR/p70S6K pathway, and VEGF production and its receptor
activity (21–23). These reports suggest that celastrol
might be associated with the regulation of HIF-1. In the present
study, we identified celastrol as a potent inhibitor of HIF-1α
activation and investigated how this compound suppressed HIF-1α
activation. The expression of HIF-1α is tightly regulated through
both protein degradation and protein synthesis. Our results showed
that celastrol strongly inhibited HIF-1α protein synthesis without
affecting the expression level of HIF-1α mRNA or degradation of
HIF-1α protein, indicating it acted as an inhibitor of HIF-1α mRNA
translation.
HIF-1α protein translation has emerged as an
important regulatory mechanism of HIF-1α-inhibitory compounds. In
the present study, we found that treatment of Hep3B cells with
celastrol suppressed phosphorylation of mTOR, phosphorylation of
p70S6K, phosphorylation of eIF4E and phosphorylation of ERK1/2.
Earlier studies have shown that HIF-1α protein translation is
mediated partly by regulation of free eIF4E levels through Akt/mTOR
and ERK pathways (24–26). In the mTOR pathway, the mammalian
target of rapamycin (mTOR), a 289-kDa serine/threonine kinase,
controls protein translation by the phosphorylation of downstream
effectors: the p70S6K that activates 4E-BP1, which in turn binds to
eIF4E and inhibits eIF4E function. Hyperphosphorylation of 4E-BP1
disrupts this binding, releasing eIF4E to be phosphorylated at Ser
209. Phosphorylation of eIF4E increases its affinity for the cap of
mRNA and may also favor its entry into initiation complexes
(27,28). In the ERK pathway, ERK1/2 can
directly phosphorylate eIF4E at Ser 209 (29,30).
Thus, the inhibition of eIF4E phosphorylation by celastrol could
play an important role in its downregulation of HIF-1α protein
synthesis. A number of studies indicate that deregulation of
protein synthesis is a major contributor in cancer initiation and
metastatic progression (31,32).
For example, eIF4E overexpression has been demonstrated in a
variety of human tumors, and has been related to disease
progression. Overexpression of eIF4E in experimental models
markedly alters cellular morphology, enhances proliferation and
induces cellular transformation, tumorigenesis and metastasis.
Conversely, blocking eIF4E function by expression of antisense RNA,
suppresses cellular transformation, tumor growth, tumor
invasiveness and metastasis. It was reported that mTOR inhibitor
rapamycin increased eIF4E phosphorylation through PI3K-dependent
and MEK-mediated mechanisms, indicating mTOR-targeted cancer
therapy may confer a resistance through a negative feedback
mechanism (33,34). In this sense, celastrol could be a
valuable lead compound for further development of a new potent
anticancer agent in combination with mTOR inhibitors.
In summary, this study shows, for the first time,
that celastrol inhibits the mTOR/p70S6K/eIF4E and ERK signaling
pathways, and HIF-1 activity in Hep3B cells. Thus, we have
elucidated important mechanisms of the anticancer activity of
celastrol, related to cell invasion and angiogenesis, which are
essential for the adaptation of cancer cells to microenvironmental
hypoxia and, hence, for tumor progression. These mechanisms may
partly explain the broad spectrum of celastrol anticancer effects,
and provide a rationale for the development of celastrol as an
anticancer drug.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China, nos. 81160250 and 81360496 and was
partially supported by the Jilin province Science and Technology
Development Plan item, no. 20130101161JC. This study also received
assistance from The Thousand Peoples Plan by the Foreign Expert
Bureau, China.
Abbreviations:
|
HIF-1
|
hypoxia-inducible factor-1
|
|
VEGF
|
vascular endothelial growth factor
|
|
topo-I
|
topoisomerase-I
|
|
EPO
|
erythropoietin
|
|
ERK1/2
|
extracellular signal-regulated
kinase-1/2
|
|
mTOR
|
mammalian target of rapamycin
|
|
eIF4E
|
eukaryotic initiation factor 4E
|
|
p70S6K
|
ribosomal protein S6 kinase
|
|
HRE
|
hypoxia response element
|
References
|
1
|
Harris AL: Hypoxia - a key regulatory
factor in tumour growth. Nat Rev Cancer. 2:38–47. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Semenza GL: HIF-1 and tumor progression:
pathophysiology and therapeutics. Trends Mol Med. 8:S62–S67. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bruick RK and McKnight SL: A conserved
family of prolyl-4-hydroxylases that modify HIF. Science.
294:1337–1340. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Epstein AC, Gleadle JM, McNeill LA, et al:
C. elegans EGL-9 and mammalian homologs define a family of
dioxygenases that regulate HIF by prolyl hydroxylation. Cell.
107:43–54. 2001. View Article : Google Scholar
|
|
5
|
Ivan M, Kondo K, Yang H, et al: HIFalpha
targeted for VHL-mediated destruction by proline hydroxylation:
implications for O2 sensing. Science. 292:464–468. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jaakkola P, Mole DR, Tian YM, et al:
Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation
complex by O2-regulated prolyl hydroxylation. Science.
292:468–472. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jeong JW, Bae MK, Ahn MY, et al:
Regulation and destabilization of HIF-1alpha by ARD1-mediated
acetylation. Cell. 111:709–720. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lu X and Kang Y: Hypoxia and
hypoxia-inducible factors: master regulators of metastasis. Clin
Cancer Res. 16:5928–5935. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mole DR, Blancher C, Copley RR, et al:
Genome-wide association of hypoxia-inducible factor (HIF)-1alpha
and HIF-2alpha DNA binding with expression profiling of
hypoxia-inducible transcripts. J Biol Chem. 284:16767–16775. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xia X, Lemieux ME, Li W, et al:
Integrative analysis of HIF binding and transactivation reveals its
role in maintaining histone methylation homeostasis. Proc Natl Acad
Sci USA. 106:4260–4265. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Semenza GL: Targeting HIF-1 for cancer
therapy. Nat Rev Cancer. 3:721–732. 2003. View Article : Google Scholar
|
|
12
|
Giaccia A, Siim BG and Johnson RS: HIF-1
as a target for drug development. Nat Rev Drug Discov. 2:803–811.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Belozerov VE and Van Meir EG: Hypoxia
inducible factor-1: a novel target for cancer therapy. Anticancer
Drugs. 16:901–909. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Salminen A, Lehtonen M, Paimela T and
Kaarniranta K: Celastrol: molecular targets of thunder god vine.
Biochem Biophys Res Commun. 394:439–442. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hervouet E, Cizkova A, Demont J, et al:
HIF and reactive oxygen species regulate oxidative phosphorylation
in cancer. Carcinogenesis. 29:1528–1537. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jin X, Jin HR, Lee D, Lee JH, Kim SK and
Lee JJ: A quassinoid 6alpha-tigloyloxychaparrinone inhibits
hypoxia-inducible factor-1 pathway by inhibition of eukaryotic
translation initiation factor 4E phosphorylation. Eur J Pharmacol.
592:41–47. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jin HR, Jin SZ, Cai XF, et al:
Cryptopleurine targets NF-κB pathway, leading to inhibition of gene
products associated with cell survival, proliferation, invasion,
and angiogenesis. PLoS One. 7:e403552012.PubMed/NCBI
|
|
18
|
Hagen T, Taylor CT, Lam F and Moncada S:
Redistribution of intracellular oxygen in hypoxia by nitric oxide:
effect on HIF1alpha. Science. 302:1975–1978. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Choi YK, Kim CK, Lee H, et al: Carbon
monoxide promotes VEGF expression by increasing HIF-1alpha protein
level via two distinct mechanisms, translational activation and
stabilization of HIF-1alpha protein. J Biol Chem. 285:32116–32125.
2010. View Article : Google Scholar
|
|
20
|
Lee JH, Koo TH, Yoon H, et al: Inhibition
of NF-kappa B activation through targeting I kappa B kinase by
celastrol, a quinone methide triterpenoid. Biochem Pharmacol.
72:1311–1321. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pang X, Yi Z, Zhang J, et al: Celastrol
suppresses angiogenesis-mediated tumor growth through inhibition of
AKT/mammalian target of rapamycin pathway. Cancer Res.
70:1951–1959. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang Y, Zhou Y, Fan Y and Zhou D:
Celastrol inhibits the growth of human glioma xenografts in nude
mice through suppressing VEGFR expression. Cancer Lett.
264:101–106. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang H, Chen D, Cui QC, Yuan X and Dou QP:
Celastrol, a triterpene extracted from the Chinese “thunder of god
vine,” is a potent proteasome inhibitor and suppresses human
prostate cancer growth in nude mice. Cancer Res. 66:4758–4765.
2006.
|
|
24
|
Laughner E, Taghavi P, Chiles K, Mahon PC
and Semenza GL: HER2 (neu) signaling increases the rate of
hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel
mechanism for HIF-1-mediated vascular endothelial growth factor
expression. Mol Cell Biol. 21:3995–4004. 2001. View Article : Google Scholar
|
|
25
|
Thomas GV, Tran C, Mellinghoff IK, et al:
Hypoxia-inducible factor determines sensitivity to inhibitors of
mTOR in kidney cancer. Nat Med. 12:122–127. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hart S, Novotny-Diermayr V, Goh KC, et al:
VS-5584, a novel and highly selective PI3K/mTOR kinase inhibitor
for the treatment of cancer. Mol Cancer Ther. 12:151–161. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lachance PE, Miron M, Raught B, Sonenberg
N and Lasko P: Phosphorylation of eukaryotic translation initiation
factor 4E is critical for growth. Mol Cell Biol. 22:1656–1663.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fukuda R, Hirota K, Fan F, Jung YD, Ellis
LM and Semenza GL: Insulin-like growth factor 1 induces
hypoxia-inducible factor 1-mediated vascular endothelial growth
factor expression, which is dependent on MAP kinase and
phosphatidylinositol 3-kinase signaling in colon cancer cells. J
Biol Chem. 277:38205–38211. 2002. View Article : Google Scholar
|
|
29
|
Ueda T, Watanabe-Fukunaga R, Fukuyama H,
Nagata S and Fukunaga R: Mnk2 and Mnk1 are essential for
constitutive and inducible phosphorylation of eukaryotic initiation
factor 4E but not for cell growth or development. Mol Cell Biol.
24:6539–6549. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu LP, Ho RL, Chen GG and Lai PB:
Sorafenib inhibits hypoxia-inducible factor-1alpha synthesis:
implications for antiangiogenic activity in hepatocellular
carcinoma. Clin Cancer Res. 18:5662–5671. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bjornsti MA and Houghton PJ: Lost in
translation: dysregulation of cap-dependent translation and cancer.
Cancer Cell. 5:519–523. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
De Benedetti A and Graff JR: eIF-4E
expression and its role in malignancies and metastases. Oncogene.
23:3189–3199. 2004.PubMed/NCBI
|
|
33
|
Wang X, Yue P, Chan CB, et al: Inhibition
of mammalian target of rapamycin induces phosphatidylinositol
3-kinase-dependent and Mnk-mediated eukaryotic translation
initiation factor 4E phosphorylation. Mol Cell Biol. 27:7405–7413.
2007. View Article : Google Scholar
|
|
34
|
Wendel HG, De Stanchina E, Fridman JS, et
al: Survival signalling by Akt and eIF4E in oncogenesis and cancer
therapy. Nature. 428:332–337. 2004. View Article : Google Scholar : PubMed/NCBI
|