Introduction
Telomere maintenance by telomerase has been proposed
as an essential prerequisite for cell immortalization and
tumorigenesis. Human telomerase reverse transcriptase, hTERT, is
the catalytic subunit of telomerase, and its expression has been
proven to be the rate-limiting step for telomerase activity
(1). Studies have revealed that
hTERT is expressed in over 85% of human tumors, whereas most normal
human somatic tissues exhibit little or no expression (2,3). The
differential hTERT expression between tumor tissues and healthy
normal tissues makes this gene a reasonable candidate for
association analysis of tumorigenesis.
The c-Myc proto-oncogene plays an important role in
the regulation of many cellular programs, including proliferation,
differentiation, self-renewal and apoptosis. Aberrant expression of
c-Myc is believed to be closely associated with cell
immortalization and tumorigenesis (4). Thirty years ago, when c-Myc was
initially classified as an oncogenic protein that cooperates with
Ras to transform primary embryonic rat cells, it was already
recognized that c-Myc activation contributes to cell
immortalization (5). Consistent
with this, some years later hTERT was identified as a direct
transcriptional target of c-Myc, and the transcriptional induction
is independent of additional protein synthesis (6,7). These
findings define a pathway of cell immortalization in which
oncogenic c-Myc activation induces expression of hTERT, and
enhanced hTERT expression leads to cell immortalization.
Evidence from numerous studies has clearly shown
that c-Myc-dependent hTERT transcription is one of the major
mechanisms for maintained expression of telomerase and apparently
contributes to tumorigenesis. However, not all incidences of
increased c-Myc expression in normal cells will result in
activation of oncogenic sequences including hTERT (8). E2F transcription factor 1 (E2F1) is a
member of the E2F family of transcription factors and displays
multiple activities that could be involved in cell differentiation,
development and apoptosis. E2F1 was determined to be
transcriptional activated by c-Myc, and has been shown to crosstalk
extensively with c-Myc signaling, which is now believed to control
cell fate decisions (9,10). Of special interest to us, as a
transcriptional regulator, E2F1 was found to be able to inhibit
hTERT gene transcription (11–13).
Therefore, we speculated that E2F1 may be a potential suppressor of
c-Myc-induced hTERT activation, which exhibits a negative feedback
regulation in normal cells upon c-Myc oncogenic signals.
The miR-17-92 gene cluster is frequently
overexpressed in human cancers, and has been shown to promote
several aspects of oncogenic transformation (14). Recent evidence has demonstrated that
E2F1 is inhibited at the post-transcriptional step by members of
miR-17-92 (miR-20a and miR-17-5p) (15–17).
However, both E2F and Myc can induce the transcription of
miR-17-92, thus forming a complicated negative feedback system in
the interaction network (18–20).
At present, some scholars believe that miR-17-92 represents a
molecular switch for cell proliferation and apoptosis via
determining the levels of these two proteins (21,22).
Based on these findings, we speculated that miR-17-92 plays a
critical mediating role in the expression of hTERT via regulating
c-Myc/E2F1 networks.
In this study, we chose to use human embryonic
fibroblast (HEF) cells as the model system, and performed
experiments to investigate whether E2F1 can act as a negative
feedback regulator of hTERT transcription in response to c-Myc
activation. Furthermore, we analyzed the effects of the
upregulation of miR-17-92 (miR-20a and miR-17-5p) expression on the
crosstalk of c-Myc/E2F1 and downstream hTERT expression.
Materials and methods
Cell culture and reagents
The HEF cells (passage 10) used in this study have
been described previously (23).
Cells were cultured in DMEM supplemented with 10% FCS (Invitrogen
Corp., Carlsbad, CA, USA), at 37°C in a humidified atmosphere
containing 5% CO2. An adenovirus (type 5) expressing
c-Myc (rAd-c-Myc) was produced by subcloning full-length c-Myc into
the vector pShuttle, and the recombinant adenovirus was produced by
overlap recombination. The HA-E2F1 plasmid and hTERT promoter
plasmid were constructed by standard genetic manipulations. The
mutant hTERT promoter construct was generated by the Site-Directed
Mutagenesis kit (Stratagene, Santa Clara, CA, USA) in accordance
with the manufacturer’s standard procedure using the wild-type
hTERT promoter construct as a template. The antisense
oligonucleotide directed to E2F1 mRNA (5′-TAGATC
CGATCCAGCTCAGTGACA-3′) was designed and provided by Sangon
Biological Co., Shanghai, China. The full-length backbone was
modified to avoid enzymatic cleavage by DNases.
Quantitative RT-PCR analysis
For quantitative real-time PCR (qRT-PCR) analysis,
total-RNA was extracted with TRIzol (Takara, Dalian, China), and
cDNA was prepared using the PrimerScript reverse transcriptase kit
(Takara). The mRNA expression levels of the targeted genes were
analyzed by qRT-PCR using Applied Biosystems 7500 Real-Time PCR and
SYBR-GreenER™ reagent system from Invitrogen. The primers used in
this study are listed as following: c-Myc, F:
5′-TCACCAGCACAACTACGCCG-3′ and R: 5′-CAGG ATGTAGGCGGTGGCTT-3′;
E2F1, F: 5′-AGGGGTGTGG GGTTGATACC-3′ and R: 5′-TCAGACACTGCAGGAGG
GAC-3′; hTERT, F: 5′-CGGAAGAGTGTCTGGAGCAA-3′ and R:
5′-GGATGAAGCGGAGTCTGGA-3′. Expression of the reference β-actin gene
was used to normalize the expression of the mRNA for each targeted
gene. Data were analyzed using the 2-ΔΔCt method.
Western blot analysis
Total cell lysates were prepared in 1X SDS buffer.
Proteins were separated by SDS-PAGE and transferred onto PVDF
membranes. Membranes were then blotted with monoclonal antibodies
specific to c-Myc (Santa Cruz Biotechnology, Santa Cruz, CA, USA),
E2F1 (Abcam, Cambridge, MA, USA), hTERT (Santa Cruz) and β-actin
(Santa Cruz). Antigen-antibody complexes were visualized with
enhanced chemiluminescence (Thermo Scientific, Rockford, IL,
USA).
Telomerase assay
To quantitatively detect changes in telomerase
levels, cells were assayed for telomerase activity using the
telomerase polymerase chain reaction-enzyme-linked immunosorbent
assay (PCR-ELISA) (Roche Diagnostics, Mannheim, Germany) according
to the manufacturer’s directions and as performed previously
(24). Briefly, cells were washed
with Dulbecco’s phosphate-buffered saline, lysed in CHAPS lysis
buffer and then assayed for protein using the Bio-Rad DC protein
assay (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s
instructions. Cell extracts equivalent to 3 mg of protein were
used. Following PCR-ELISA, telomerase activity was detected using a
Dynex-MRX plate reader and recorded as absorbance units.
Apoptosis analysis
Detection of apoptotic cells by flow cytometry was
performed as described previously (25). HEF cells cultured in 6-well plates
were infected with recombinant adenoviruses at various MOIs for 48
h before cells were harvested, and then the Annexin V/propidium
iodide binding assay was performed using a flow cytometer (XL-MCL;
Coulter Epics, Miami, FL, USA).
Reporter assay
HEF cells were transfected with the indicated
plasmids using Lipofectamine 2000 (Invitrogen) for 48 h, and the
luciferase activity was measured using the dual luciferase system
(Promega, Madison, WI, USA) as described previously (26). The relative luciferase activity was
expressed as a ratio of the firefly luciferase activity to the
Renilla luciferase activity.
ChIP and ChIP-re-ChIP assays
Chromatin immunoprecipitation (ChIP) experiments
were performed using a ChIP assay kit (Upstate Biotechnology,
Millipore, CA, USA) according to the manufacturer’s instructions.
Cross-linked proteins were immunoprecipitated using an anti-E2F1
(Abcam), and normal mouse IgG (Santa Cruz). For ChIP-re-ChIP
experiments, primary immunocomplexes obtained with anti-E2F1 or
anti-c-Myc were washed and eluted with 10 mM dithiothreitol, and
eluates were diluted in IP dilution buffer to perform re-ChIP with
anti-c-Myc or anti-E2F1. The recovered DNA was amplified with
specific primer pairs and analyzed by qRT-PCR. Normal IgG and input
DNA values were used to subtract and normalize values from ChIP and
ChIP-re-ChIP samples. The percent input method was used to
quantitate the values of the immunoprecipitated DNA. Primers used
for the hTERT promoter region were as follows: forward,
5′-GAGCAGCTGCGCTGT-3′ and reverse, 5′-AGCTGGAAGGTGAAG-3′.
miRNA expression level assays
The expression levels of mature miRNAs were measured
by TaqMan MicroRNA assays kit (Applied Biosystems, Foster City, CA,
USA) following the manufacturer’s protocol. The U6 primers were
used as a normalization control. For relative expression levels,
the 2−ΔΔCt method was used as previously described
(27). Experiments were carried out
in triplicate for each data point, and data analysis was carried
out using Bio-Rad IQ software.
Statistical analysis
The data are expressed as means ± SD. Differences
were compared by one-way ANOVA analysis followed by LSD t-test. All
statistical analyses were performed using SPSS 17.0 software (SPSS
Inc., Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Co-expression of E2F1 increases the
apoptosis of HEF cells following regulation of c-Myc-dependent
hTERT induction
Both E2F1 and hTERT have been determined to be the
direct transcriptional targets of c-Myc (6,7,9,10).
In this study, we initially verified whether c-Myc activation was
sufficient to induce expression of both E2F1 and hTERT in the HEF
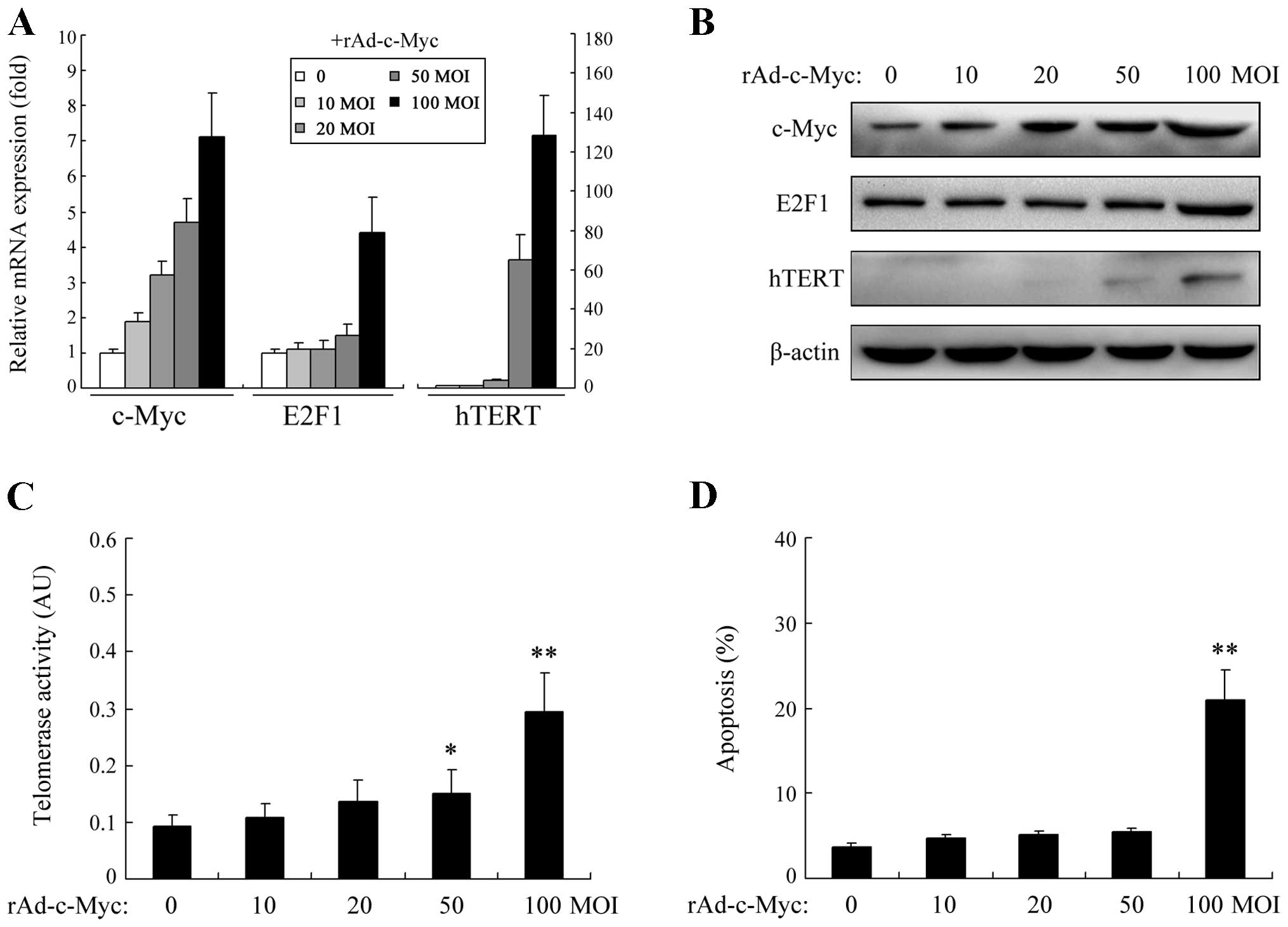
cells. As illustrated in Fig. 1A and
B, an increase in c-Myc activity by rAd-c-Myc transfection
enhanced the expression of both E2F1 and hTERT at the mRNA and
protein levels. However, intriguingly, no dose-dependent effect on
E2F1 expression was observed. Upon severe c-Myc activation,
significant E2F1 upregulation was induced, whereas following mild
and moderate c-Myc activation, no significant E2F1 upregulation was
noted. Regarding hTERT, an increase in the expression level was
observed in a dose-dependent manner but was restricted to a limited
amount even when c-Myc activity was greatly increased. We further
tested the telomerase activity of HEF cells upon c-Myc activation.
As shown in Fig. 1C, only
moderate/high c-Myc inputs could result in a significant increase
in telomerase activity. Since it has been determined that in normal
cells elevated E2F1 expression participates in many aspects of the
apoptotic process, we further detected the apoptosis of HEF cells.
As shown in Fig. 1D, in the
presence of high levels of c-Myc activation, the apoptosis rate of
HEF cells was also significantly increased.
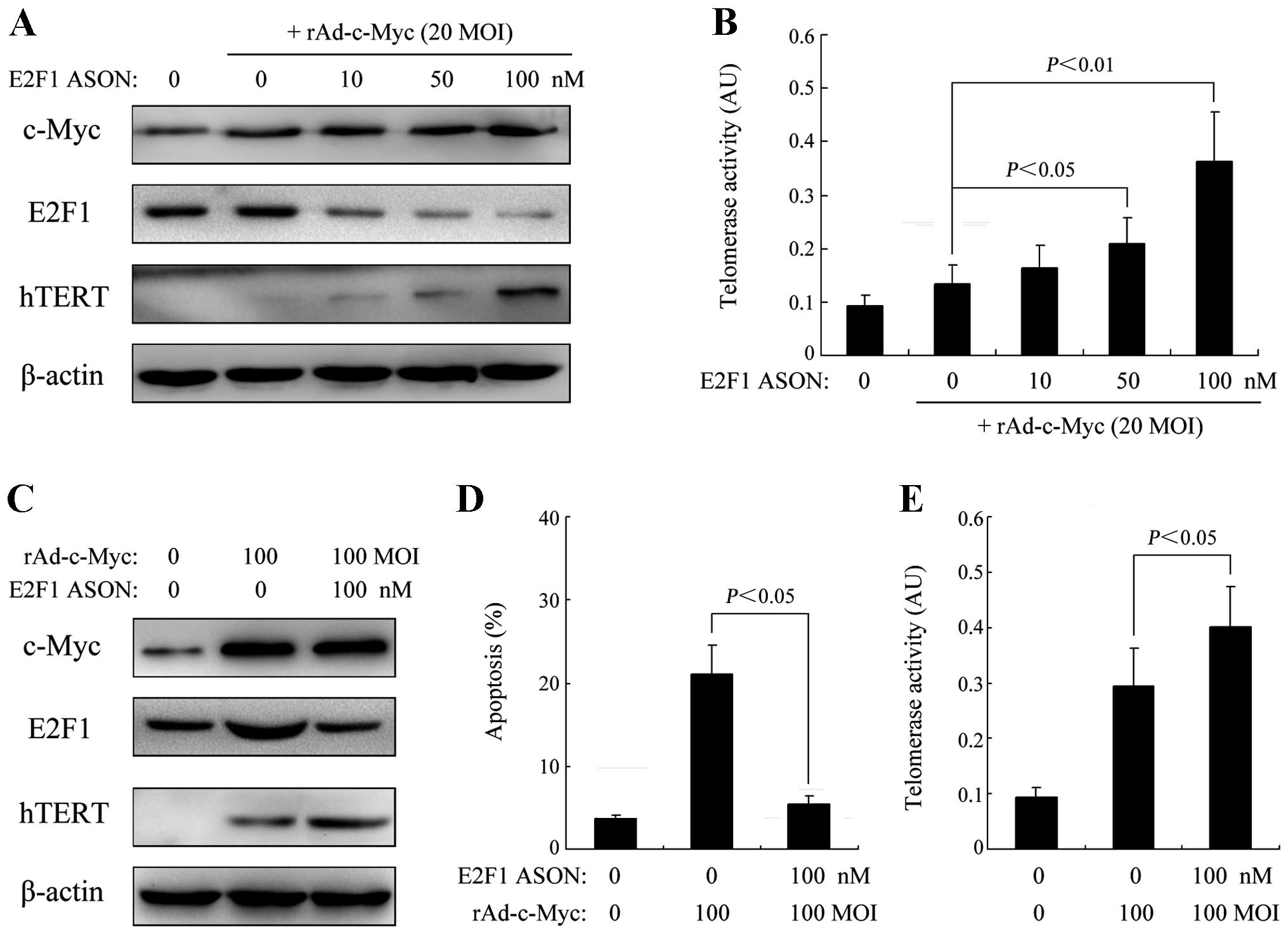
Specific inhibition of E2F1 expression
leads to elevated c-Myc-induced hTERT transcription
Although the expression of hTERT after c-Myc
activation in HEF cells was observed to be dose-dependent, it was
restricted to a low level, and when hTERT expression was markedly
increased, it was accompanied by an increased E2F1 expression of a
magnitude that could induce significant apoptosis, suggesting a
negative feedback loop restricting hTERT activity during c-Myc
oncogenesis. Therefore, we subsequently examined whether E2F1
inhibition has an effect on c-Myc-induced hTERT expression. As
shown in Fig. 2A, a decrease in
E2F1 expression by specific antisense oligonucleotide (ASON) at
concentrations of 50 and 100 nM led to a significant increase in
hTERT expression after moderate c-Myc activation (20 MOI
rAd-c-Myc), while as shown in Fig.
2B, the telomerase activity of HEF cells was also significantly
increased. In addition, the enhanced apoptosis of HEF cells in
response to a high level c-Myc activation (100 MOI rAd-c-Myc) was
inhibited after specific blockage of E2F1 expression, while the
increased telomerase activity of HEF cells was further enhanced by
inhibiting E2F1 expression (Fig.
2C–E).
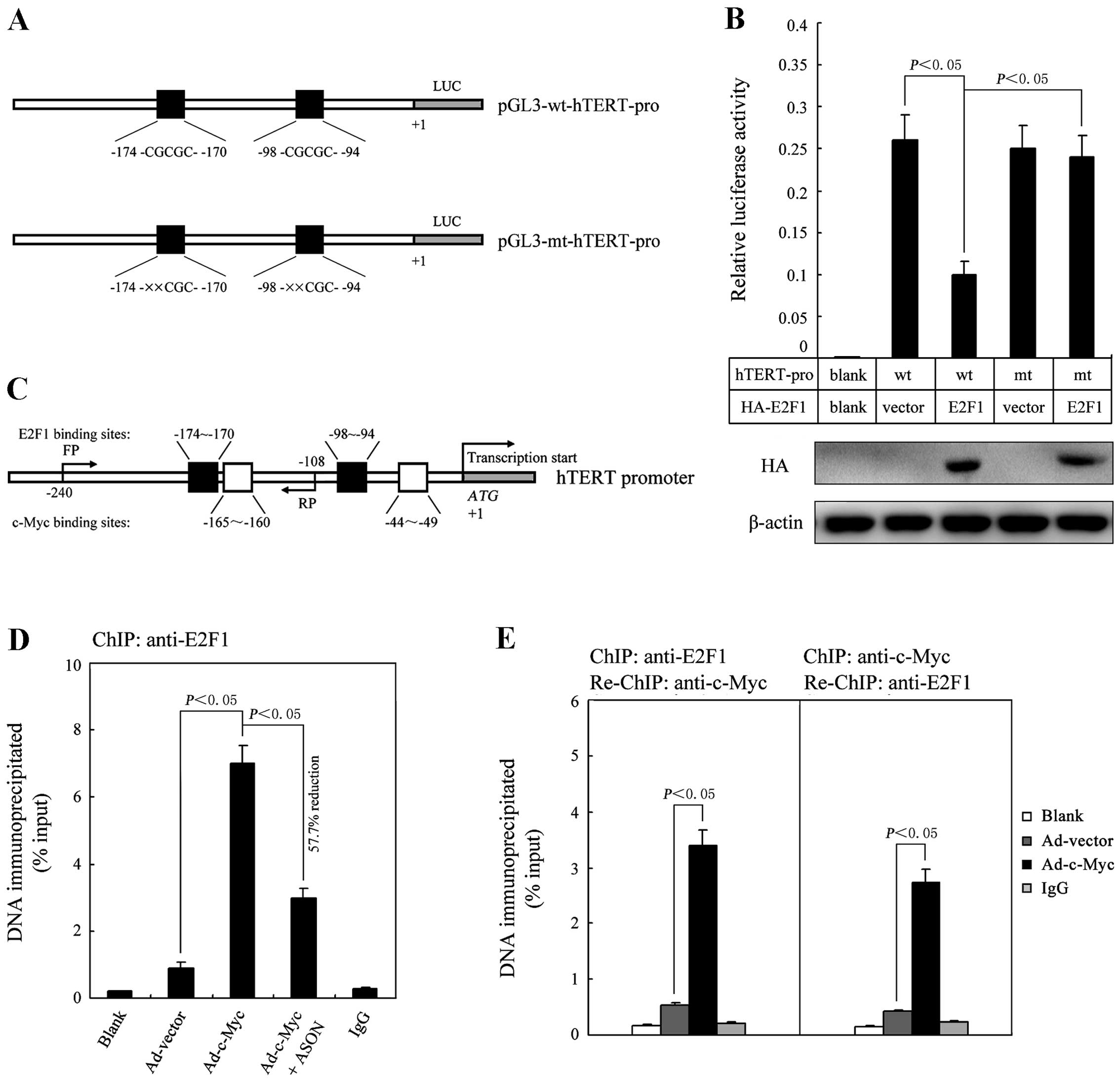
E2F1 is a transcriptional repressor for
hTERT gene expression induced by c-Myc activation
E2F1 has been found to be able to bind the hTERT
promoter (at −174 and −98 bp) and repress its expression in several
human cell lines (11–13). We therefore hypothesized that in
normal cells E2F1 may act as a transcriptional repressor that
restricts hTERT gene expression in response to oncogenic c-Myc
stimulation. To test this hypothesis, we performed a luciferase
reporter assay to examine whether E2F1 regulates hTERT promoter
activity in HEF cells and found that overexpression of E2F1
significantly reduced the hTERT promoter activity (Fig. 3A and B). To detect the direct
binding of E2F1 to the hTERT promoter, we next performed ChIP assay
using the anti-E2F1 antibody in HEF cells transfected with 20 MOI
rAd-c-Myc. The qRT-PCR results indicated that E2F1 could directly
bind to the hTERT promoter region (Fig.
3C and D). Furthermore, when E2F1 expression was inhibited by a
specific ASON, the qRT-PCR expression levels of the
immunoprecipitated hTERT promoter region was observed to have a
measurable reduction (Fig. 3D).
Finally, to demonstrate that E2F1 controls c-Myc-induced hTERT gene
expression, we performed ChIP-re-ChIP assay in
rAd-c-Myc-transfected HEF cells by using anti-E2F1 and anti-c-Myc
antibodies. As shown in Fig. 3E,
the qRT-PCR data indicated that E2F1 and c-Myc simultaneously
associated on the hTERT promoter region.
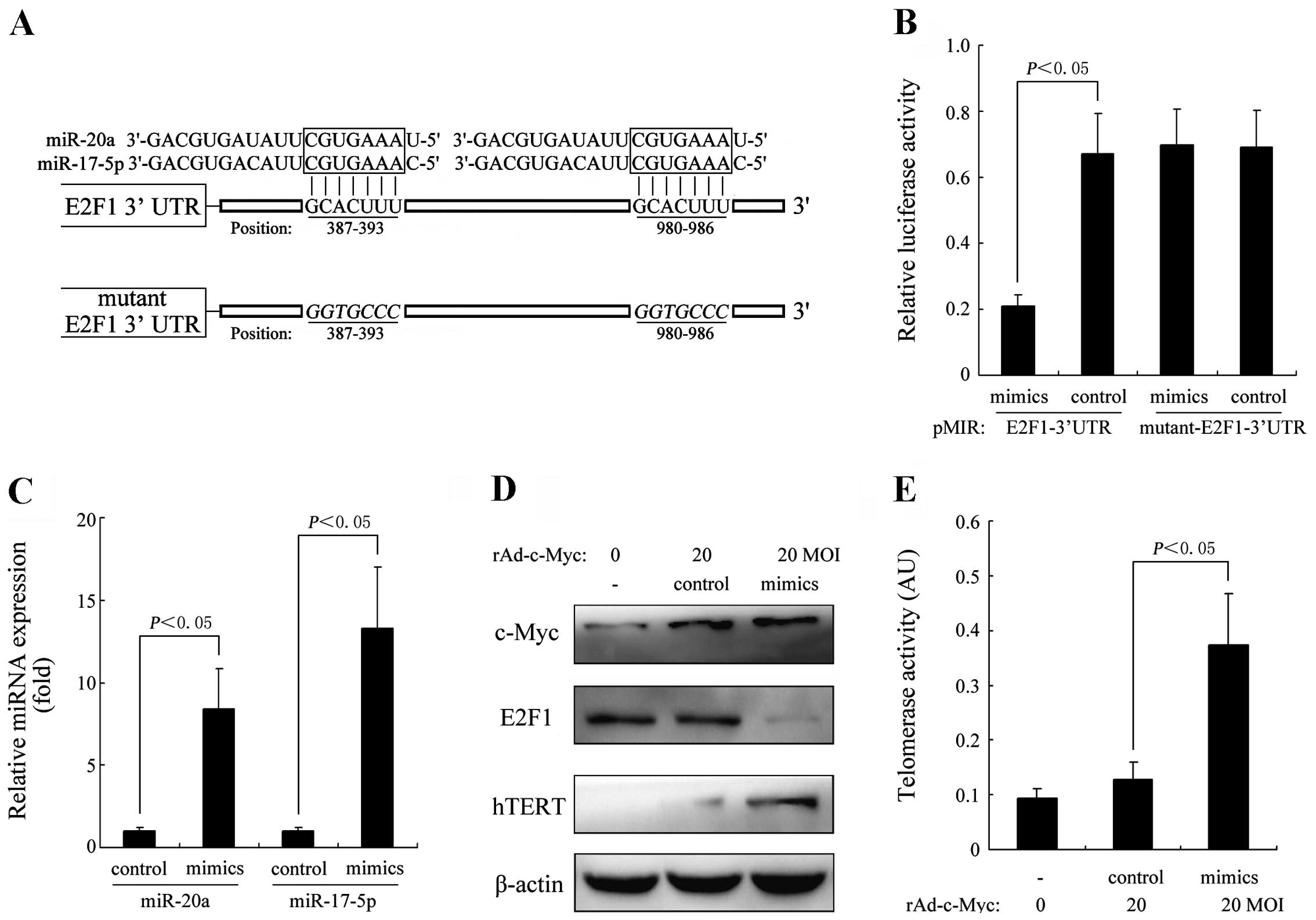
Overexpression of the miR-17-92 cluster
attenuates the E2F1-dependent inhibition of hTERT in
c-Myc-activated HEF cells
Considerable evidence indicates that members of the
miR-17-92 cluster, miR-20a and miR-17-5p, play critical roles in
the regulation of c-Myc/E2F1 networks (15–22).
In this study, we further investigated the effect of
miR-20a/miR-17-5p overexpression on E2F1-dependent inhibition of
hTERT in c-Myc-activated HEF cells. To ascertain whether
miR-20a/miR-17-5p directly regulate E2F1 expression through the
target sites in the 3′UTR of E2F1 mRNA, we constructed a luciferase
reporter vector with the two putative E2F1 3′UTR target sites for
miR-20a/miR-17-5p downstream of the luciferase gene
(pMIR-E2F1-3′UTR) and the mutant version with a mutation of 6 bp
from the site of perfect complementarity (pMIR-mutant-E2F1-3′UTR)
(Fig. 4A). Luciferase reporter
vector together with the miR-20a/miR-17-5p mimics or mimic control
were transfected into the HEF cells. As shown in Fig. 4B, a significant decrease in relative
luciferase activity was noted when pMIR-E2F1-3′UTR was
co-transfected with miR-20a/miR-17-5p mimics, but not in the
controls (P<0.05). In addition, we co-transfected HEF cells with
20 MOI rAd-c-Myc and miR-20a/miR-17-5p mimics. As shown in Fig. 4C, the expression levels of miR-20a
and miR-17-5p were expected to be greatly increased after
transfection. The expression levels of c-Myc, E2F1 and hTERT were
detected by western blot analysis. As indicated in Fig. 4D, the protein expression of E2F1 was
greatly suppressed in the HEF cells transfected with
miR-20a/miR-17-5p mimics, while the expression of hTERT was greatly
increased, accompanied by a significant increase in telomerase
activity (Fig. 4E).
Discussion
Numerous studies indicate that there is extensive
crosstalk between E2F1 and c-Myc signal transduction pathways, and
this interaction is vital to cell viability and function.
Rounbehler et al using K5 c-Myc-transgenic mice, showed that
inactivation of E2F1 not only increased the incidence of tumor
formation, but also promoted tumor initiation, indicating that E2F1
activation is a control mechanism serving to protect normal cells
from c-Myc-induced tumorigenesis (28,29).
Observations that support this hypothesis also include the finding
that i) c-Myc-mediated proliferation and lymphomagenesis are
compromised by E2F1 loss (30) and
ii) E2F1 blocks and c-Myc accelerates hepatic ploidy and
tumorigenesis in transgenic mouse models (31). These results further indicate that
E2F1 can counteract c-Myc oncogenic activation, especially in the
process of tumor initiation.
It is recognized that E2F1 has a dual function: it
triggers both proliferation and apoptosis via activation of
downstream target genes (32).
Overexpression of E2F1 causes apoptosis via p53-dependent and
p53-independent pathways, and this is an effective mechanism for
suppressing tumorigenesis (33,34).
In this study, we observed that high expression of the c-Myc
oncoprotein induced significant apoptosis of HEF cells, and when
E2F1 expression was specifically blocked, the enhanced apoptosis
was markedly attenuated, suggesting the involvement of
E2F1-mediated apoptosis. Of note, Leone et al revealed that
E2F1 is required for c-Myc-induced apoptosis in mouse embryonic
fibroblast cells (35). However,
Rounbehler et al reported that in E2F1-null transgenic mice,
tumorigenesis was accelerated while the c-Myc-induced apoptosis and
proliferation were unaffected by the status of E2F1, indicating
that E2F1 has other functions with which to suppress the oncogenic
transformation by c-Myc (28,29).
Induction of hTERT expression and subsequent
telomerase activation constitute a critical step in tumorigenesis,
especially in the process of tumor initiation (4). Studies have shown that c-Myc plays a
critical role in the induction of hTERT gene transcription through
the E-box located within the core promoter region (at −165 and −49
bp) (6,7). However, in normal cells, not all c-Myc
upregulation leads to the expression of hTERT and subsequent
telomerase activation (8). In
addition, studies have found that in normal cells including
embryonic fibroblast cells, c-Myc induction is a common event which
is required for the cellular response to various types of stress
but detectable hTERT protein expression is very rare (36–38),
indicating that the c-Myc-induced hTERT transcription is tightly
controlled and limited in non-oncogenic cells.
Although E2F1 has been proven to be a suppressing
regulator of hTERT expression, little is known concerning the
influence of E2F1 on c-Myc-induced hTERT activation. In this study,
we present evidence that E2F1 negatively regulates c-Myc-dependent
hTERT transcription in HEF cells through direct binding to the
promoter region. As E2F1 is also a transcriptional target of c-Myc,
it thus establishes a negative feedback pathway in which responsive
E2F1 protein expression inhibits hTERT expression and acts as an
apoptotic trigger, thus counteracting the c-Myc oncogenic signal
transduction in normal cells. Of note, we performed the same
experiments in hTERT-negative human osteosarcoma U2OS cells but no
expression changes of hTERT and E2F1 were observed upon rAd-c-Myc
transfection (data not shown), indicating that the negative
feedback pathway for suppression of hTERT transcription is no
longer functional when tumorigenesis has occurred. Our study
therefore extends the explanation of how E2F1 counteracts c-Myc
oncogenic activation, and provides a clue to the understanding of
mechanisms of hTERT activation in the process of tumor
initiation.
The miR-17-92 cluster, also called oncomir-1, is
among the first miRNAs to be validated as showing oncogenic
potential. Genomic amplification and elevated expression of
miR-17-92 were both found in various human malignances, and its
enforced expression exhibits strong tumorigenic activity in
multiple mouse tumor models (39).
miR-17-92 carries out pleiotropic functions during both normal
development and malignant transformation, as it acts to promote
proliferation, inhibit differentiation, increase angiogenesis, and
sustain cell survival (39).
Considerable evidence indicates that the miR-17-92 cluster is
critically involved in the regulation of c-Myc/E2F1 networks
(15–22). E2F1 was found to be inhibited at the
post-transcriptional level by members of the miR-17-92 cluster
(miR-20a and miR-17-5p), while as transcription factors both c-Myc
and E2F1 can induce the expression of microRNAs within the
miR-17-92 cluster. Since both miR-17-92 and E2F1 can be
transcriptionally activated by c-Myc, this establishes an unusual
network in which c-Myc activates the transcription of E2F1 while
simultaneously inhibiting its translation. Over the past few years,
based on observations using transgenic mouse models and
mathematical models, the regulation of miR-17-92 on c-Myc/E2F1
networks was thought to represent a molecular switch for cell
proliferation and apoptosis. Therefore, we speculated that
miR-17-92 also plays a regulatory role in the expression of hTERT
via determining the relative protein level of c-Myc and E2F1. In
this study, by performing a dual-luciferase reporter assay, we
determined that in non-oncogenic HEF cells miR-17-92
(miR-20a/miR-17-5p) can directly inhibit E2F1 expression through
the target sites in the 3′UTR of E2F1 mRNA. Furthermore, we
investigated the effects of the upregulation of miR-20a/miR-17-5p
expression on hTERT transcription in HEF cells with moderated c-Myc
activation, and observed that the hTERT expression and subsequent
telomerase activity were significantly enhanced. This result
indicates that miR-17-92 is involved in the regulation of crosstalk
between c-Myc/E2F1, and its upregulation can attenuate the
restriction of E2F1 on c-Myc-induced hTERT expression, and as a
result, boost the risk of tumorigenesis. In addition, we also
hypothesized that mutants of the hTERT promoter region might be
another mechanism by which it escapes from the transcriptional
suppression of E2F1 and due to this fact, normal cells can easily
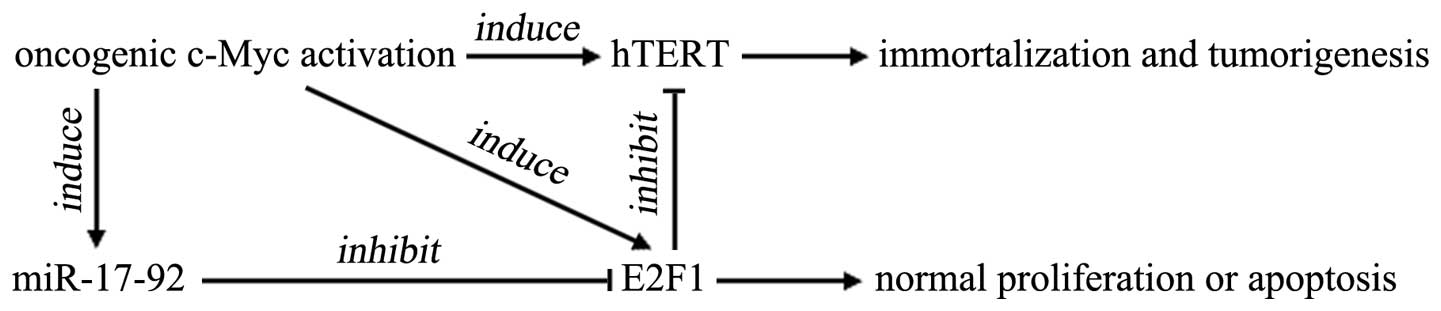
be transformed by oncogenic c-Myc activation. Fig. 5 is a schematic diagram of how E2F1
negatively regulates c-Myc-induced hTERT transcription and the
involvement of the miR-17-92 cluster (40).
Taken together, in the present study, we showed that
E2F1 acts as a negative feedback regulator of c-Myc-induced hTERT
transcription. If this negative regulating mechanism of E2F1 is
attenuated or abrogated by miR-17-92 upregulation, hTERT is easily
activated by c-Myc overexpression, thereby leading to cell
immortalization and tumorigenesis.
Acknowledgements
This study was supported by the National Foundation
of Natural Sciences, China (nos. 81101533, 81071727, 81170356 and
81270450) and China Postdoctoral Science Foundation (nos.
20100481468 and 201104755).
References
|
1
|
Wojtyla A, Gladych M and Rubis B: Human
telomerase activity regulation. Mol Biol Rep. 38:3339–3349. 2011.
View Article : Google Scholar
|
|
2
|
Harley CB: Telomerase and cancer
therapeutics. Nat Rev Cancer. 8:167–179. 2008. View Article : Google Scholar
|
|
3
|
Kim NW, Piatyszek MA, Prowse KR, et al:
Specific association of human telomerase activity with immortal
cells and cancer. Science. 266:2011–2015. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stewart SA and Weinberg RA: Telomeres:
cancer to human aging. Annu Rev Cell Dev Biol. 22:531–557. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Land H, Parada LF and Weinberg RA:
Tumorigenic conversion of primary embryo fibroblasts requires at
least two cooperating oncogenes. Nature. 304:596–602. 1983.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu KJ, Grandori C, Amacker M, et al:
Direct activation of TERT transcription by c-MYC. Nat Genet.
21:220–224. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Greenberg RA, O’Hagan RC, Deng H, et al:
Telomerase reverse transcriptase gene is a direct target of c-Myc
but is not functionally equivalent in cellular transformation.
Oncogene. 18:1219–1226. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kyo S, Takakura M, Fujiwara T and Inoue M:
Understanding and exploiting hTERT promoter regulation for
diagnosis and treatment of human cancers. Cancer Sci. 99:1528–1538.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Matsumura I, Tanaka H and Kanakura Y: E2F1
and c-Myc in cell growth and death. Cell Cycle. 2:333–338. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Coller HA, Forman JJ and Legesse-Miller A:
‘Myc’ed messages’: myc induces transcription of E2F1 while
inhibiting its translation via a microRNA polycistron. PLoS Genet.
3:e1462007.
|
|
11
|
Crowe DL, Nguyen DC, Tsang KJ and Kyo S:
E2F-1 represses transcription of the human telomerase reverse
transcriptase gene. Nucleic Acids Res. 29:2789–2794. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Won J, Chang S, Oh S and Kim TK:
Small-molecule-based identification of dynamic assembly of
E2F-pocket protein-histone deacetylase complex for telomerase
regulation in human cells. Proc Natl Acad Sci USA. 101:11328–11333.
2004. View Article : Google Scholar
|
|
13
|
Lacerte A, Korah J, Roy M, Yang XJ, Lemay
S and Lebrun JJ: Transforming growth factor-beta inhibits
telomerase through SMAD3 and E2F transcription factors. Cell
Signal. 20:50–59. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Esquela-Kerscher A and Slack FJ: Oncomirs
- microRNAs with a role in cancer. Nat Rev Cancer. 6:259–269. 2006.
View Article : Google Scholar
|
|
15
|
O’Donnell KA, Wentzel EA, Zeller KI, Dang
CV and Mendell JT: c-Myc-regulated microRNAs modulate E2F1
expression. Nature. 435:839–843. 2005.PubMed/NCBI
|
|
16
|
He L, Thomson JM, Hemann MT, et al: A
microRNA polycistron as a potential human oncogene. Nature.
435:828–833. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nagel S, Venturini L, Przybylski GK, et
al: Activation of miR-17-92 by NK-like homeodomain proteins
suppresses apoptosis via reduction of E2F1 in T-cell acute
lymphoblastic leukemia. Leuk Lymphoma. 50:101–108. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chang TC, Yu D, Lee YS, et al: Widespread
microRNA repression by Myc contributes to tumorigenesis. Nat Genet.
40:43–50. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sylvestre Y, De Guire V, Querido E, et al:
An E2F/miR-20a autoregulatory feedback loop. J Biol Chem.
282:2135–2143. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li Y, Li Y, Zhang H and Chen Y:
MicroRNA-mediated positive feedback loop and optimized bistable
switch in a cancer network involving miR-17-92. PLoS One.
6:e263022011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wong JV, Yao G, Nevins JR and You L:
Viral-mediated noisy gene expression reveals biphasic E2f1 response
to MYC. Mol Cell. 41:275–285. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Aguda BD, Kim Y, Piper-Hunter MG, Friedman
A and Marsh CB: MicroRNA regulation of a cancer network:
consequences of the feedback loops involving miR-17-92, E2F, and
Myc. Proc Natl Acad Sci USA. 105:19678–19683. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liang GP, Luo XD and Yang ZC: Influence of
human telomerase reverse transcriptase gene transfection on the
proliferation of human embryonic fibroblasts. Zhonghua Shao Shang
Za Zhi. 21:30–32. 2005.(In Chinese).
|
|
24
|
Yang SM, Fang DC, Yang JL, Chen L, Luo YH
and Liang GP: Antisense human telomerase reverse transcriptase
could partially reverse malignant phenotypes of gastric carcinoma
cell line in vitro. Eur J Cancer Prev. 17:209–217. 2008. View Article : Google Scholar
|
|
25
|
Zhang YF, Li XH, Shi YQ, et al: CIAPIN1
confers multidrug resistance through up-regulation of MDR-1 and
Bcl-L in LoVo/Adr cells and is independent of p53. Oncol Rep.
25:1091–1098. 2011.PubMed/NCBI
|
|
26
|
McNabb DS, Reed R and Marciniak RA: Dual
luciferase assay system for rapid assessment of gene expression in
Saccharomyces cerevisiae. Eukaryot Cell. 4:1539–1549. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rounbehler RJ, Rogers PM, Conti CJ and
Johnson DG: Inactivation of E2f1 enhances tumorigenesis in a Myc
transgenic model. Cancer Res. 62:3276–3281. 2002.PubMed/NCBI
|
|
29
|
Rounbehler RJ, Schneider-Broussard R,
Conti CJ and Johnson DG: Myc lacks E2F1’s ability to suppress skin
carcinogenesis. Oncogene. 20:5341–5349. 2001.
|
|
30
|
Baudino TA, Maclean KH, Brennan J, et al:
Myc-mediated proliferation and lymphomagenesis, but not apoptosis,
are compromised by E2f1 loss. Mol Cell. 11:905–914. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Conner EA, Lemmer ER, Sánchez A, Factor VM
and Thorgeirsson SS: E2F1 blocks and c-Myc accelerates hepatic
ploidy in transgenic mouse models. Biochem Biophys Res Commun.
302:114–120. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Engelmann D and Pützer BM: The dark side
of E2F1: in transit beyond apoptosis. Cancer Res. 72:571–575. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Polager S and Ginsberg D: p53 and E2f:
partners in life and death. Nat Rev Cancer. 9:738–748. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wu Z and Yu Q: E2F1-mediated apoptosis as
a target of cancer therapy. Curr Mol Pharmacol. 2:149–160. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Leone G, Sears R, Huang E, et al: Myc
requires distinct E2F activities to induce S phase and apoptosis.
Mol Cell. 8:105–113. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Herold S, Herkert B and Eilers M:
Facilitating replication under stress: an oncogenic function of
MYC? Nat Rev Cancer. 9:441–444. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Park JK, Chung YM, Kang S, et al: c-Myc
exerts a protective function through ornithine decarboxylase
against cellular insults. Mol Pharmacol. 62:1400–1408. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lü MH, Liao ZL, Zhao XY, et al:
hTERT-based therapy: A universal anticancer approach (Review).
Oncol Rep. 28:1945–1952. 2012.PubMed/NCBI
|
|
39
|
Olive V, Jiang I and He L: mir-17–92, a
cluster of miRNAs in the midst of the cancer network. Int J Biochem
Cell Biol. 42:1348–1354. 2010.
|
|
40
|
Zhang Y and Fang D, Yang S and Fang D:
E2F1: a potential negative regulator of hTERT transcription in
normal cells upon activation of oncogenic c-Myc. Med Sci Monit.
18:RA12–RA15. 2012. View Article : Google Scholar : PubMed/NCBI
|