Introduction
Hepatocellular carcinoma (HCC) is the fifth leading
cause of all cancer-related deaths worldwide, and most of these
deaths occur in developing countries (1). In recent years, studies regarding the
pathogenesis of HCC, from microbial metabolites, a new type of
oncogene to microRNAs (1–4), have weaved an intricate network.
However, little progress in curing this cancer has been achieved.
HCC is also characterized by its insensitivity to chemotherapy and
radiotherapy. Although several targeted therapeutic drugs have been
discovered (5), their clinical
effects on HCC still require further observation. For example,
sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor
angiogenesis, and induces tumor cell apoptosis (6). Based on a previous study (7), PJ34, an inhibitor of PARP-1, a nuclear
enzyme that not only responds to DNA damage and facilitates DNA
repair, but also mediates cell death through a caspase-independent
pathway (8), was found to
effectively suppress proliferation of HepG2 cells. The inhibitory
effect of minocycline at nanomolar concentrations on PARP-1 was
revealed nearly 10 years ago (9).
Minocycline, a semisynthetic tetracycline, is a
highly lipophilic molecule capable of infiltrating tissues and
blood (10). As a replacement of
earlier tetracycline, minocycline, which has a low propensity to
produce antibiotic resistance, is commonly used to treat many types
of infections. With further exploration of its functions and
mechanisms, particular aspects of anti-inflammation (11) and protection of the nervous system
(12–14), minocycline has found a wider and
deeper utilization. Only recently has research examined how
minocycline inhibits proliferation and promotes apoptosis (15). The dosage required to induce
effective apoptosis was tens of folds higher than the dosage used
for treatment of inflammation. Because of this, there is
substantial toxicity associated with minocycline treatment
(16). Therefore, the combination
of chemotherapeutics with minocycline is a new method by which to
combat the toxicity issues associated with high dosages of
minocycline alone.
Cisplatin crosslinks DNA in several different ways,
interfering with mitotic cell division. Cells undergo DNA damage
responses that induce apoptosis when repair is not possible.
Cisplatin elicits a complex response in the cell, including
apoptosis, DNA repair and drug resistance involving the ATR, p53,
p73 and MAPK pathways (17). The
effects of cisplatin treatment on apoptosis and DNA repair are
enhanced when used in combination with minocycline. Cisplatin
treatment is also limited by toxicity (18,19).
Herein, we present our in vitro and in
vivo results to show that minocycline treatment induces cell
cycle arrest and apoptosis. Additionally, when low-dose cisplatin
was used in combination with minocycline, the suppression of HCC
cell proliferation and apoptosis was enhanced above the level
presented when using a single agent alone.
Materials and methods
Cell lines, cell culture and transient
transfection
The human embryonic liver cell line L02 and HCC cell
lines Huh7 and HepG2 were purchased from the Typical Training
Content Preservation Committee Cell Bank, of the Chinese Academy of
Sciences (China). The cells were cultured in Dulbecco’s modified
Eagle’s medium (DMEM; Gibco-BRL, USA) supplemented with 10% fetal
bovine serum (FBS) and 1% penicillin/streptomycin at 37°C in a
humidified atmosphere with 5% CO2.
p27 siRNAs (RiboBio Co. Ltd., China) were used to
transiently transfect HepG2 cells. The transfection was performed
using Lipofectamine® 2000 (Invitrogen, Carlsbad, CA,
USA), according to the manufacturer’s instructions. Cells were
seeded at a density of 5×104 per well in a 6-well
plate.
Cell proliferation assay
Cells were seeded in 6-cm plates at 5×104
cells per well. The cells were cultured with minocycline and/or
cisplatin at different concentrations. Cell numbers were counted on
days 3, 6, and 9 after seeding. The assay was repeated three
times.
Soft agar colony formation assay
A soft agar colony formation assay was performed for
the transfected and control cells. Briefly, 1×103 cells
were seeded into a 6-well plate in a medium containing 0.3% noble
agar and cultured for 14 days. The number of colonies was
determined by direct counting using an inverted microscope (Nikon,
Japan).
Flow cytometry to analyze the cell cycle
and apoptosis
For cell cycle analysis, 1×105 HepG2
cells were washed three times in PBS and serum starved for 48 h.
The cells were stimulated with DMEM containing 10% FBS for 24, 48,
or 72 h. Cells (1×104) were analyzed from each sample on
a FACSCalibur flow cytometer (Becton Dickinson).
For the apoptosis analysis, 1×105 cells
per well were seeded in 6-well plates and incubated with DMEM
containing 10% FBS. After 48 h, cells were stained for 15 min with
FITC-Annexin V and propidium iodide in binding buffer and then
analyzed by flow cytometry within 1 h.
Xenograft assay
Male Balb/c nude mice (n =18) were provided by the
Experimental Animal Center of the Tongji Medical College. HepG2
cells (2×106) were injected into each of the bilateral
flanks of the mice. On day 14 post injection, the mice were
randomly divided into three groups: the minocycline, minocycline
and cisplatin, and control groups.
Minocycline at 6 mg/kg in 0.l ml was injected
intraperitoneally, and cisplatin was injected intraperitoneally at
0.3 mg/kg. The same volume of saline was injected into the control
group. The tumors were measured every 3 days and tumor sizes were
calculated on day 21.
TUNEL assay
Sections of tumor tissues were deparaffinized in
xylene, rehydrated, washed with PBS, and treated with 20 μg/ml of
Proteinase K (Roche) for 10 min at room temperature. Then reactions
were conducted using the TUNEL Apoptosis Assay Kit®
(Roche) according to the manufacturer’s instructions, and detected
using DAB. The percentage of TUNEL-positive cells was assessed in
five randomly selected fields for each tissue section.
Western blotting
Western blotting was performed with specific primary
antibodies against CDK2, CDK4, p53, PARP-1, p21, caspase-3,
casapase-8, ku80 and p27 (rabbit anti-human antibody; 1:1000;
Epitomics, Burlingame, CA, USA), followed by the appropriate
secondary HRP-conjugated antibodies (1:5000; Pierce, Rockford, IL,
USA). The protein bands were visualized using an enhanced
chemiluminescence detection system (Pierce).
Statistical analysis
ANOVA was performed to determine the statistical
significance among the groups. A P-value <0.05 was considered to
indicate a statistically significant result. All experimental data
were analyzed using the SPSS statistical software (version
16.0).
Results
Minocycline inhibits the proliferation of
HCC cell lines, but not the normal liver cell line
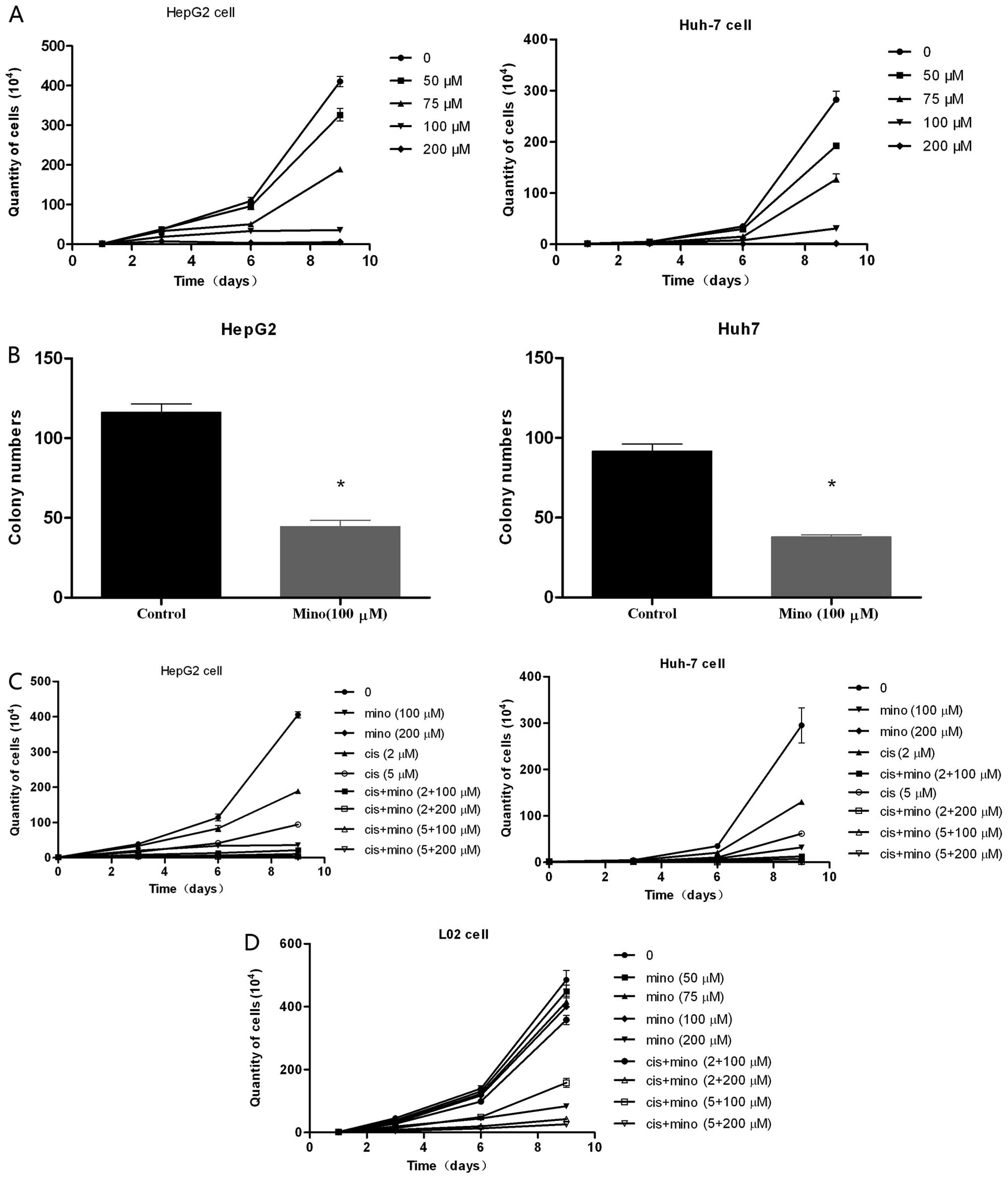
When HepG2 or Huh7 cells were cultured with
minocycline at concentrations of 50, 75, and 100 μM, cell
proliferation was significantly suppressed compared to the control
cells cultured without minocycline (Fig. 1A). The suppressive effects of
minocycline were dose-dependent, and the number of cells in the
treated group was significantly lower than the number of cells in
the control group on days 6 and 9. However, when the normal human
liver cell line L02 was cultured with minocycline at 100 μM, no
obvious suppressive effect was observed on days 3, 6 and 9
(Fig. 1D). At a concentration of
200 μM, minocycline strongly suppressed the proliferation of HCC
cells, but it also weakly suppressed L02 cell proliferation.
Furthermore, the soft agar assay revealed that minocycline
treatment significantly inhibited colony formation by 50.5–64.5%
compared to the control clones (Fig.
1B).
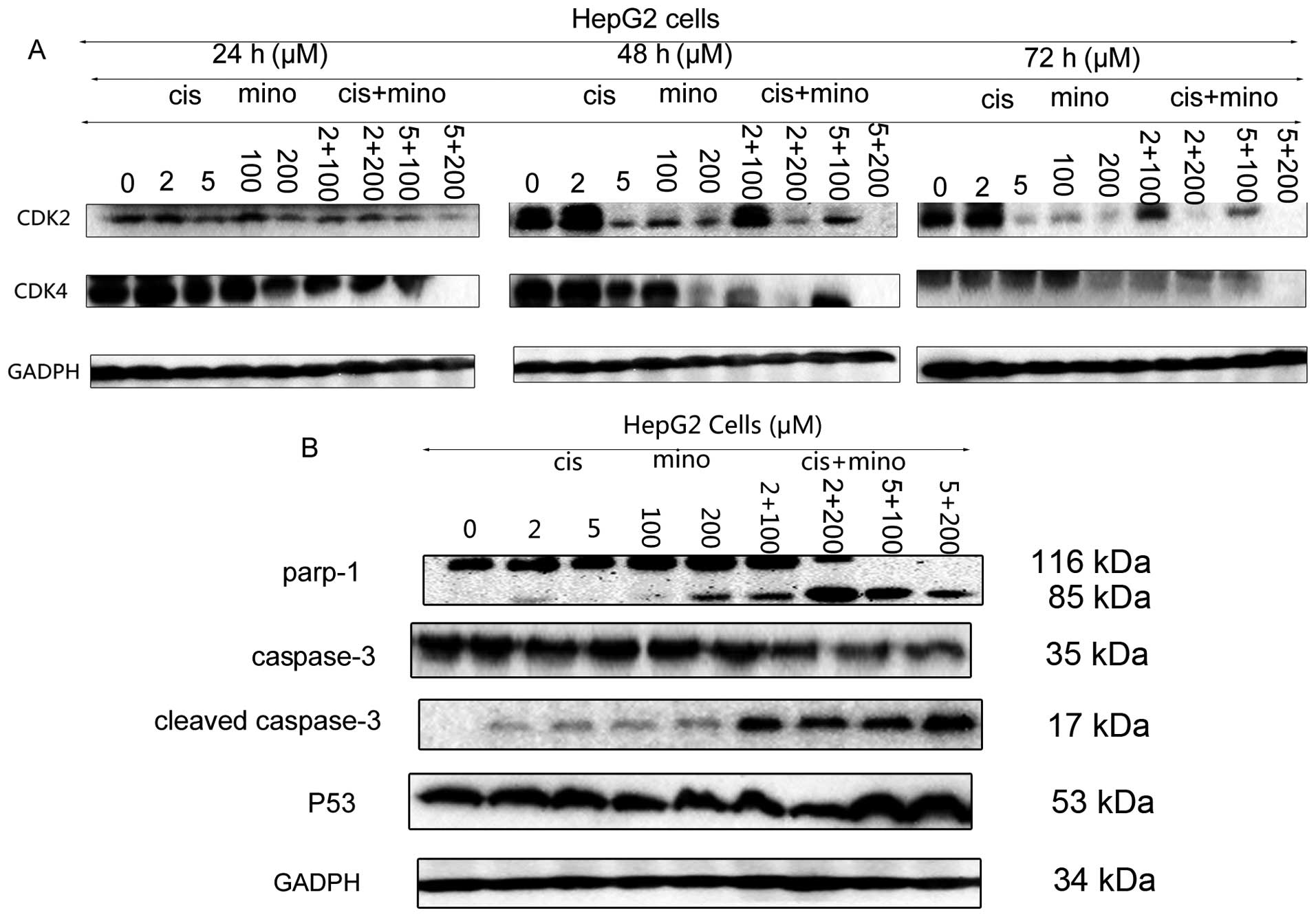
Inhibition of HCC cell proliferation by
minocycline is enhanced by low-dose cisplatin
When HepG2 cells were cultured with minocycline at a
concentration of 100 μM in combination with 2 μM or 5 μM cisplatin,
the level of suppression of cell proliferation was comparable to
that of treatment with 200 μM minocycline. The suppression was
greater than with treatment of 2 μM or 5 μM cisplatin alone
(Fig. 1C). There was no effect on
L02 cells. Although treatment of HepG2 cells with a concentration
of 200 μM minocycline in combination with 2 μM or 5 μM cisplatin
decreased proliferation even further, this concentration also
suppressed the proliferation of L02 cells.
Cisplatin enhances the effects of
minocycline in regards to increased cell apoptosis and cell cycle
arrest
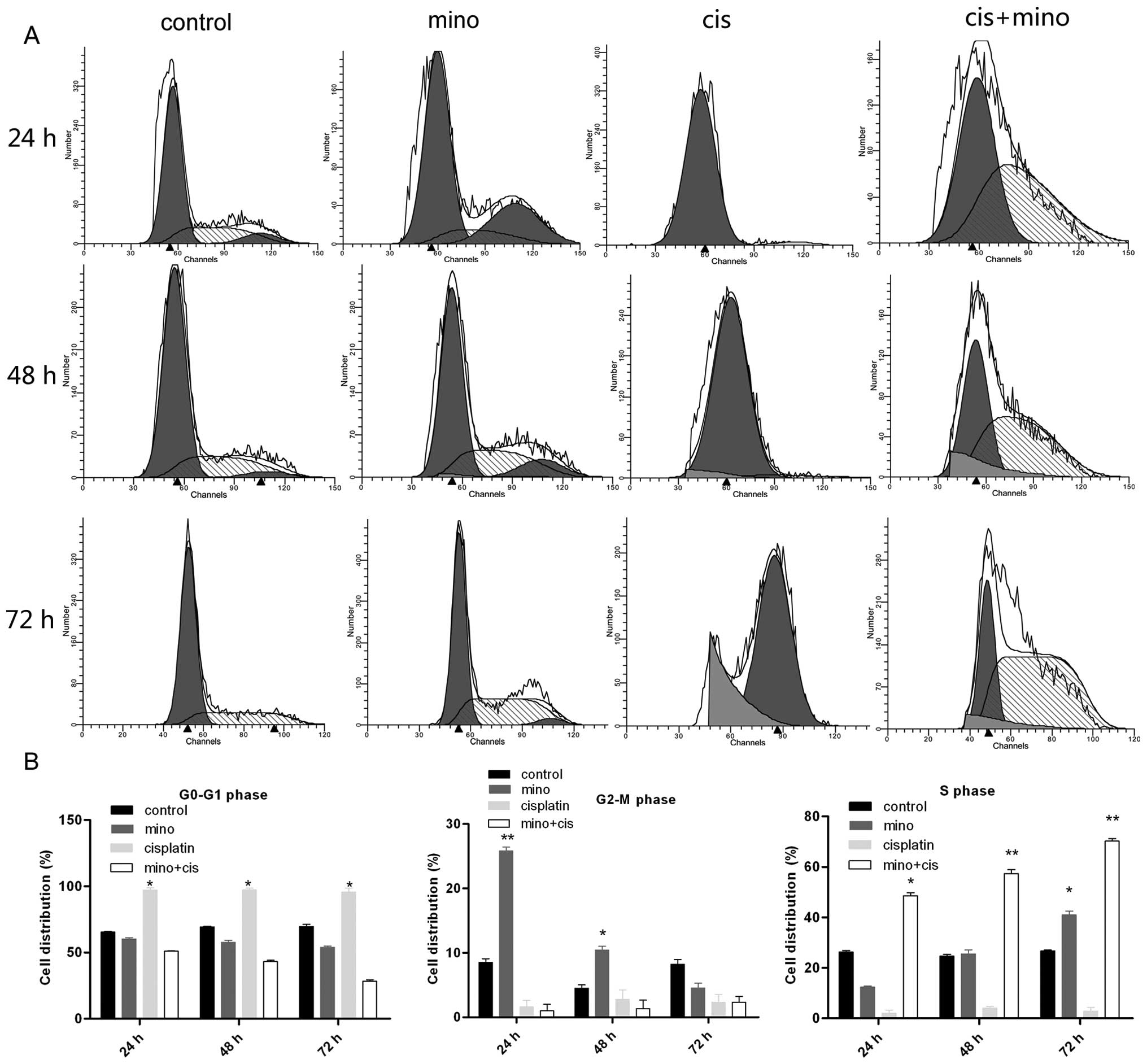
HCC cells were divided into four treatment groups:
the control; 100 μM minocycline; 2 μM cisplatin; and 100 μM
minocycline and 2 μM cisplatin groups. At 24 h, minocycline had no
effect on cell cycle arrest. However, at 48 and 72 h, more cells
were arrested in the S phase, with distribution rates of 49.1±0.54,
57.6±0.39 and 71.2±1.2% at the three time points, respectively.
Moreover, low-dose cisplatin induced G0/G1 cell cycle arrest in
almost all cells. The combination of minocycline and cisplatin
significantly accelerated the S phase arrest at all three time
points (Fig. 2A and B). Minocycline
treatment alone only induced S phase arrest after 72 h (40±0.94%),
while the combination of minocycline and cisplatin induced S phase
arrest after 24 h (49.1±0.54%). After 72 h, the combination
treatment induced S phase arrest at a rate of 71.2±1.2%.
Additionally, the G0/G1 phase arrest induced by cisplatin treatment
was eliminated with combination therapy, with rates decreasing from
97.6±1.38 to 4.1±1.22%.
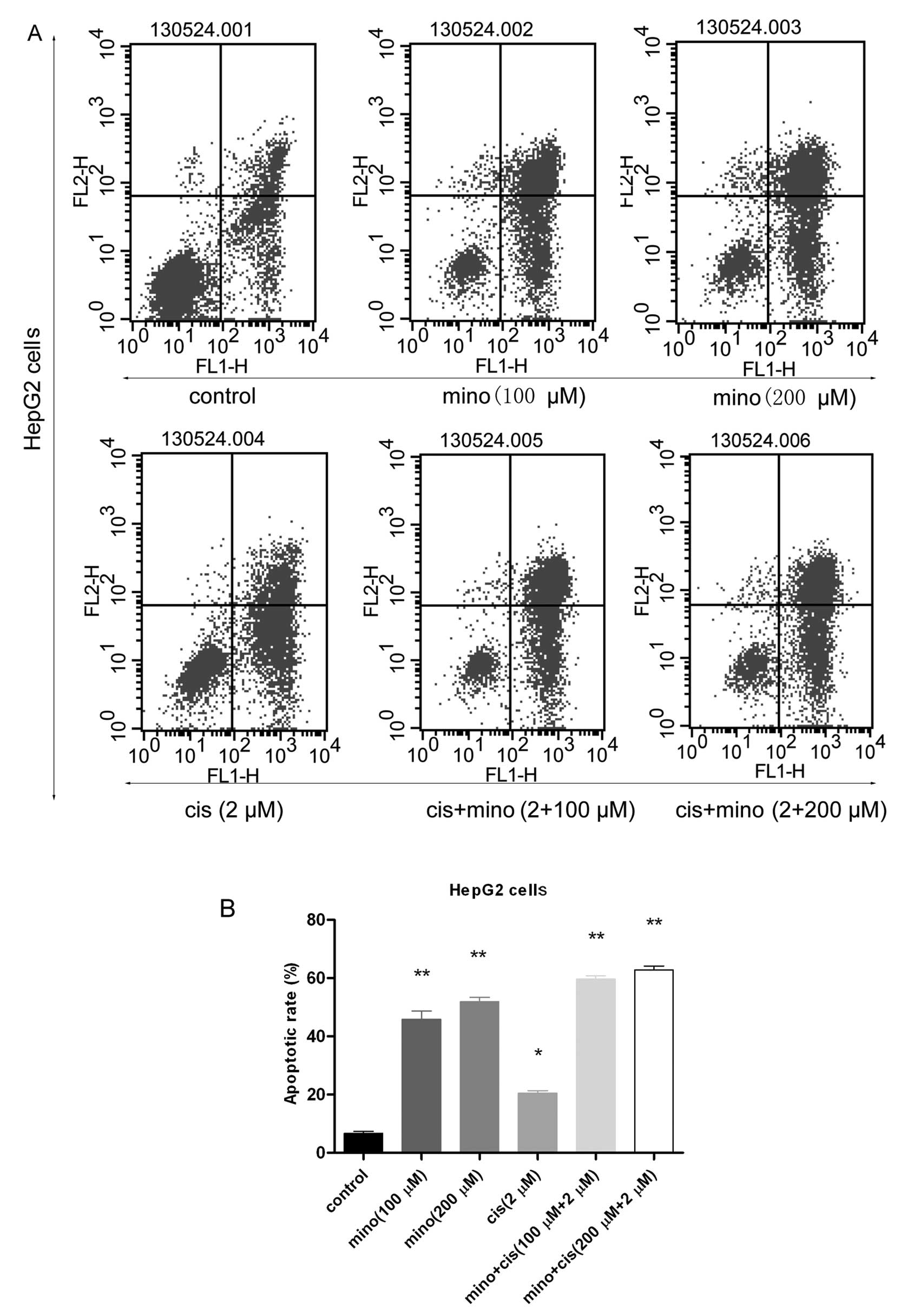
Flow cytometry was used to analyze the induction of
apoptosis in these cells. The apoptotic rates in the control, 100
μM minocycline, 200 μM minocycline, 2 μM cisplatin, 100 μM
minocycline and 2 μM cisplatin, and 200 μM minocycline and 2 μM
cisplatin groups were 5.81±0.15, 43.83±0.25, 51.44±1.52,
20.31±0.44, 60.26±1.72 and 67.34%, respectively (Fig. 3A and B). Late stage apoptotic rates
in the groups treated with 100 μM minocycline, 200 μM minocycline,
or 2 μM cisplatin were much higher than the rate in the control
group. In the group treated with 200 μM minocycline, the apoptotic
rate reached 51.44±1.52%, but in the group that was treated with
the combination of 100 μM minocycline and 2 μM cisplatin, the
apoptotic rate reached 60.26±1.72%. The group that was treated with
combination therapy also had a significantly higher rate of
apoptosis than the control group.
Minocycline induces apoptosis in HCC
cells through a mitochondrial-independent pathway and by
suppressing the DNA repair process
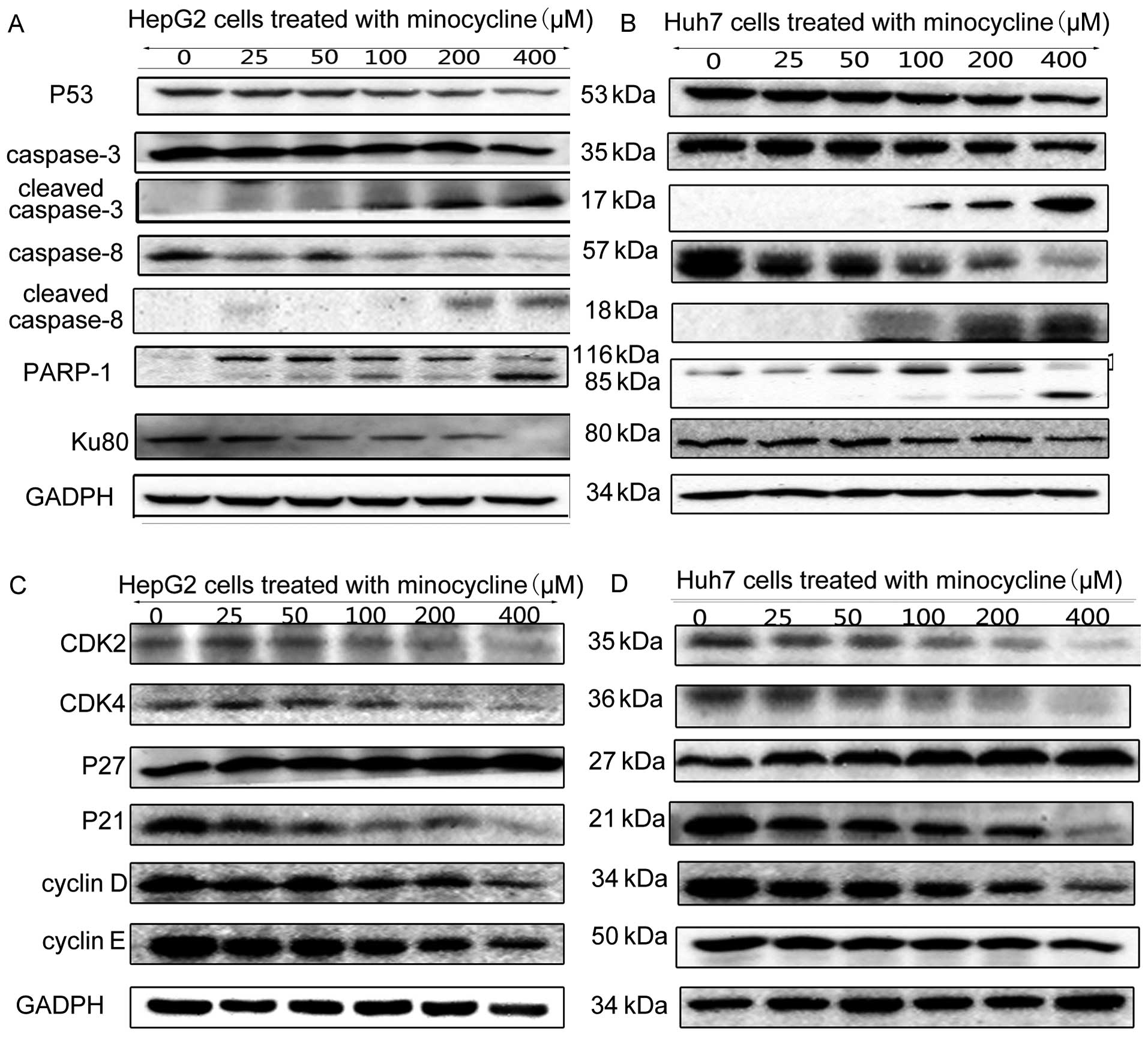
Cleaved caspase-3 was upregulated in HCC cells
treated with minocycline. Since cleaved caspase-8 was also
upregulated in HCC cells treated with minocycline, the extrinsic
apoptosis pathway was activated. The downregulation of PARP-1
elucidated another pathway associated with apoptosis induced by
minocycline. In addition to PARP-1, the expression of another DNA
repair factor, Ku80, was suppressed by minocycline treatment
(Fig. 4A and B).
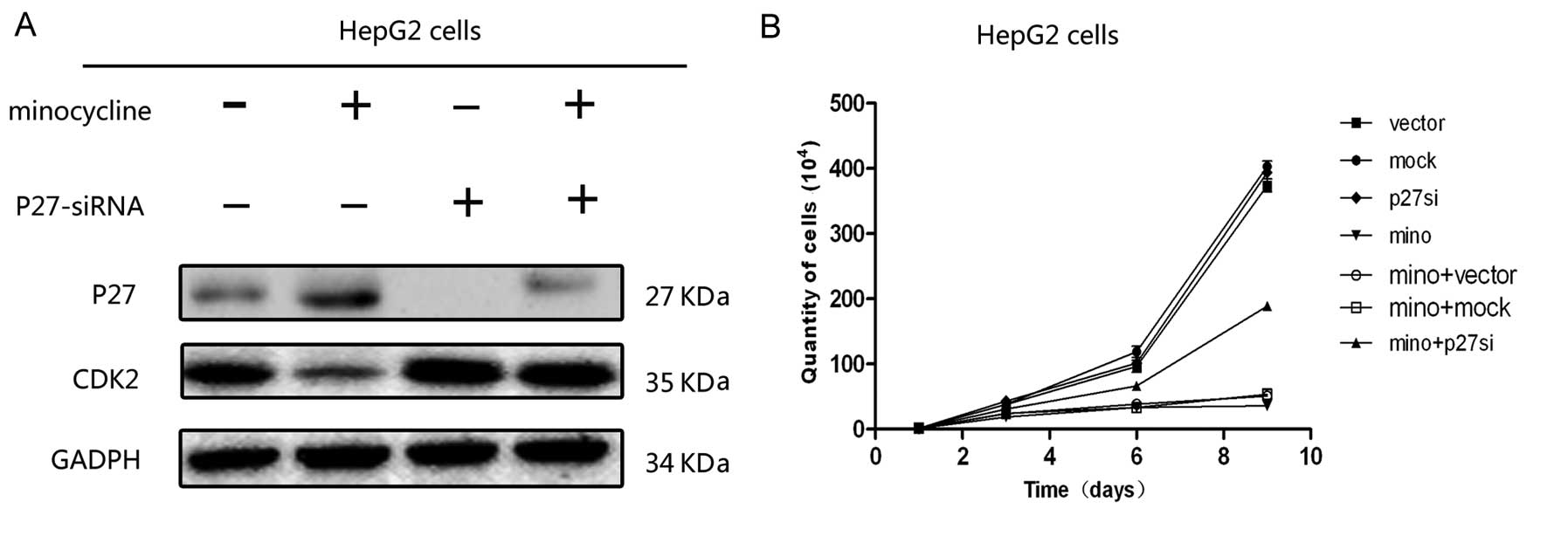
Minocycline induces HCC cell cycle arrest
through the p27 pathway
In HepG2 cells, the expression levels of CDK2, CDK4,
cyclin D, and cyclin E were significantly decreased in cells
treated with minocycline (Fig. 4C and
D), while minocycline treatment had no effect on the expression
of p53 and p21. However, the expression of p27 was dose-dependently
upregulated with minocycline treatment (Fig. 4C and D). To verify this result, p27
expression was knocked down by siRNA, When the p27 expression was
blocked, the minocycline-induced suppression of downstream factors,
such as CDK2, was reversed (Fig.
5A).
Cell proliferation assay demonstrated that the
proliferation of the HepG2 cells treated with minocycline was
significantly increased after transfection with p27-siRNA compared
with the mock-transfected cells or the vector-transfected control
cells on days 6 and 9 of culture (Fig.
5B). There was no significant difference in cell numbers
between the mock-, vector- and p27-siRNA-transfected cells which
were not treated by minocycline at all time points after
transfection.
Cisplatin enhances the effect of
minocycline on cell cycle arrest and apoptosis
Cisplatin can induce apoptosis through both the
extrinsic and intrinsic pathways. Cisplatin can also increase the
expression of p53, which activates p21 to induce cell cycle arrest
in the G0/G1 phase, and it also can downregulate the expression of
PARP-1 and suppress the expression of p27. In vitro assays
showed that the combination of cisplatin and minocycline enhanced
the levels of apoptosis in HCC cells when compared with the
application of a single drug. The expression of cleaved caspase-3
and p53 was upregulated following treatment with the combination
therapy when compared to that induced by treatment with minocycline
alone (Fig. 6A and B). Although
CDK2 and CDK4 expression levels were decreased, that was not enough
to induce the suppressive effect on HCC cells observed at early
time points. Therefore, we hypothesized that the S phase arrest
induced by the combination of the two drugs was caused mainly by
suppression of the PARP-1 DNA repair pathway. The downregulation of
PARP-1 verified our deduction (Fig. 6A
and B).
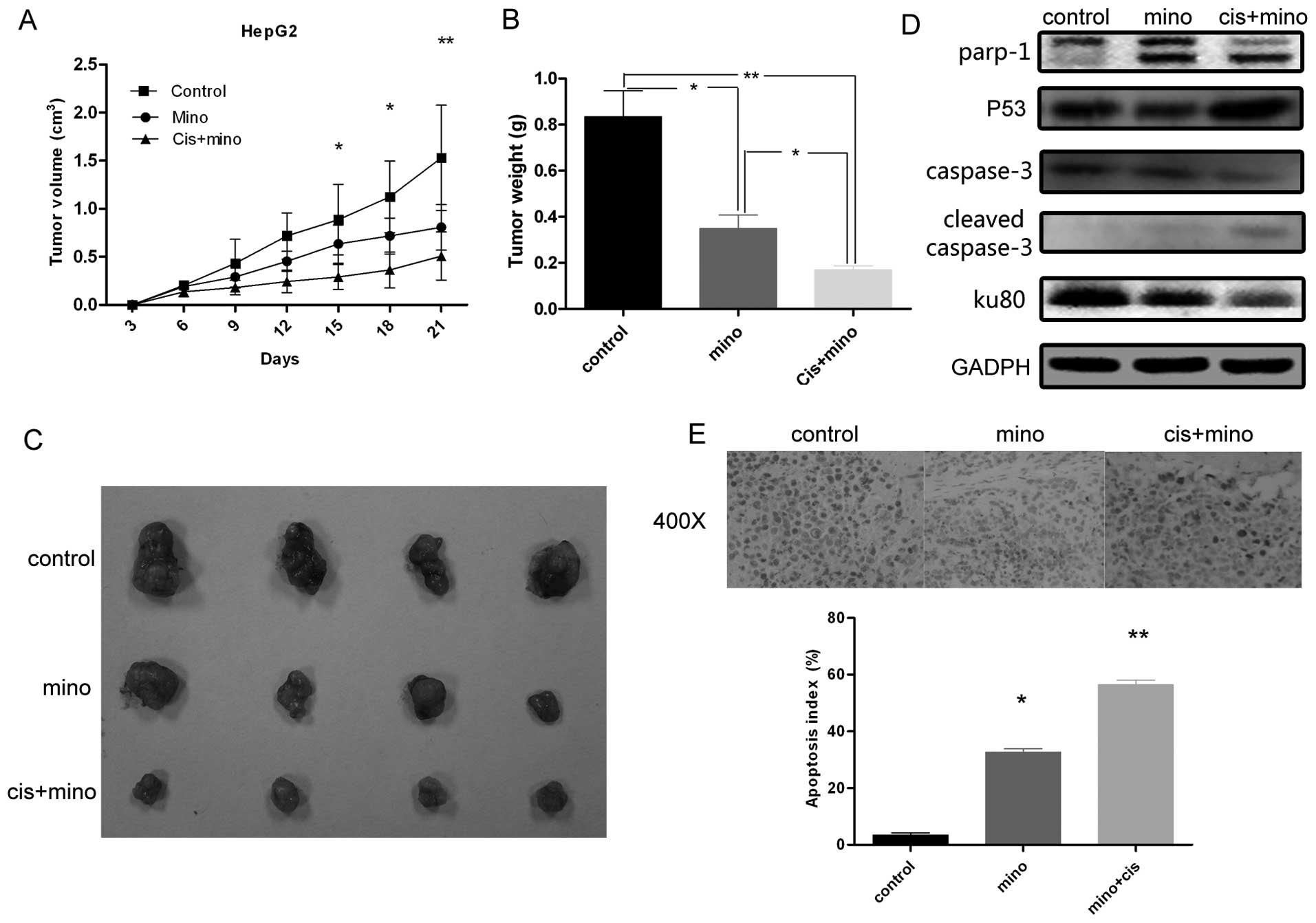
Minocycline and cisplatin treatment
inhibits HepG2 tumor growth in nude mice
Nude mice received injections of 2×106
HepG2 cells in each flank. Tumors formed after two weeks, and
tumor-bearing mice were randomly divided into three groups. As
shown in Fig. 7A, the mean volumes
of the tumors from mice treated with minocycline (0.70
cm3) or cisplatin in combination with minocycline (0.45
cm3) were significantly smaller than those from the
control group (1.66 cm3), and tumors from mice treated
with the combination therapy were significantly smaller than tumors
from mice treated with minocycline alone. In addition, when
compared to tumors from the control group, tumors from mice that
received any treatment grew at significantly slower rates and were
smaller at all time points examined from day 24 post-injection
(Fig. 7A). The mass of tumors from
mice that received minocycline alone or combination therapy was
significantly lower than that of tumors from mice in the control
group (Fig. 7B and C), and the mass
of tumors from the mice that received combination therapy was
significantly smaller than that from tumors from the mice treated
with minocycline alone. Western blot analysis (Fig. 7D) verified the effect of minocycline
and/or cisplatin on the expression of PARP-1, caspase-3, and Ku80
in vitro. As shown by TUNEL assay, the rate of apoptosis was
calculated. As shown in Fig. 7E,
treatment with minocycline alone or minocycline in combination with
cisplatin resulted in a significantly higher apoptotic index (32.7
and 56.4% respectively), when compared to the control group.
Discussion
Minocycline is a second-generation tetracycline
antibiotic with favorable pharmacological properties. It is also
used as an anti-inflammatory and neuroprotective agent. In this
report, we present novel and direct evidence that minocycline
markedly induces apoptosis and cell cycle arrest in liver cancer
cells.
Minocycline inhibits PARP-1 expression at nanomolar
concentrations (9), and this
research suggests that minocycline could be useful to treat HCC.
Based on a previous study (7), an
inhibitor of PARP-1 suppressed the proliferation of HCC cells. We
hypothesized that minocycline could function similarly, and should
be investigated further given its ability to infiltrate tissues and
blood. We treated several HCC cell lines with minocycline at
different concentrations. We found that the proliferation of liver
cells was suppressed when the suitable dosage of minocycline was
applied. However, minocycline had no suppressive effect on the
proliferation of L02 cells at the same dosage. Moreover,
minocycline treatment changed the morphology of HepG2 and Huh7
cells from smooth and plump to shrinking. The cell nucleus also
appeared to undergo distinctive changes, including pyknosis and
karyolitic and apoptotic body formation, a typical feature of
apoptosis (20). Using flow
cytometry, we determined that minocycline treatment induced S phase
arrest with an increase in concentration, especially after 72 h.
Moreover the late stage apoptotic cells increased with its
concentration.
Western blot analysis was used to explore the
underlying mechanism responsible for this phenomenon. Treatment
with increasing concentrations of minocycline increased the
expression levels of CDK2, CDK4, cyclin D, and cyclin E, which
explains the observed cell cycle arrest noted with treatment
(21). We then examined the
expression of the upstream factors of CDK2 and CDK4: p53, p21 and
p27. Minocycline treatment had no effect on the expression of p53
or p21, the downstream factor of p53 (22). However, p27, which also regulates
CDKs and cyclins (23,24), was induced following minocycline
treatment, and the suppression of CDKs and cyclins by minocycline
was reversed by blocking p27 expression. We, therefore, concluded
that minocycline regulates cell cycle arrest by activating the p27
pathway, but the mechanism by which minocycline induced apoptosis
of HCC cell lines remain unclear.
We then tested the effects of minocycline treatment
on other apoptotic factors induced by mitochondria (25,26),
Minocycline did not affect the intrinsic mitochondrial apoptotic
pathway. Minocycline treatment induced both the expression of
cleaved caspase-3 and cleaved caspase-8, which indicated the
activation of the extrinsic apoptotic pathway (27,28).
A decrease in PARP-1 expression, as noted with
minocycline treatment, has been shown to affect both cell cycle
arrest and apoptosis in other pathways (8,29,30).
Through suppressed PARP-1, minocycline induced HCC cells into
apoptosis directly.
Although minocycline induces apoptosis and cell
cycle arrest, the high doses needed also have toxicity to normal
cells (16). Therefore, we wanted
to use another drug in combination with minocycline so that we
could reduce the dosage but still have a similar effect on cancer
cell death. Cisplatin can induce apoptosis through both the
extrinsic and intrinsic apoptotic pathways (31,32),
which has some overlap with the functions of minocycline. Then we
chose it to continue our research. The combination of cisplatin and
minocycline enhanced the apoptosis of HCC cells when compared with
treatment with either minocycline or cisplatin alone. The
expression of p53 was also increased, suggesting that the
combination of these two drugs may have synergy in inducing
apoptosis.
Based on the findings of other studies, cisplatin
induced the expression of p21 while decreasing p27 expression
(33), thereby antagonizing the
suppressive effects of minocycline on CDK2 and CDK4 complexes.
Cisplatin treatment induced G0/G1 arrest, but the combination of
minocycline and cisplatin induced S phase arrest. Minocycline
induced S phase arrest after 72 h, thus we concluded that p27
upregulation was not the primary mechanism. The strong suppressive
effect of this combination on PARP-1 expression blocked the repair
of HCC cells and inhibited damaged cells from entering the G2/M
phase, thus inducing cell accumulation in the S phase and
apoptosis. This synergy of drugs was enhanced over time. In the
animal model, subcutaneous tumors were implanted in Balb/c nude
mice. The tumors in the group treated with the combination therapy
were much smaller than the tumors in both groups treated with a
single drug and the control group, by volume and weight. Western
blotting and TUNEL assay of tumors showed the same effect on
apoptosis and proliferation with in vitro experiments.
Then KLF17, one of the typical factors associated
with HCC metastasis (34), was
tested by western blotting. Minocycline also upregulated its
expression (data not shown). Minocycline also had some effect on
inhibiting HCC metastasis.
In conclusion, minocycline inhibited HCC cell
proliferation by inducing apoptosis and cell cycle arrest through
extrinsic apoptosis, P27 and PARP-1. In addition, a low dose of
cisplatin enhanced minocycline apoptosis capacity. In addition,
cisplatin enhanced S phase arrest induced by minocycline through
suppressed PARP-1. Finally, cisplatin upregulated the expression of
P53 which was not affected by minocycline. These data need further
evaluation in clinical conditions and in other types of cancer,
particularly in cancers that have a tight relation with PARP-1
expression.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Yoshimoto S, Loo TM, Atarashi K, et al:
Obesity-induced gut microbial metabolite promotes liver cancer
through senescence secretome. Nature. 499:97–101. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Min L, Ji Y, Bakiri L, et al: Liver cancer
initiation is controlled by AP-1 through SIRT6-dependent inhibition
of survivin. Nat Cell Biol. 14:1203–1211. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xu X, Fan Z, Kang L, et al: Hepatitis B
virus X protein represses miRNA-148a to enhance tumorigenesis. J
Clin Invest. 123:630–645. 2013.PubMed/NCBI
|
|
5
|
Maluccio M and Covey A: Recent progress in
understanding, diagnosing, and treating hepatocellular carcinoma.
CA Cancer J Clin. 62:394–399. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu L, Cao Y, Chen C, et al: Sorafenib
blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and
induces tumor cell apoptosis in hepatocellular carcinoma model
PLC/PRF/5. Cancer Res. 66:11851–11858. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang SH, Xiong M, Chen XP, Xiao ZY, Zhao
YF and Huang ZY: PJ34, an inhibitor of PARP-1, suppresses cell
growth and enhances the suppressive effects of cisplatin in liver
cancer cells. Oncol Rep. 20:567–572. 2008.PubMed/NCBI
|
|
8
|
Yu S-W, Wang H, Poitras MF, et al:
Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by
apoptosis-inducing factor. Science. 297:259–263. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Alano CC, Kauppinen TM, Valls AV and
Swanson RA: Minocycline inhibits poly(ADP-ribose) polymerase-1 at
nanomolar concentrations. Proc Natl Acad Sci USA. 103:9685–9690.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yong VW, Wells J, Giuliani F, Casha S,
Power C and Metz LM: The promise of minocycline in neurology.
Lancet Neurol. 3:744–751. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Le CH, Morales A and Trentham DE:
Minocycline in early diffuse scleroderma. Lancet. 352:1755–1756.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhu S, Stavrovskaya IG, Drozda M, et al:
Minocycline inhibits cytochrome c release and delays progression of
amyotrophic lateral sclerosis in mice. Nature. 417:74–78. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zink MC, Uhrlaub J, DeWitt J, et al:
Neuroprotective and anti-human immunodeficiency virus activity of
minocycline. JAMA. 293:2003–2011. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Stefanova N, Bücke P, Duerr S and Wenning
GK: Multiple system atrophy: an update. Lancet Neurol. 8:1172–1178.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pourgholami MH, Ataie-Kachoie P, Badar S
and Morris DL: Minocycline inhibits malignant ascites of ovarian
cancer through targeting multiple signaling pathways. Gynecol
Oncol. 129:113–119. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bouwman P and Jonkers J: The effects of
deregulated DNA damage signalling on cancer chemotherapy response
and resistance. Nat Rev Cancer. 12:587–598. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Siddik ZH: Cisplatin: mode of cytotoxic
action and molecular basis of resistance. Oncogene. 22:7265–7279.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fuertes MA, Alonso C and Pérez JM:
Biochemical modulation of Cisplatin mechanisms of action:
enhancement of antitumor activity and circumvention of drug
resistance. Chemical Rev. 103:645–662. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang D and Lippard SJ: Cellular processing
of platinum anticancer drugs. Nat Rev Drug Discov. 4:307–320. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Haubrich WS: Apoptosis. Gastroenterology.
119:18052000. View Article : Google Scholar
|
|
21
|
Lord CJ and Ashworth A: The DNA damage
response and cancer therapy. Nature. 481:287–294. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Abbas T and Dutta A: p21 in cancer:
intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nakayama K, Nagahama H, Minamishima YA, et
al: Targeted disruption of Skp2 results in accumulation of cyclin E
and p27(Kip1), polyploidy and centrosome overduplication. EMBO J.
19:2069–2081. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
le Sage C, Nagel R, Egan DA, et al:
Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222
promotes cancer cell proliferation. EMBO J. 26:3699–3708.
2007.PubMed/NCBI
|
|
25
|
Penninger JM and Kroemer G: Mitochondria,
AIF and caspases - rivaling for cell death execution. Nat Cell
Biol. 5:97–99. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Virag L, Robaszkiewicz A, Vargas JM and
Oliver FJ: Poly(ADP-ribose) signaling in cell death. Mol Aspects
Med. 34:1153–1167. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Günther C, Martini E, Wittkopf N, et al:
Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and
terminal ileitis. Nature. 477:335–339. 2011.PubMed/NCBI
|
|
28
|
Jin Z, Li Y, Pitti R, et al: Cullin3-based
polyubiquitination and p62-dependent aggregation of caspase-8
mediate extrinsic apoptosis signaling. Cell. 137:721–735. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Stilmann M, Hinz M, Arslan SÇ, Zimmer A,
Schreiber V and Scheidereit C: A nuclear poly(ADP-Ribose)-dependent
signalosome confers DNA damage-induced IkappaB kinase activation.
Mol Cell. 36:365–378. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Midorikawa R, Takei Y and Hirokawa N: KIF4
motor regulates activity-dependent neuronal survival by suppressing
PARP-1 enzymatic activity. Cell. 125:371–383. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vondálová Blanárová O, Jelinková I, Szöor
A, et al: Cisplatin and a potent platinum(IV) complex-mediated
enhancement of TRAIL-induced cancer cells killing is associated
with modulation of upstream events in the extrinsic apoptotic
pathway. Carcinogenesis. 32:42–51. 2011.PubMed/NCBI
|
|
32
|
Barton C, Davies D, Balkwill F and Burke
F: Involvement of both intrinsic and extrinsic pathways in
IFN-gamma-induced apoptosis that are enhanced with cisplatin. Eur J
Cancer. 41:1474–1486. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Qin LF and Ng IO: Induction of apoptosis
by cisplatin and its effect on cell cycle-related proteins and cell
cycle changes in hepatoma cells. Cancer Lett. 175:27–38. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu FY, Deng YL, Li Y, et al:
Down-regulated KLF17 expression is associated with tumor invasion
and poor prognosis in hepatocellular carcinoma. Med Oncol.
30:4252013. View Article : Google Scholar : PubMed/NCBI
|