Introduction
Numerous factors involved in the most deadly
consequences of carcinogenesis and metastasis, are poorly
understood. Interactions between motile stem cells and other cell
types have been proposed to contribute to the mechanisms of
carcinogenesis and acquisition of metastatic ability. Cell-specific
in vivo models to explore these mechanisms could provide
valuable insights into potential therapeutic targets. A
well-established multi-step hypothesis has emerged from
characterization of the hallmarks and genetic interactions
associated with cancer; that accumulation of specific cellular
genetic mutations can lead to carcinogenesis, malignancy and
metastasis (1,2). Although mutations surely play a role
in carcinogenesis, clearly the initiation and progression of cancer
involves more than genetic alterations alone. Other critical
factors include the tumor microenvironment, inflammation (3), interactions with tumor stromal cells
(4), [including myofibroblasts
(5)] and recruitment of mesenchymal
stem cells (MSCs) to the tumor microenvironment (6,7). The
recent discoveries of stem cells and cancer stem cells and
investigations of their potential roles in cell fusion,
carcinogenesis and metastasis have led to novel perspectives and
approaches concerning the mechanisms of metastasis.
He et al (8)
proposed a stem cell fusion model in which altered pre-malignant
cells (including benign tumor cells) fuse with bone marrow-derived
MSCs, to form a hybrid cancer cell with the hallmarks of metastatic
cancer (2), and expressing other
common phenotypes associated with malignancy. This hypothesis
suggested that cell fusion and gene mutations are both important
components of comprehensive carcinogenesis mechanisms. The
hypothesis provided explanations for: i) the remarkable
similarities between malignant cells and stem cells; ii) the
ability of non-mutagens to be carcinogens; iii) the ability of
non-mutagenic processes, such as wound healing or chronic
inflammation, to promote malignant transformation and iv) the
generation of aneuploidy and other common characteristics of
malignant cancer cells. The observation of spontaneous or induced
fusion of highly motile macrophages, monocytes, tissue stem cells
or bone marrow-derived mesenchymal cells with differentiated or
tumor cells, yielding hybrid cells with altered properties, has
been reported (9). The fusion of
various types of cells as a driver of metastatic cancer has been
discussed in recent excellent reviews including Lu and Kang
(10,11), Bjerkvig et al (12), and Pawelek and Chakraborty (13).
In the present study, we performed in vitro
and in vivo experiments to explore the acquisition of
enhanced malignant characteristics by cells derived from the
polyethylene glycol (PEG)-induced fusion of rat bone marrow-derived
MSCs and HepG2 cells. To distinguish between the parental cells,
rat MSCs were labeled with DiI, and HepG2 cells were labeled by
transfection with a plasmid expressing enhanced green fluorescent
protein. The resulting dual-labeled fused progeny cells were
distinguished from parental cells and collected by flow cytometry.
Several phenotypic characteristics of malignant cells [aneuploidy,
in vitro migratory and invasive ability,
epithelial-mesenchymal transition (EMT) markers and regulatory
factors, matrix metalloproteinase (MMP)2 and 9 activity and in
vivo formation of metastases] were compared in the parental and
fused cells. The experimental observations reported in this study
support our previously proposed mechanism and the role of the
fusion of stem cells with altered cells in the development of
metastatic ability.
Materials and methods
Mesenchymal stem cell isolation,
culturing and identification
Ten male Sprague-Dawley rats, 1 month in age,
(Experimental Animal Center, Nanjing Medical University Center)
with a body mass of 80–100 g were sacrificed and sterilized in 75%
ethanol for 10 min. Bone marrow was isolated under sterile
conditions from femurs and tibias as previously described (14). To select a purified population of
MSCs, briefly, bones were isolated and marrow was flushed out with
serum-free (s-f) L-DMEM and filtered through a 0.1-μm filter. Cells
were propagated by incubation in standard growth medium.
Non-adherent cells were removed after 4 and 24 h and every 2–3 days
thereafter by gently washing with medium. Cultures were passed at
80–90% confluency 3–4 times by detachment with 0.25% trypsin for 1
min. Flow cytometry was performed on trypsin-detached cells after
washing and re-suspension with phosphate-buffered saline (PBS) 3
times. Antibody reagents for cell surface markers, including CD34,
CD45, CD90 and CD105 (eBioscience, USA) were prepared according to
the manufacturer’s instructions and added to separate cell
suspensions. The mixtures were incubated 20 min in the dark at room
temperature (rt). The presence of specific cell markers was
identified by flow cytometric analysis.
DiI labeling of MSC membranes
Confluent monolayers of MSCs were labeled for 3 h at
37°C with 400 μg of DiI (Beyotime, China) in 20 ml Dulbecco’s
modified Eagle’s medium (DMEM), following the manufacturer’s
instructions. DiI is a lipophilic carbon cyanine dye characterized
by specificity, high sensitivity, and strong fluorescence on
binding to plasma membranes, facilitating the observation of whole
cells. The labeled cells were washed in PBS and used in the cell
fusion protocol.
HepG2 cell culture and transfection
Human HepG2 cells (Shanghai Institutes for
Biological Sciences, CAS) have a low metastatic phenotype. They
were cultured in high glucose DMEM (H-DMEM, 4.5 g/l glucose; Gibco,
USA), 10% fetal bovine serum (FBS), 2 mM L-glutamine and 100 U/ml
penicillin/streptomycin. The cells were transfected with pEGFP-N1
plasmid, which encodes a gene for green fluorescent protein
(HepG2-eGFP). Transfected cells were selected for 4 weeks in 500
μg/ml G418 (Invitrogen, USA). The fluorescence, monitored every 3
days, was retained during the selection and subsequent experiments.
Then the HepG2-eGFP cells were identified and collected by flow
cytometry.
Cell fusion and selection
A modification of the procedure of Köhler and
Milstein was used (15). Briefly,
5×105 MSCs were mixed with 1×105 HepG2 cells
before washing 3 times by re-suspension in 30 ml s-f medium and
centrifugation for 5 min at 1,000 rpm at rt. One milliliter of
polyethylene glycol, PEG2000 (Sigma, USA) was slowly added to the
washed cell pellet. Thirty milliliters of s-f DMEM was added to the
cell-PEG2000 mixture after 1 min. The suspension was incubated in a
37°C water bath for 10 min to allow fusion. The cells were
pelleted, re-suspended in serum-containing DMEM, plated in standard
growth medium, cultured 3 days, and sorted by flow cytometry to
identify and collect the fused cells containing both GFP and DiI.
Fused cells in culture retained their viability for 1 month. The
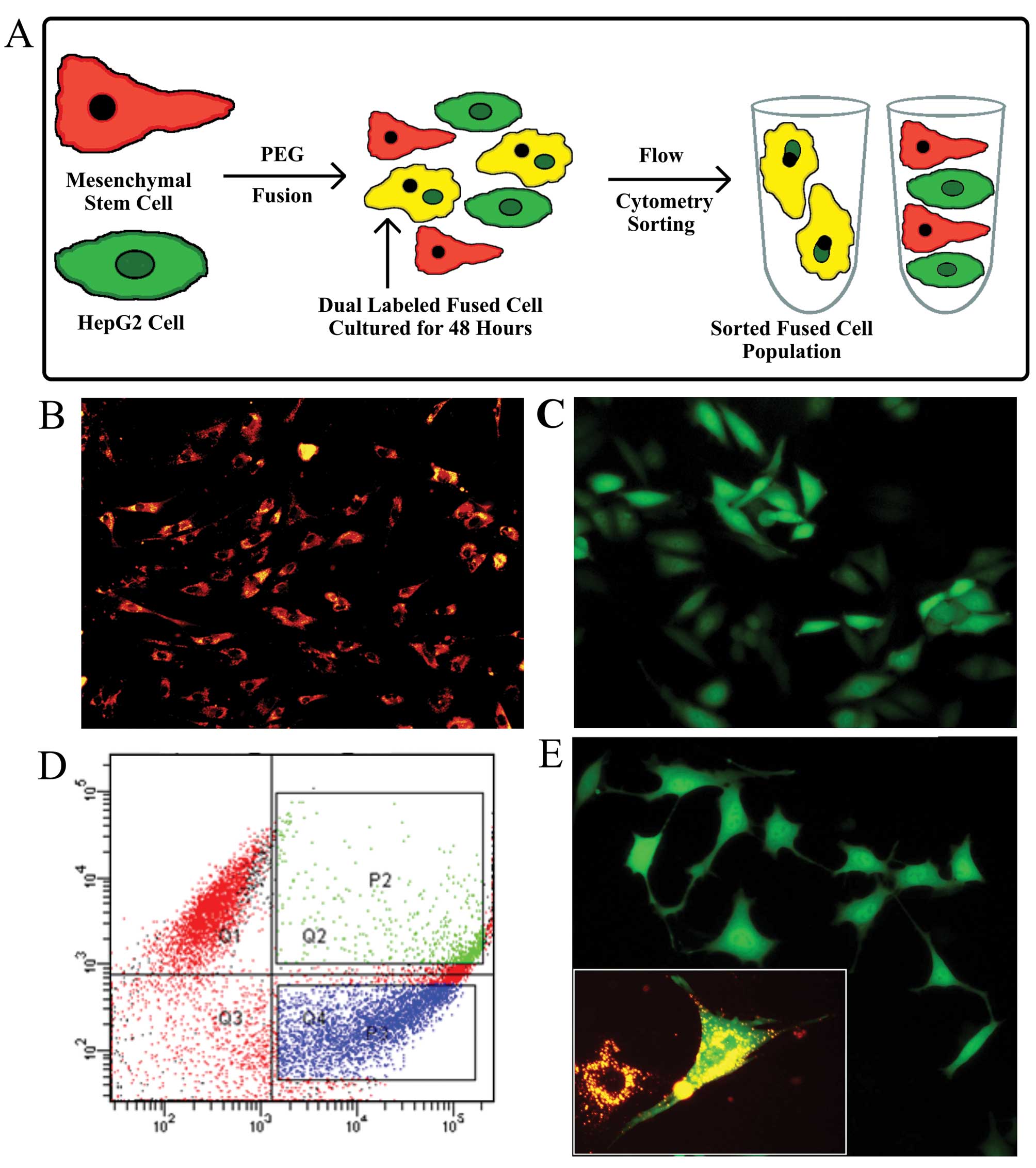
cell fusion protocol used in this report is illustrated in Fig. 1A.
Chromosome analysis
Log phase HepG2-eGFP cells, MSCs and fused cells,
suspended in s-f medium, were treated with 0.2 μg/ml colchicine and
incubated for 3 h. The cells were then treated with 75 mM KCl for
30 min in a 37°C water bath, fixed with methanol:acetic acid (3:1)
mixture for 1 h at rt, dried on pre-cooled slides and stained with
Giemsa solution (Invitrogen). Ten mitotic images were selected for
each cell type, and the number of chromosomes in each image was
counted using an inverted microscope (Olympus) with a ×10 objective
and a ×100 oil immersion lens. The average chromosome number/cell
was calculated for each cell type at 2 and 4 weeks after
fusion.
Assays of cellular invasion and
migration
Assays were performed in triplicate in Transwell
dishes with 8 μm pore size inserts. Standard growth medium
containing 20% FBS was placed in the bottom chambers of the
Transwell dishes. Migrating or invading cells were counted at ×100
magnification. One-way ANOVA was used to calculate p-values for the
cell counts.
Migration assays
A suspension of 20,000 HepG2 cells, MSCs or fused
cells in s-f DMEM was layered on uncoated inserts seated in
Transwell dishes (Millipore, USA). Dishes were incubated for 16 h
at 37°C. The medium was removed from the upper and bottom chambers.
The inserts were fixed 1 h with 4% paraformaldehyde and stained
with 0.1% crystal violet at rt. Cells remaining on top of the
insert were removed by scraping. Seven randomly selected fields of
the invaded cells on the bottom of the insert were chosen and the
number of invasive cells was counted.
Invasion assays
Transwell inserts were coated with 45 μl of a 1:8
dilution of Matrigel (Becton-Dickinson, USA) in s-f DMEM and
incubated for 1 h to gel. A suspension of 40,000 cells in s-f DMEM
was layered onto the inserts as described above and incubated for
36–48 h. The invading cells on the bottom of the inserts were
stained, and seven randomly selected microscopic fields were
counted.
Western blot assay
The fused cells, HepG2 cells (included for
comparison with the HepG2-eGFP cells), HepG2-eGFP cells and MSCs
were harvested and lysed on ice for 10 min in RIPA lysis buffer (50
mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.2 mM EDTA, 1 mM
phenylmethylsulfonyl fluoride, 1% NP-40 and 0.25% deoxycholate;
Beyotime, China). Protein concentration was assayed with a BCA
assay kit (Pierce, USA) according to the manufacturer’s
instructions. The assay was as previously described (16) with the following modifications.
Fifty micrograms of proteins from each cell type were denatured 5
min at 95°C in sample buffer (250 mM Tris, pH 6.8, 10% SDS, 50%
glycerol, 5% mercaptoethanol and 0.5 mM bromphenol blue) and
separated by electrophoresis on 10% SDS-PAGE gels. Equiloading of
samples was detected with a 1:1,000 dilution of β-actin antibody
(Cell Signaling), as per the manufacturer’s protocol. Proteins were
transferred onto PVDF membranes (Millipore). Membranes were blocked
with 5% fat-free milk at rt for 2 h and cut. Sections were then
incubated at 4°C overnight with a 1:1,000 dilution of one of the
following primary monoclonal antibodies: anti-vimentin (GeneTex),
anti-E-cadherin, anti-Twist1, anti-Snail and with anti-β-actin (all
from Cell Signaling) at 4°C. After three washes for 10 min in PBS
supplemented with 0.05% Tween-20 (PBST), the membrane was incubated
with a peroxidase-conjugated secondary antibody (Zhongshan Golden
Bridge Biotechnology) for 2 h at rt. Enhanced chemiluminescence
reagent (Pierce) was used for detection as per the manufacturer’s
instructions. Gel images were scanned into a file and processed
with PowerPoint software.
Gelatin zymography assays
For each of the three cell types, 2×105
cells/well in a 12-well plate were incubated for 12 h at 37°C. The
medium was removed, and 500 μl s-f DMEM was added to each well.
After incubation for 24 h, the medium from each plate was harvested
and pooled and separated by electrophoresis on a 10% SDS-PAGE gel
containing 1 mg/ml gelatin to detect MMP2 and 9 activities. After
electrophoresis, the gels were equilibrated in 2.5% Triton X-100
and incubated in renaturing buffer [50 mM Tris-HCl (pH 7.5), 10 mM
CaCl2, 150 mM NaCl, 1 mM ZnCl2 and 0.02%
NaN3] for 42 h at 37°C. The gel was stained with
Coomassie R250 and destained until clear bands of MMP activity were
visible against the dark blue background. Activities of MMP2 and 9
were identified by comparing band mobility with molecular weight
standards. Images were obtained by scanning gels with Alpha
Innotech gel imaging systems.
Xenograft assay
Six-week-old BALB/c nude mice were purchased from
Shanghai Laboratory Animal Company. Mice were maintained in a
pathogen-free environment, under temperature-controlled conditions.
Cages, bedding and drinking water were autoclaved and changed
regularly. Food was sterilized by irradiation. Mice were maintained
at a daily cycle of 12-h periods of alternating light and dark.
The fused cells, HepG2 cells and MSCs were
harvested, counted and centrifuged. For each cell type,
2.4×107 cells were suspended in 240 μl of Matrigel
(Becton-Dickinson), and placed on ice. Twenty-one BALB/c nude male
mice were randomly divided into three groups of seven mice, one
group for each cell type. Before inoculation, all mice were
anesthetized by intraperitoneal injection with 1% pentobarbital
sodium (10 μl/g body weight) (Sigma, Germany). The peritoneal
cavity was opened and the left liver lobe was exposed. The left
liver lobe of each mouse was injected with 30 μl of Matrigel
containing 3×106 cells. Implantation was finished within
6 h. The mice received gentamicin in drinking water (80,000 U/l) up
to 1 week following implantation. Body mass and survival rate were
calculated each week (data not shown). Surviving mice were
sacrificed after 10 weeks and necropsied to assess metastatic tumor
formation. The livers, lungs, kidneys and brains of mice in each
group were isolated.
Histological preparation of the
specimens
The liver, lung, brain and kidney were removed and
fixed in 10% formalin, dehydrated and embedded in paraffin.
Sections of 4-μm thickness were cut and stained with hematoxylin
and eosin (H&E) for microscopic observation by a
pathologist.
Results
Fusion of HepG2-eGFP cells with
DiI-labeled MSCs generates dual-labeled progeny cells
MSCs were selected from the total population of bone
marrow-derived cells based upon their differential adherent
properties in culture (14). Since
MSCs do not express a unique marker on their surfaces, their
identification often involves screening for multiple surface
markers such as CD34, CD45, CD90 and CD105. Flow cytometric
analysis of MSCs showed that cell surface markers were
characteristically positive for CD90 and CD105 and
characteristically negative for CD34 and CD45 (data not shown)
indicating that MSCs were not contaminated by hematopoietic cell
lineages. As liver cancer cells can express some of the same CD
markers as MSCs, we labeled the MSCs with DiI and HepG2 cells with
the plasmid eGFP.
Rates of spontaneous fusion can vary between 1 in
102 to 1 in 106 in vitro (9) and in vivo (17). Flow cytometric analysis, gated to
select for only dual labeled cells, detected both labels in 6.9% of
the total cell population after fusion was induced by PEG2000
treatment, an indicator of the efficiency of fusion (Fig. 1D). The sorted fused cells were
collected and cultured. They remained viable until use in the
experiments 2 weeks later. The fused cells retained their GFP
plasmid-induced green fluorescent properties for at least 1 month
after fusion. DiI red fluorescence labeling was maintained in
cultures for ~1 week. Fluorescence microscopy of both types of
labeled progenitor cells [(Fig. 1B and
C, and the fusion hybrid 1 month after fusion (Fig. 1E)], showed homogeneous labeling. In
the Fig. 1E insert image of the
fused cells, cells with more than one nucleus are visible. Many
fused cells contained two nuclei initially, but after 36–48 h,
contained only one nucleus. The marked mesenchymal morphology of
long cellular extrusions, shallow cell bodies, and prominent nuclei
observed in Fig. 1E, suggest that
an EMT occurred.
Fused cells are aneuploids
If fusion occurs, the fused cells should initially
contain more chromosomes per cell than either progenitor cell type.
The average numbers of Giemsa-stained chromosomes/cell were
compared for each type at 2 and 4 weeks after fusion, and are shown
in Table I. The average chromosome
count was markedly increased in the fused cells, and was not a
multiple of the haploid chromosome number. This observation is
characteristic of aneuploidy, and indicates that the cells
contained chromosomes obtained from both MSCs and HepG2-GFP cells.
There was no extensive loss of chromosomes in the progenitor cells
over one month, the duration of observation, but the average
chromosome counts for fused cells decreased 18% between 2 and 4
weeks, indicating that some chromosomal instability was
present.
 | Table IAverage number of chromosomes. |
Table I
Average number of chromosomes.
| Cells | Two weeks after
fusion | Four weeks after
fusion | Change (%) |
|---|
| MSCs | 42.3±1.1 | 41.3±4.1 | −0.02 |
| HepG2-GFP | 86.0±8.4 | 89.6±16.0 | +0.04 |
| Fused cells | 157.5±17.5 | 128.9±21.3 | −18.20 |
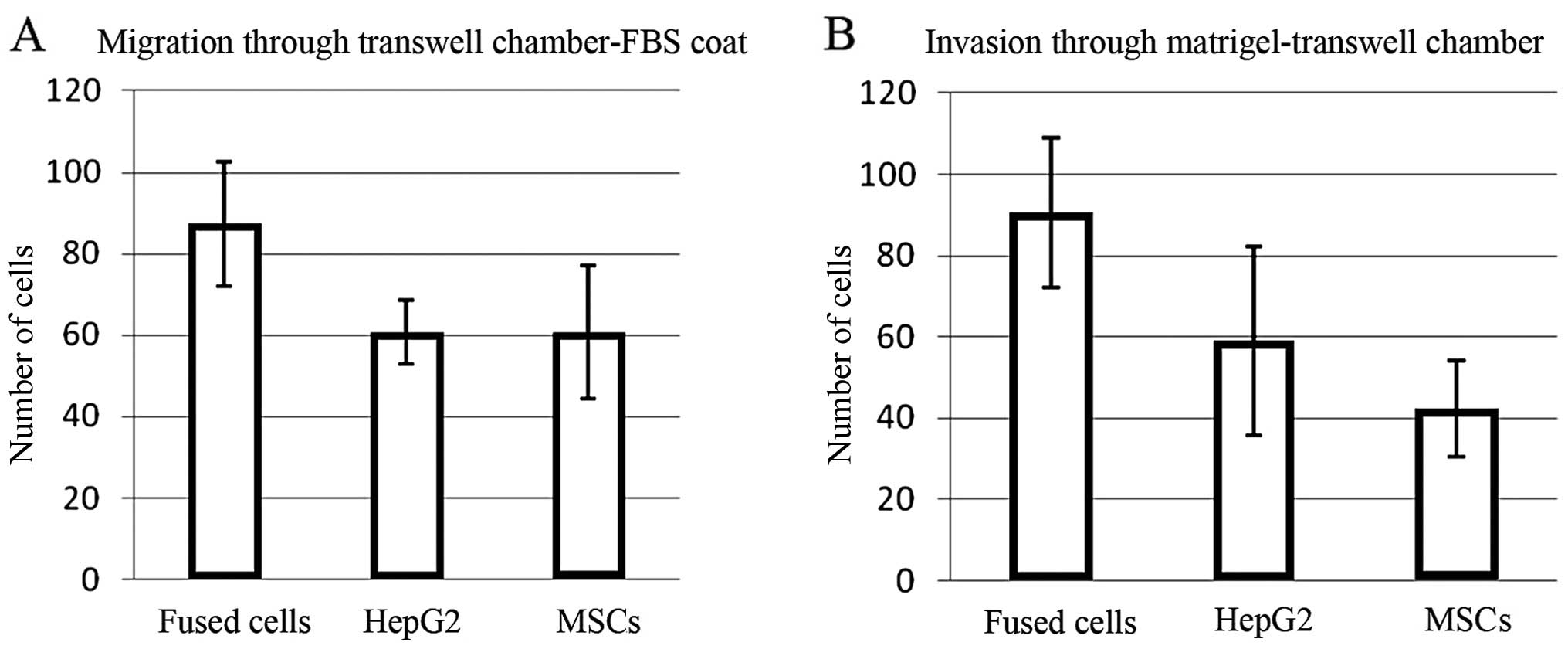
Fused cells exhibit increased migration
and invasion
The number of fused cells migrating through the
uncoated Transwell membranes was ~50% greater than the numbers of
the MSCs or HepG2 cells (p=0.015431 and 0.000613, respectively)
(Fig. 2A). The fused cells were
also ~50% more invasive through Matrigel-coated Transwell membranes
than the HepG2 cells, and twice as invasive as the MSCs (p=0.014211
and 0.007125, respectively) (Fig.
2B). Error bars represent the standard deviation of the cell
number obtained for each field counted.
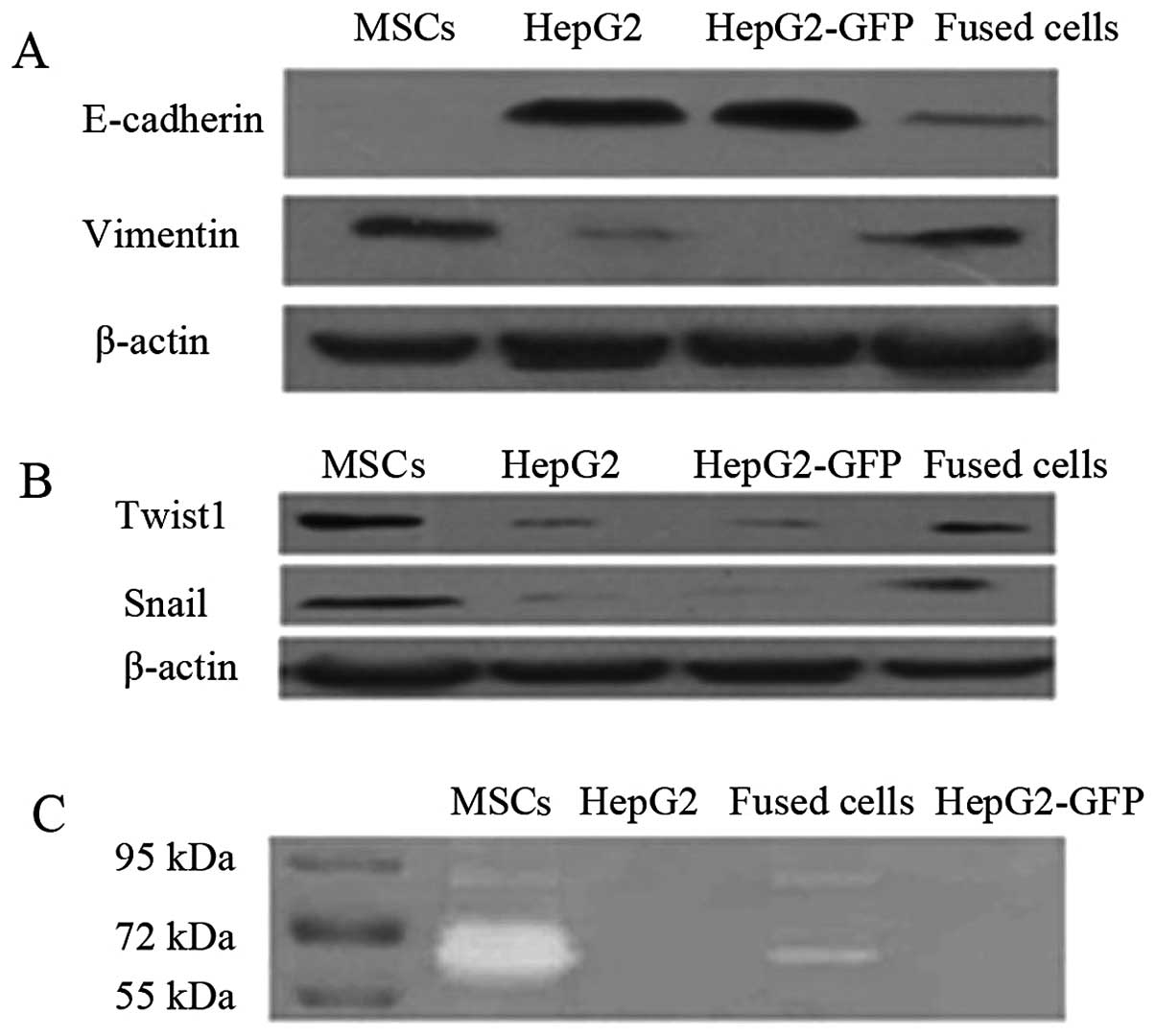
Fused cells exhibit increased EMT marker
expression and increased levels of active MMP2 and MMP9
After 1 month in culture, the fused cells exhibited
morphology typical of mesenchymal cells, unlike their progenitors
(Fig. 1E). Epithelial cells
undergoing EMT typically lose expression of epithelial markers such
as E-cadherin and increase expression of mesenchymal markers
including vimentin. Western blot assays detected reduced expression
of E-cadherin and increased expression of vimentin in the fused
cells when compared to HepG2 cells (Fig. 3A). The EMT regulatory factors Twist1
and Snail were also highly expressed in the fused cells and MSCs,
when compared to levels in the HepG2/HepG2-eGFP cells (Fig. 3B). Gelatin zymography detected
higher activities of secreted MMP2 and MMP9 in the fused cells when
compared with the activity levels in the HepG2 and HepG2-eGFP cells
(Fig. 3C).
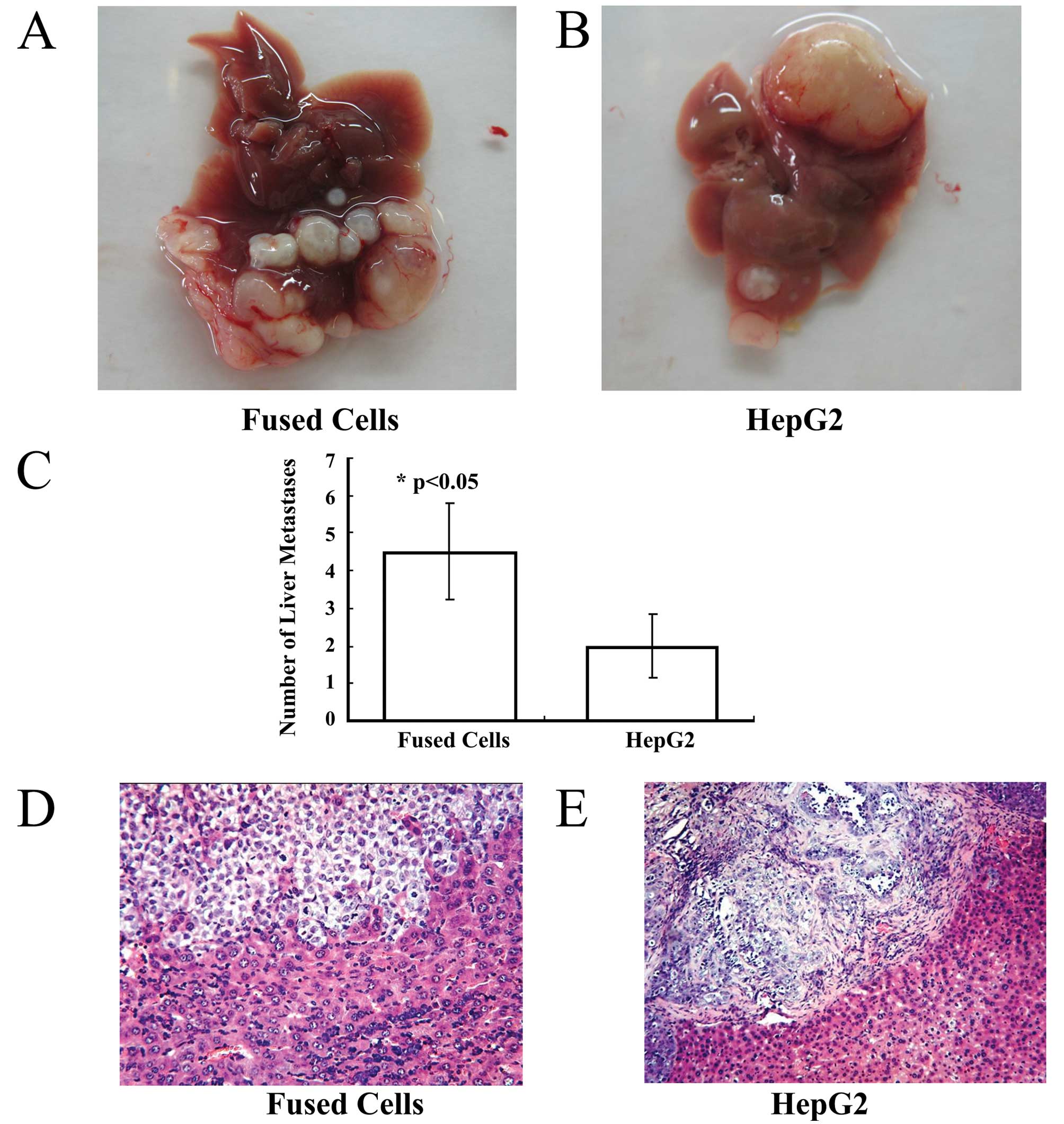
Fused cells are more metastatic in vivo
than MSCs or HepG2 cells
The fused cells, HepG2-eGFP cells and MSCs were
orthotopically injected into the left liver lobe of three separate
groups of nude mice. After 10 weeks, the survival rate was 28.58%
in the fusion group and 57.14% in the HepG2-eGFP group.
Gross examination
One hepatocellular carcinoma lesion at the site of
orthotopic injection occurred in the left liver lobe of each mouse
of the fusion group and the HepG2-eGFP group. Malignant
hepatocellular carcinoma lesions were observed around the
orthotopic lesion in the injected liver lobes (Fig. 4A and B). The average number of liver
malignant lesions was 4.50±1.29 in the fusion group, and was
2.0±0.82 in the HepG2-eGFP group.
Metastatic carcinoma lesions were present in the
lungs of the fusion group and in the HepG2 group. The number of
liver metastases showed a statistically significant difference
between the 2 groups (Fig. 4C)
(p<0.05). No lesions were found in the livers and lungs of the
mice inoculated with the MSCs.
Histological examination
Histological staining and microscopic observation of
sections of liver from the mice inoculated with fused cells
revealed a hepatocellular carcinoma invading normal liver tissue.
At high magnification, a few residual hepatocytes were noted in the
carcinoma tissue (Fig. 4D).
However, in tissue sections from the HepG2-eGFP-inoculated group, a
well-defined hepatocellular carcinoma was observed. Fibrous
connective tissue surrounded the hepatocellular carcinoma, and it
did not invade into the normal liver tissue (Fig. 4E).
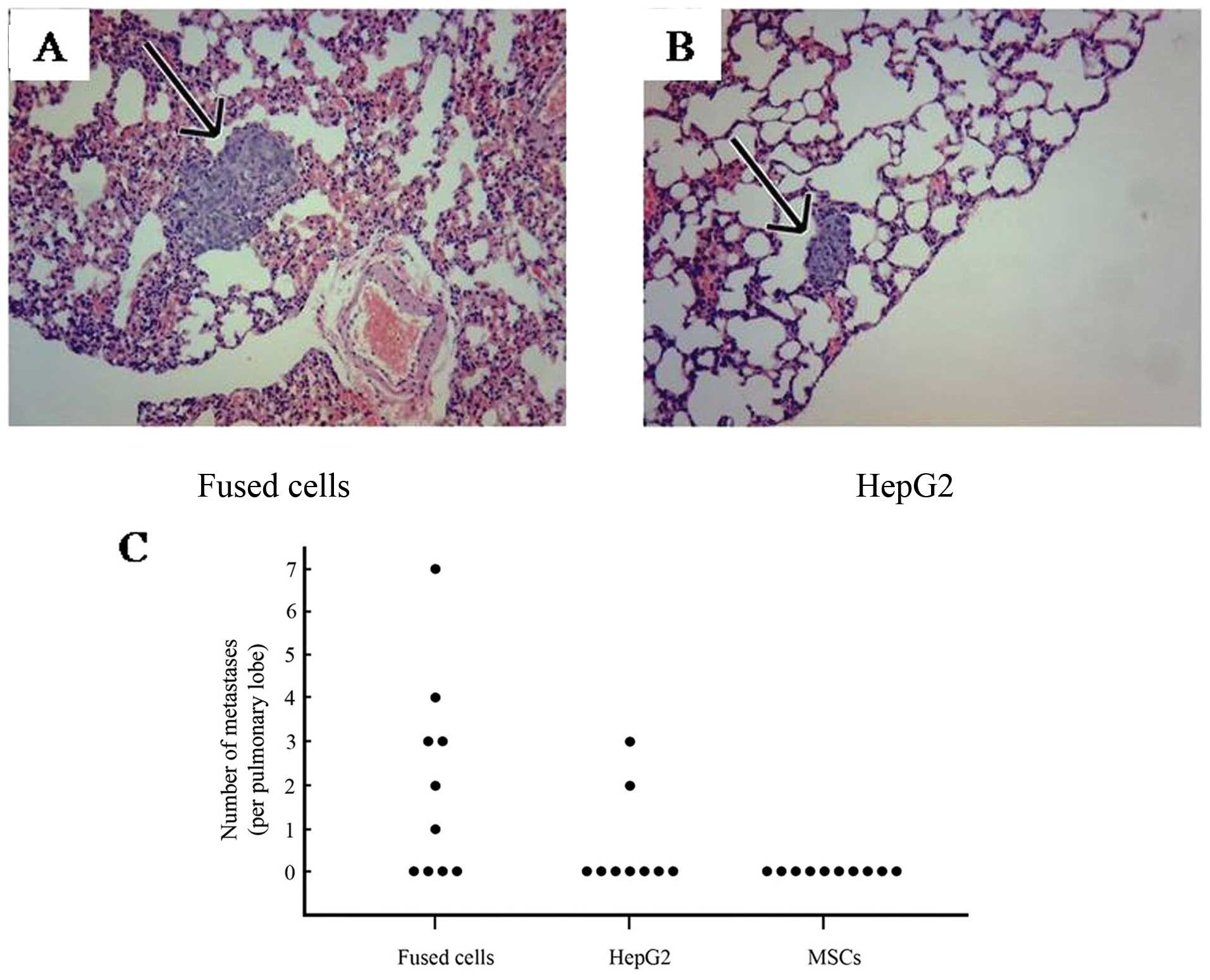
Many lung metastatic lesions were observed.
Representative lung tissue sections from fused cells (Fig. 5A) and HepG2 cells (Fig. 5B) are shown. The average number of
lung metastases present was 3.29±1.89 per pulmonary lobe in the
tumors generated by the fused cells, and was 1.25±0.75 in the
HepG2-eGFP inoculated mice. As shown in Fig. 5C, the numbers of metastases detected
in lung tissue from mice inoculated with the parental and progeny
cells are compared. A total of 20 metastatic lesions were detected
in lungs of the mice inoculated with fused cells compared to 5
detectable lesions generated in the mice inoculated with the HepG2
cells. No metastases were found in the kidney or brain in any mice.
All organs were normal in the mice receiving MSCs. These results
indicate that the fusion between the HepG2-eGFP cells and MSCs
enhanced tumor metastasis in vivo.
Discussion
It has been argued that the introduction of
wild-type tumor suppressors into a recipient cell by fusion should
reverse the loss of heterozygosity present in the recipient cell
which caused a more malignant phenotype, and should thus, make the
recipient cell less transformed and less malignant. The stem cell
fusion hypothesis (8) predicts the
opposite, that the fused cells would be both more migratory and
more invasive. To test this hypothesis, we fused HepG2
low-tumorigenic cancer cells and MSCs and compared the fused cells
to each of the parental cell types. These initial experiments
support our hypothesis, and indicate that despite our use of cells
from two different species to generate the fused cells for our
in vivo experiments, the mechanisms of generation of
metastatic tumorigenic cells may have common features in different
species. Although additional characterization and identification of
rat and human DNA in the tumors from fused cells should be
conducted, the clear differences in the properties of the parental
and progeny cells in these initial experiments indicate that those
differences are not simply due to the contamination of fused
progeny cells with the HepG2 parent. In most comparisons described
in this report, the fused cells possessed more
malignancy-associated factors than either parental cell type.
Similarly, fusing human gastric epithelial GES-1 cells with human
umbilical cord-derived MSCs was found to result in malignant
transformation of the progeny cells (unpublished data, personal
communication from Dr Xianghui He).
Aneuploidy is commonly observed in solid tumor
cells, and is considered a hallmark of malignancy. It is also a
predicted consequence of the stem cell fusion hypothesis. Numerous
reviews and articles have presented data and theories supporting
aneuploidy as a cause of cancer [for example (18)]. Aneuploidy may not always be
associated with a pathological state. Aneuploid cells are found in
the embryonic brain (19) and in
the liver (20), and do not seem to
always create detrimental effects. Several studies (21) support the variability and complexity
of the effects and interactions of cellular aneuploidy, chromosomal
instability, the presence of other mutations that suppress
proliferation (such as p53), or amplify the cellular stresses
induced by aneuploidy. Although it has been often suggested that
aneuploidy is the event driving the ultimate pre-cancerous to
metastatic conversion, the possibility that fusion actually drives
the aneuploidy has received less attention. The aneuploidy we
observed in the fused cells was maintained for at least 1 month,
even though nuclear fusion was observed in the cultured fused cell
population after <1 month. The 18% decrease in the average
number of chromosomes after 1 month in the fused cells is
indicative of chromosomal instability. In fact, an earlier
application of the stem cell fusion model by Stein-Werblowsky and
Ablin (22) demonstrated
hyperchromasia in prostate cancer cells as a consequence of fusion
of spermatozoa with normal prostatic epithelial cells. Our findings
in this report support the predictions of the cell fusion model,
and suggest many additional experiments which could clarify the
relationship between fusion, aneuploidy, cancer progression and
metastasis.
Since the cultured fused cells had irregular
morphology and protruding processes, characteristics of cells
undergoing EMT, we compared the expression of markers associated
with EMT, a developmental process in which epithelial cells reduce
intercellular adhesion and acquire mesenchymal properties,
including downregulation of the epithelial marker E-cadherin and
upregulation of the mesenchymal markers vimentin, Twist and Snail
(23). In certain tumors, EMT
results from tumor cell-MSC-induced fusion (24). Our western blot assays of fused cell
total protein detected a decrease in expression of E-cadherin and
an increase in expression of vimentin, supporting the occurrence of
an EMT. In addition, both Twist and Snail were highly expressed in
the fused cells. These alterations in EMT markers are also
associated with invasion and metastasis. The matrix
metalloproteinases (MMPs) are a family of ECM-degrading enzymes
involved in EMT and cellular migration and invasion. Several MMPs,
including MMP2 and MMP9, are induced in HepG2 cells by Snail
(25). MMP9 transcription and
cellular invasion are induced by overexpression of Snail in MDCK
cells (26). Increased expression
of Snail and secretion of active MMP2 and 9 were observed in the
fused cells.
The lack of adequate in vivo models presents
a major challenge to both basic research understanding of
metastasis and therapeutic development (27,28).
Fusion between genetically altered cells and stem cells and their
use in a tumorigenic and metastatic in vivo assay can be a
flexible approach to the development of specific genetically
defined and diverse models of metastasis. A recent report described
specific genetic alterations of normal human lung epithelial cells
which transformed them into tumorigenic cells (29). These cells were tumorigenic, but not
metastatic in a mouse model. The observations made in this in
vivo xenograft assay with orthotopic injection of fused cells
into livers of nude mice establish further support for an increased
metastatic phenotype in fused cells. Increased local invasiveness
of the fused cells compared to the HepG2 cells was manifested as
more numerous liver tumors as well as more metastatic lung lesions.
Multiple models consisting of different cell types with specific
genetic alterations fused with stem cells could be developed and
evaluated using the Materials and methods described in this
manuscript. Such model systems may allow for more accurate
screening and testing of therapeutic targets for prevention of the
most deadly consequence of carcinogenesis: acquisition of
metastatic capacity.
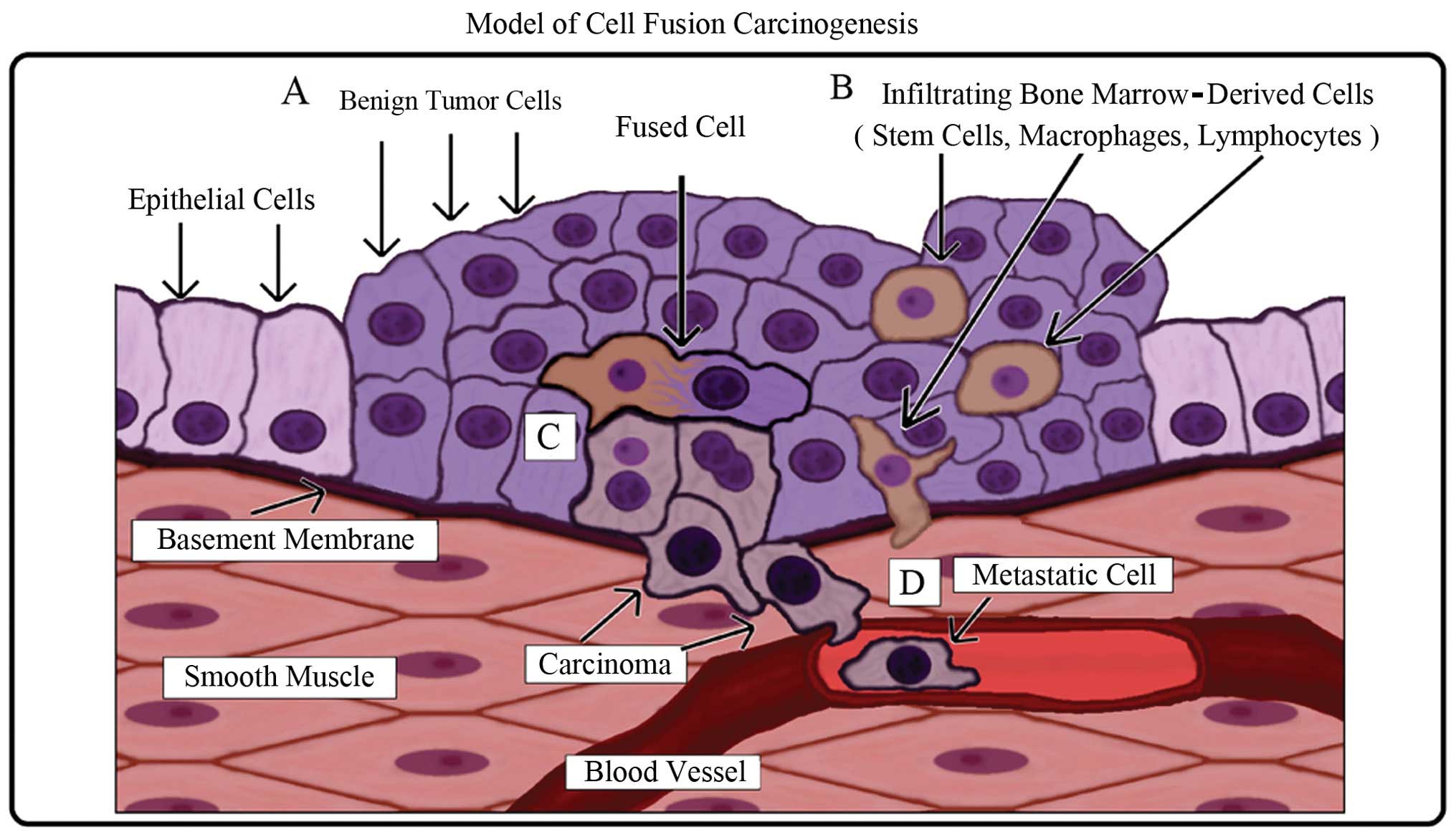
A schematic of the model is illustrated in Fig. 6. Carcinogenesis is a multi-step
process in which genetic mutations can induce an altered
hyperplastic cell phenotype which then forms a benign neoplasm and
subsequent local tissue damage (Fig.
6A). Bone marrow-derived cells (stem cells, macrophages and
lymphocytes) are recruited to repair the damaged tissue (Fig. 6B). In the process of normal repair,
fusion between an altered cell and a stem cell occurs (Fig. 6C). The resulting hybrid cell
acquires epigenetic properties from the stem cell such as
self-renewal, plasticity, and the capability to migrate to and
survive in circulation (metastasis) while retaining both epithelial
characteristics and mutations from the original tumor cell
(Fig. 6D).
The fusion event is a potential mechanism for
transferring stem cell-like properties to the altered cell. It is
important to note that traits and qualities of metastatic cells
overlap significantly with those of stem cells. This is no
coincidence and its importance cannot be overstated. The individual
mechanistic elements imparting ‘stemness’ may include a variety of
protein, RNA mediated and/or epigenetic controls. However,
regardless of the molecular mechanism(s) responsible, the stem cell
may be a wellspring from which the altered cell perverts stem cell
potency into metastatic activity.
These experiments were undertaken to test the
previously proposed hypothesis that fusion between an altered,
potentially premalignant cell, with little or no metastatic
ability, and a bone marrow-derived mesenchymal stem cell, could
form a hybrid cell with increased metastatic capacity and increased
levels of phenotypic markers associated with cancer cells. We
demonstrated that i) viable hybrid cells can be generated from
fusion of a human liver cancer tumor cell line and rat MSCs, and
identified by a double fluorescent label approach. ii) Fused
liver-MSC hybrid cells, when orthotopically inoculated into nude
mice, formed more tumors at both the liver injection site and at a
distant site, the lung, than did control cells from either parental
type. iii) The fused cells, compared to parental cells, were
aneuploid, expressed higher levels of two matrix metalloproteinases
associated with invasion and metastasis, expressed higher levels of
markers and regulatory factors associated with an epithelial to
mesenchymal transition, and exhibited an enhanced motility in
Transwell assays, all phenotypes characteristic of malignant cells.
iv) In histological comparisons of representative sections of the
nodules formed by fused cells or the liver cancer cell line, tumors
arising after fused cell inoculation invaded normal tissue, while
those formed after inoculation of the low-metastatic HepG2 cells
did not. v) We propose that this system is a very adaptable model
for investigation of the mechanisms of metastasis. Specifically,
altered cells with known genotypes when fused with MSCs, would
yield multiple systems for investigation. We are continuing to
expand and develop this approach to the generation of much needed,
specific and useful models of metastasis.
Acknowledgements
We thank Yong Li, Xialing Zhang and Yuhua Li for
assistance with this project. We are grateful to Drs G. Tim Bowden,
Rein Kilkson and Richard J. Ablin for their teachings, and to our
colleagues Drs Yu Duan, Jian Wang and Amy H.T. Davis for their
helpful discussions. We also appreciate Clifton Leaf’s book for
continuing to inspire us. This study was supported by Cure Cancer
Worldwide Company of USA and the Natural Science Foundation of
Shandong Province (no. ZR2013HQ018).
Abbreviations:
|
rt
|
room temperature
|
|
s-f
|
serum-free
|
|
MSC
|
mesenchymal stem cell
|
|
MMP
|
matrix metalloproteinase
|
References
|
1
|
Cairns J: Cancer, Science and Society.
Freeman; San Francisco: 1978
|
|
2
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Guthrie GJ, Charles KA, Roxburgh CS,
Horgan PG, McMillan DC and Clarke SJ: The systemic
inflammation-based neutrophil-lymphocyte ratio: experience in
patients with cancer. Crit Rev Oncol Hematol. 88:218–230. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Casazza A, Di Conza G, Wenes M,
Finisguerra V, Deschoemaeker S and Mazzone M: Tumor stroma: a
complexity dictated by the hypoxic tumor microenvironment.
Oncogene. Apr 22–2013.(Epub ahead of print). View Article : Google Scholar
|
|
5
|
De Wever O, Demetter P, Mareel M and
Bracke M: Stromal myofibroblasts are drivers of invasive cancer
growth. Int J Cancer. 123:2229–2238. 2008.PubMed/NCBI
|
|
6
|
Mishra PJ, Mishra PJ, Glod JW and Banerjee
D: Mesenchymal stem cells: flip side of the coin. Cancer Res.
69:1255–1258. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hass R and Otte A: Mesenchymal stem cells
as all-round supporters in a normal and neoplastic
microenvironment. Cell Commun Signal. 10:262012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
He X, Tsang TC, Pipes BL, Ablin RJ and
Harris DT: A stem cell fusion model of carcinogenesis. J Exp Ther
Oncol. 5:101–109. 2005.PubMed/NCBI
|
|
9
|
Ding J, Jin W, Chen C, Shao Z and Wu J:
Tumor associated macrophage x cancer cell hybrids may acquire
cancer stem cell properties in breast cancer. PLoS One.
7:e419422012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lu X and Kang Y: Cell fusion as a hidden
force in tumor progression. Cancer Res. 69:8536–8539. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lu X and Kang Y: Cell fusion hypothesis of
the cancer stem cell. Adv Exp Med Biol. 714:129–140. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bjerkvig R, Tysnes BB, Aboody KS, Najbauer
J and Teris AJ: Opinion: the origin of the cancer stem cell:
current controversies and new insights. Nat Rev Cancer. 5:899–904.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pawelek JM and Chakraborty AK: Fusion of
tumour cells with bone marrow-derived cells: a unifying explanation
for metastasis. Nat Rev Cancer. 8:377–386. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Soleimani M and Nadri S: A protocol for
isolation and culture of mesenchymal stem cells from mouse bone
marrow. Nat Protoc. 4:102–106. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Köhler G and Milstein C: Continuous
cultures of fused cells secreting antibody of predefined
specificity. Nature. 256:495–497. 1975.
|
|
16
|
Yang MH, Chang SY, Chiou SH, Liu CJ, Chi
CW, Chen PM, et al: Overexpression of NBS1 induces
epithelial-mesenchymal transition and co-expression of NBS1 and
Snail predicts metastasis of head and neck cancer. Oncogene.
26:1459–1467. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Duelli D and Lazebnik Y: Cell fusion: a
hidden enemy? Cancer Cell. 3:445–448. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gerling M, Nousiainen K, Hautaniemi S,
Krüger S, Fritzsche B, Homann N, et al: Aneuploidy-associated gene
expression signatures characterize malignant transformation in
ulcerative colitis. Inflamm Bowel Dis. 19:691–703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Luzzo KM, Wang Q, Purcell SH, Chi M,
Jimenez PT, Grindler N, et al: High fat diet induced developmental
defects in the mouse: oocyte meiotic aneuploidy and fetal growth
retardation/brain defects. PLoS One. 7:e492172012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hanna MO, Zayed NA, Darwish H and Girgis
SI: Asynchronous DNA replication and aneuploidy in lymphocytes of
hepatocellular carcinoma patients. Cancer Genet. 205:636–643. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pfau SJ and Amon A: Chromosomal
instability and aneuploidy in cancer: from yeast to man. EMBO Rep.
13:515–527. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Stein-Werblowsky R and Ablin RJ:
Consideration of the possible etiology of prostatic cancer. II
International Congress of Andrology, Tel Aviv, Israel. Israel J Med
Sci. 17:abs. 743. 1981.
|
|
23
|
Fan F, Samuel S, Evans KW, Lu J, Xia L,
Zhou Y, et al: Overexpression of Snail induces
epithelial-mesenchymal transition and a cancer stem cell-like
phenotype in human colorectal cancer cells. Cancer Med. 1:5–16.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gavert N and Ben-Ze’ev A:
Epithelial-mesenchymal transition and the invasive potential of
tumors. Trends Mol Med. 14:199–209. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Horikawa T, Yoshizaki T, Kondo S, Furukawa
M, Kaizaki Y and Pagano JS: Epstein-Barr virus latent membrane
protein 1 induces Snail and epithelial-mesenchymal transition in
metastatic nasopharyngeal carcinoma. Br J Cancer. 104:1160–1167.
2011. View Article : Google Scholar
|
|
26
|
Jordà M, Olmeda D, Vinyals A, Valero E,
Cubillo E, Llorens A, et al: Upregulation of MMP-9 in MDCK
epithelial cell line in response to expression of the Snail
transcription factor. J Cell Sci. 118:3371–3385. 2005.PubMed/NCBI
|
|
27
|
Parsons JT, Zetter B and Mohla S: Shifting
paradigms in tumor metastasis: challenges and opportunities. Cancer
Biol Ther. 1:582–585. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Goldstein RH, Weinberg RA and Rosenblatt
M: Of mice and (wo)men: mouse models of breast cancer metastasis to
bone. J Bone Miner Res. 25:431–436. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sasai K, Sukezane T, Yanagita E, Nakagawa
H, Hotta A, Itoh T and Akagi T: Oncogene-mediated human lung
epithelial cell transformation produces adenocarcinoma phenotypes
in vivo. Cancer Res. 71:2541–2549. 2011. View Article : Google Scholar : PubMed/NCBI
|