Introduction
Oral squamous cell carcinoma (OSCC) accounts for
approximately 4% of all carcinomas in men and 2% in women
worldwide, with geographical variation in frequency (1). Although advances in early diagnosis
and multimodal treatments, including surgery, chemotherapy and
irradiation have been achieved, the 5-year survival rate of OSCC
patients has remained at 50–60% due to recurrence and metastasis
(2). Since conventional cytotoxic
therapies act upon rapidly dividing normal cells as well as
malignant cells, which results in the significant morbidity in
patients with solid tumors including OSCC, this method is of
limited benefit for survival (3).
Therefore, the development of improved anticancer therapies that
effectively and specifically target epithelial tumor cells while
minimizing the toxic side effects commonly associated with
conventional cytotoxic therapies is urgently needed.
With the enhanced understanding of key cellular
pathways involved in tumor growth, progression and cell death,
molecular targeted therapies have been exploited (4). Recently, novel treatments aimed to
target specific molecules, such as epidermal growth factor receptor
(EGFR), aberrantly expressed in OSCC, have been investigated and
tested in clinical trials at several research centers with
promising results (5). For many
years, the EGFR has been investigated as a major target for the
treatment of uncontrolled tumor growth (6). The EGFR, a glycosylated transmembrane,
is involved in regulating cell growth, differentiation and the
survival of cells (7). Increasing
evidence has shown that EGFR is often overexpressed in human
malignancies such as gastrointestinal and abdominal carcinomas,
lung carcinomas, carcinomas of the reproductive tract, melanomas,
glioblastomas and thyroid carcinomas, as well as OSCC (8,9), yet
absent in hematopoietic cells (10). Although data are inconsistent,
overexpression is often associated with an aggressive tumor
phenotype and a poor clinical prognosis. Growth factor-induced EGFR
signaling is important in many normal cellular processes with
effects ranging from apoptosis to migration and differentiation
(10,11). To target tumor cell proliferation or
growth via EGFR, monoclonal antibodies (mAbs) against this receptor
have been developed (11–13). These mAbs are highly specific with
few side effects and may result in synergistic effects when
combined with chemotherapy and radiation (14). Among these agents cetuximab (CET)
(Erbitux®, C225; ImClone LLC) is a human-murine chimeric
IgG mAb that has been approved by the Food and Drug Administration
(FDA) for treatment of colorectal cancer as a single drug or in
combination with chemotherapy and for HNSCC in combination with
radiation therapy or as a monotherapy after failure of
platinum-based therapy (2004 approval). CET, a 152-kDa molecule, is
composed of two 449-amino-acid heavy chains and of two
214-amino-acid light chains interfaced both by covalent (disulfide)
and non-covalent bonds (15). It
competitively binds to the extracellular domain of EGFR, preventing
tyrosine kinase activation, inhibiting cell growth and inducing
apoptosis (16). Preclinical
studies suggest that CET inhibits the proliferation of cancer cell
lines expressing EGFR, and increases the cytotoxic activity of
chemotherapy and radiation (14,17).
Unfortunately, only 10–20% of cancer patients were responsive to
and clinically benefited from anti-EGFR mAbs due to intrinsic and
acquired resistance (18). Thus,
combined treatment of CET with chemotherapy or other drugs may be a
beneficial strategy with which to increase the therapeutic effect
on OSCC.
Celecoxib (CXB) is a cyclooxygenase 2-selective
nonsteroidal anti-inflammatory drug (NSAID) that has been approved
for the treatment of adult arthritis, and has been found to exhibit
therapeutic effects on various types of cancers (19). Currently, CXB is widely being tested
in clinical trials for its therapeutic activity against various
cancers as a single agent and also in combination with other agents
(20,21). Recently, combined EGFR and COX-2
inhibition trials have been completed and demonstrate that this
combination could inhibit head and neck squamous cell carcinoma
(HNSCC) growth and decrease drug resistance (22).
Therefore, in this context, in the present study we
selected CXB as a COX-2 inhibitor in combination with CET for
suppressing EGFR and COX-2 expression and for simultaneously
reducing the doses of both drugs for the treatment of OSCC. We also
evaluated the feasibility of CXB in combination with CET in
inhibiting OSCC cell growth in vitro and in vivo, and
revealed the underlying molecular mechanisms of CET in combination
with CXB involved in the induction of apoptosis.
Materials and methods
Reagents
CXB, one type of COX-2 inhibitor, was purchased from
Pfizer Corporation Inc. (New York, NY, USA). CET was provided by
Merck Serono (Darmstadt, Germany).
Cell culture
HSC3, an OSCC cell line, was obtained from the
American Type Culture Collection (ATCC; Manassas, VA, USA). The
cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM;
Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine
serum (FBS; Invitrogen) in a humidified atmosphere of 5%
CO2 at 37°C.
Cell viability assay
HSC3 cells grown in monolayers were harvested and
dispensed in 96-well culture plates in 100 μl of DMEM at a
concentration of 5×103 cells per well. After 24 h,
different drug concentrations of CET (0–400 μg/ml), CXB (0–40 μM),
or both (0–200 μg/ml CET plus 20 μM CXB) were added to the cells.
Cell viability was assessed using 3-(4,5-dimethylthiazol-2-yl)-2,5
diphenyltetrazolium bromide (MTT) colorimetric assay at 490 nm with
minor modifications according to a previous study (23). This assay was carried out in
triplicate. The inhibition rate was calculated according to the
following formula: Inhibition rate (%) = [1 - (average absorbance
of the experimental group/average absorbance of the blank control
group)] × 100%.
Cell apoptosis assay
To evaluate the change in the apoptotic index,
terminal transferase dUTP nick end labeling (TUNEL) assay was used.
HSC3 cells (1×105 cells/ml in 6-well plates) were
cultured and treated with their respective half maximal inhibitory
concentration (IC50) values for CET, CXB, or both for 48
h. After fixation in 4% paraformaldehyde/PBS (pH 7.2) for 15 min at
room temperature and permeabilization in a permeabilization buffer
for 2–5 min, the assay was performed on cells on coverslips using a
commercially available in situ apoptosis detection kit (In
situ Cell Death Detection kit, POD, Roche Diagnostic, Branchburg,
NJ, USA). After applying anti-FITC HRP conjugate at 37°C for 30
min, diaminobenzidine (DAB) was applied to generate an insoluble
colored substrate at the site of DNA fragmentation for 10 min.
Apoptosis indices of both cell types for each treatment were
calculated as the percentage of cells displaying TUNEL labeling out
of the total number of nuclei. In addition, we also detected
caspase-3 and -8 activity by ELISA as an additional indicator of
apoptosis.
Caspase activity assay
The activity of caspase-3 and -8 was measured using
the Caspases Colorimetric Protease Assay kits (Millipore,
Billerica, MA, USA) as per the manufacturer’s instructions.
Briefly, cells were treated with their respective half maximal
inhibitory concentration (IC50) values for CET, CXB, or
both for 24 h, then washed twice with ice-cold PBS (pH 7.2) and
harvested by centrifugation at 700 × g for 10 min. The cell pellets
were then lysed in 150 μl buffer provided in the kit. Protein
concentrations of the lysates were determined using the Lowry
method. Then, an aliquot of lysates (80 μl) was incubated with 10
μl substrate of each caspase at 37°C for 2 h. Samples were analyzed
at 405 nm by a microplate reader (Thermo Fisher Scientific Inc.,
Waltham, MA, USA).
Migration assays
To assess the effect of CET combination with CXB on
cell migration, a wound-healing assay was performed. In brief,
migration chambers (8-μm pores; Corning) were coated for 1 h at
37°C with either 10 μg/ml purified plasma fibronectin or PBS and
blocked with migration buffer [3:1 of DMEM:F12 and 0.5% (w/v) BSA]
for 1 h. Cells were harvested following treatment with their
respective half maximal inhibitory concentration (IC50)
values for CET, CXB, or both for 48 h. Then, 1×105 cells
were resuspended in 100 μl of migration buffer, placed in the
chambers and incubated at 37°C for 4 h. Non-migrated cells were
removed, samples were fixed in 10% (v/v) buffered formalin, and
stained with haematoxylin and mounted on glass slides. Using an
inverted phase-contrast microscope (Leica DMR, Germany), the number
of migrated cells was determined by counting 5 randomly selected
fields.
Invasion assays
The invasiveness of the HSC3 cells following
treatment with CET, CXB or CET combined with CXB in vitro
was measured using BD BioCoat™ Matrigel invasion chambers (Becton
Dickinson Labware, Bedford, MA, USA) according to the
manufacturer’s instructions. In brief, filters were precoated on
the upper side with Matrigel provided in the kits (1 mg/ml). The
lower chamber was filled with culture media containing 10% FBS.
Cells (3×105) treated with the indicated drugs in low
serum media [3:1 of DMEM:F12 with 0.5% (v/v) FCS and 2% (w/v)
L-glutamine] in the inner chamber and KGM was used as a
chemo-attractant. Plates were incubated for 16 h at 37°C, and the
invaded cells were observed with an immunofluorescence microscope
by counting the cells that had invaded into the bottom of the cell
culture insert. We also detected MMP-9 and MMP-2 protein expression
by western blotting as an additional indicator of invasion and
migration.
Measurement of prostaglandin-E2 (PGE2)
production and VEGF expression
PGE2 synthesis was determined by competitive
enzyme-linked immunosorbent assay (ELISA) as previously described
with minor modification (23). In
brief, HSC3 cells were treated with their respective half maximal
inhibitory concentration (IC50) values for CET, CXB, or
their combination for 48 h in 12-well plates, and then the culture
media were centrifuged to remove cell debris. Cell-free culture
media were collected at the indicated time, then PGE2 levels were
measured by competitive ELISA as described by the kit manufacturer
(Cayman Chemical, Ann Arbor, MI, USA) using an ELISA reader
(μQuant; BioTek Instruments, Inc., Winooski, VT, USA).
HSC3 cells were treated with their respective half
maximal inhibitory concentration (IC50) values for CET,
CXB, or their combination for 48 h in 24-well plates, and then the
culture media were centrifuged to remove cell debris. Cell-free
culture media were collected at indicated time. Protein levels of
vascular endothelial growth factor (VEGF) in the cell supernatant
were determined by the Human VEGF ELISA kit (Yanyu, Shanghai,
China) according to the manufacturer’s instructions. Samples were
measured in triplicate and were properly diluted to ensure that the
measured values were within the concentration range of the standard
curve.
Tumor xenograft assay
To assess in vivo the combined effect of CET
and CXB on OSCC, we used an OSCC xenotransplanted nude mouse
tumorigenesis model. For inoculation of OSCC cells, the detached
HSC3 cells were diluted and emulsified with medium to a final cell
concentration of 5×107 cells/ml, and the 200 μl emulsion
was then inoculated subcutaneously into the right flank of a total
of 80 female BALB/c nude mice (6–8 weeks of age; weight ~200 mg;
Laboratory Animal Center of Jilin University, Changchun, China).
The mice were maintained in an environment complying with the NIH
guidelines for the care and use of laboratory animals, following a
protocol approved by the Ethics Committees of the Disease Model
Research Center, Jilin University. All animals were divided into 4
groups (10 mice/group) when the volume of the ensuing mass reached
75–100 mm3. The control group received 1% polysorbate
resuspended in deionized water. The other three groups were treated
with CXB (4.56 mg/kg body weight), CET (1 mg/kg body weight), or
CXB plus CET (2.5 mg/kg plus 0.5 mg/kg body weight, respectively)
intraperitoneally on alternative days for 3 weeks. Each mouse was
weighed every day to evaluate the side effects of the
administrations. The lengths and widths of the tumors were measured
with a caliper every 7 days, and the tumor volume (in cubic
millimeters) was calculated. At the end of 7 days, the animals were
euthanized using chloroform and their spleen tissues were collected
and cultured for a splenocyte surveillance study (23). In addition, each tumor was excised
and weighed when the mice were sacrificed. Parts of each tumor
tissue were wax embedded for H&E staining to study cell
apoptosis in vivo by TUNEL.
Western blot analysis
HSC3 cells were treated with their respective
IC50 values for CET, CXB or their combination for 48 h.
The cells were then homogenized in lysis buffer (Tris-HCl 50
mmol/l, EDTA 5 mmol/l, NaCl 150 mmol/l, sodium deoxycholate 1%,
Na3VO4 500 μmol/l, Triton X-100 0.5%, AEBSF
10 μmol/l, NaF 10 mmol/l) on ice. The homogenates were then
centrifuged at 14,000 rpm at 4°C for 30 min, and the supernatants
were collected for protein concentration determination using the
Bradford reagent (Sigma). Cell extracts (50 μg of protein) were
separated on a sodium dodecyl sulfate-polyacrylamide
electrophoretic gel (SDS-PAGE) and transferred to nitrocellulose
membranes, which were blocked in 3% bovine serum albumin (BSA) for
2 h. After blocking, the membranes were incubated with primary
antibodies overnight at 4°C for 2 h, and then with horseradish
peroxidase-conjugated secondary antibody for 2 h at room
temperature. Protein bands were visualized with enhanced
chemiluminescence reagent (ECL; GE Healthcare, Velizy-Villacoublay,
France). Blots were stripped and reprobed with anti-β-actin to
control for loading variations. Quantity One® Software
(Bio-Rad) was used for quantification of the protein bands. For
western blot analysis, the following antibodies were used: a mouse
monoclonal anti-β-actin, a mouse monoclonal anti-MMP-9 and a mouse
monoclonal anti-MMP-2 (Sigma Aldrich, St. Louis, MO, USA), and
mouse monoclonal anti-AKT, mouse monoclonal anti-phosphorylated
(p)-AKT, mouse monoclonal anti-EGFR, mouse monoclonal anti-p-EGFR,
mouse monoclonal anti-PI3K, mouse monoclonal anti-p-PI3K, and
horseradish peroxidase-conjugated goat anti-mouse IgG (Santa Cruz
Biotechnology, Santa Cruz, CA, USA).
Statistical analysis
Data from at least three independent experiments are
expressed as mean ± SD. Statistical comparison of more than two
groups was performed using one-way ANOVA followed by a Tukey’s post
hoc test. All statistical tests were two-sided, and a P-value of
<0.05 was considered indicative of a statistically significant
difference. Statistical analyses were undertaken using the
SPSS® Statistical Package, version 13.0 (SPSS Inc.,
Chicago, IL, USA) and GraphPad Prism, version 5.01
(GraphPad® software, San Diego, CA, USA) for
Windows.
Results
Effect of CET in combination with CXB on
OSCC proliferation
To evaluate the effect of CET, CXB, and the
combination on the cell viability of OSCC cancer cells in
vitro, HSC3 cells were treated with increasing concentrations
of CET (0–400 μg/ml), CXB (0–40 μM), or both (0–200 μg/ml CET plus
20 μM CXB). Treatment with CTX alone resulted in an IC50
value of 210.35±15.28 μg/ml. Treatment with CXB alone also resulted
in an IC50 value of 25.5±1.780 μM. Combination treatment
(0–200 ng/ml CET plus 20 μM CXB) resulted in a leftward shift in
the concentration-response curve such that the IC50
value was reduced to 100.88±7.98 μg/ml. In addition, our results
also demonstrated that CET, CXB and the combination inhibited cell
proliferation dose-dependently Based on the results, we selected
the respective IC50 values of the drugs for further
treatments throughout the study.
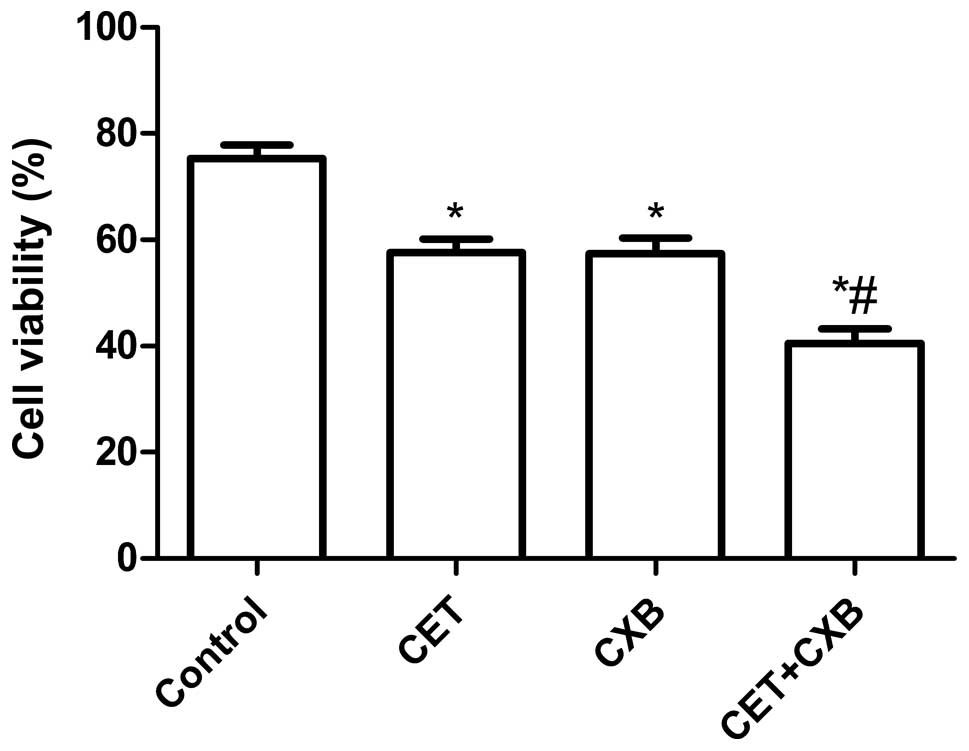
We next examined whether the combination of
relatively low concentrations of CET and CXB could additively or
synergistically inhibit OSCC cell proliferation in vitro
with their respective IC50 values for CET, CXB or the
combination. As shown in Fig. 1,
the inhibitory rate of the combination treatment group was higher
than the rates of the single drug groups (P<0.01). There was no
statistically significant difference between the CET group and the
CXB group (P>0.05). In addition, the inhibitory rate of the
combination treatment group was higher than that of the control
group (all P<0.05).
Effect of CET in combination with CXB on
OSCC apoptosis
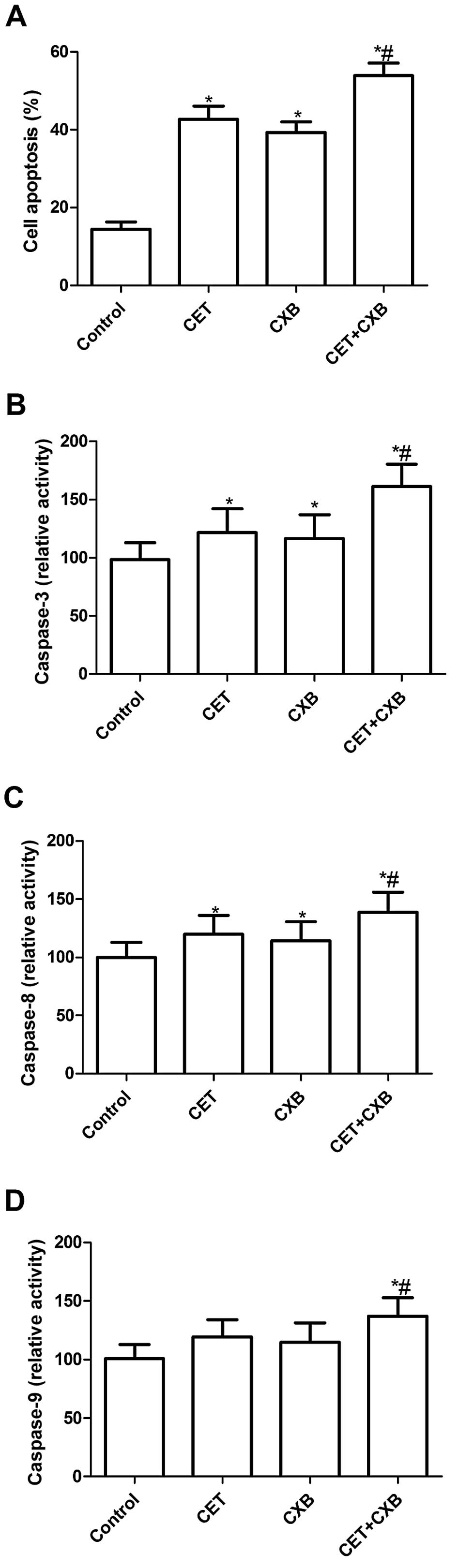
To verify whether CET and CXB exert an anticancer
effect via induction of apoptosis, apoptotic cancer cells in
vitro were detected with a light microscope using a TUNEL
technology. The assays were performed on HSC3 cells treated with
CET or CXB alone and the combination of the two agents at their
respective IC50 values. HSC3 cells treated with CET, CXB
or the combination had an increased percentage of apoptotic cells
compared with the untreated cells (P<0.05, Fig. 2A). In addition, the low-dose
combination resulted in an even higher percentage of apoptotic
cells than did the higher doses of either drug alone. There was no
significant difference between the CET group and the CXB group in
the induction of OSCC apoptosis. These data were consistent with
the results from the MTT assay, which indicated an additive effect
of CET and CXB in inducing cell death via apoptosis.
To explore the possible mechanism involved in the
induction of cell apoptosis of CTX in combination with CXB,
caspase-3, -8 and -9 activity was determined by ELISA. The results
showed that caspase-3, -8 and -9 activity was significantly
increased in the treatment group compared to the control group
(P<0.01, Fig. 2B–D).
Importantly, caspase-3, -8 and -9 activity was significantly
increased in the combination group compared to the activity in the
single treatment groups (P<0.05, Fig. 2B–D).
Effect of CET in combination with CXB on
OSCC migration and invasion
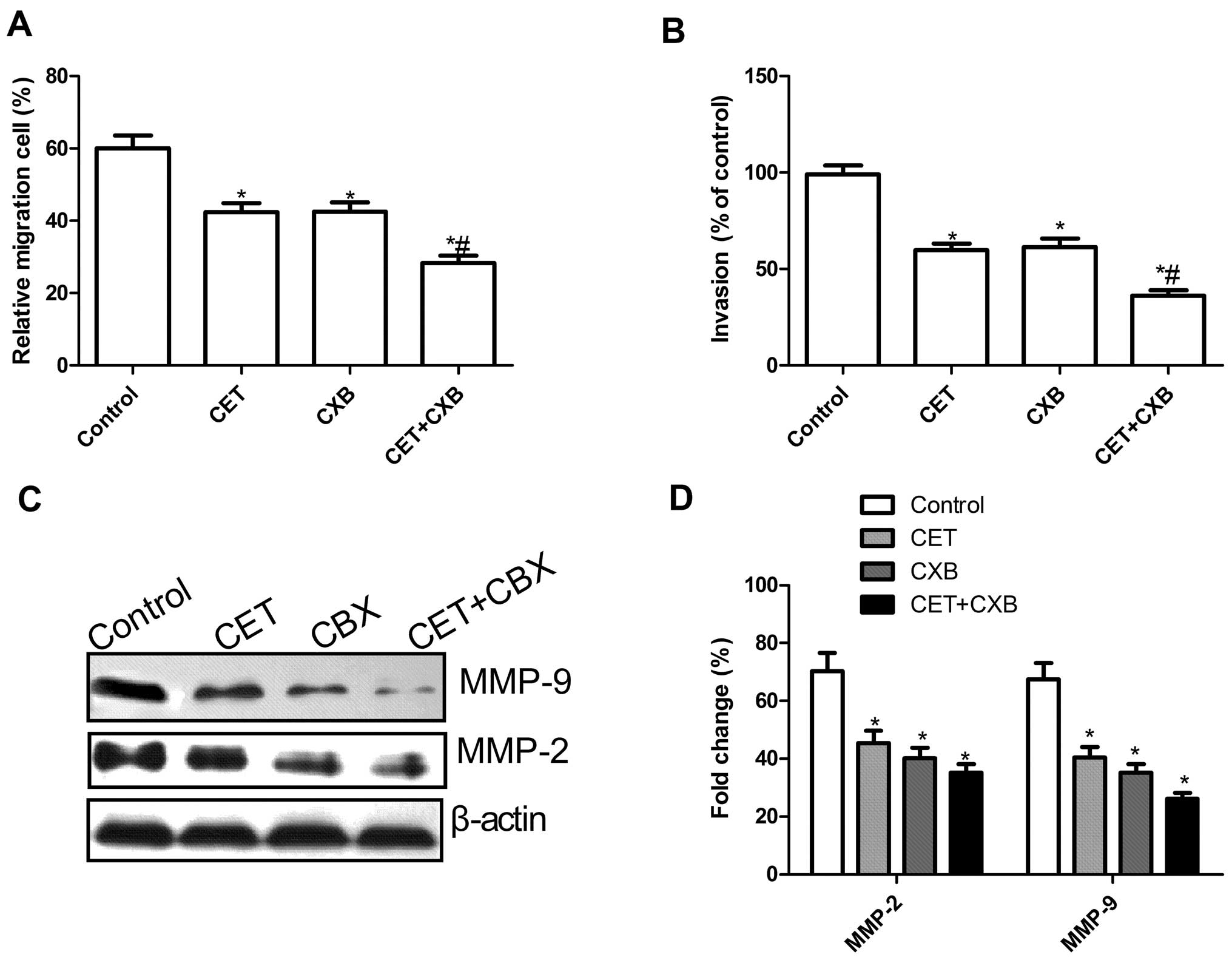
To ascertain the inhibitory effect of CET in
combination with CXB on OSCC cell motility in vitro, a
wound-healing assay was performed. After 48 h, cells in the CET
group, the CXB group and the combination CET and CXB group
exhibited a significantly reduced migration capacity compared to
the control group. Compared to the CET or the CXB group, the cells
in the combination group exhibited significantly reduced migration
of HSC3 cells (P<0.05; Fig.
3A).
The ability of CET in combination with CXB to reduce
the invasiveness of OSCC was further investigated by the Transwell
system assay. Cell invasion was also significantly decreased in the
treatment groups compared to the control group (P<0.05, Fig. 3B). Compared to the CXB or the CET
group, the combination treatment group greatly inhibited HSC3 cell
invasion (P<0.05, Fig. 3B).
To determine the potential mechanisms involved in
the inhibition of cell migration and invasion in vitro,
MMP-9 and MMP-2 protein expression was examined by western
blotting. Western blot analysis revealed a significant decrease in
MMP-2 and MMP-9 proteins in all treatment groups compared to the
control group (Fig. 3C). The
combination group showed a maximally reduced expression compared to
either the CET or CXB group (P<0.05, Fig. 3D).
Effect of CET in combination with CXB on
PGE2 production and VEGF expression in HSC3 cells
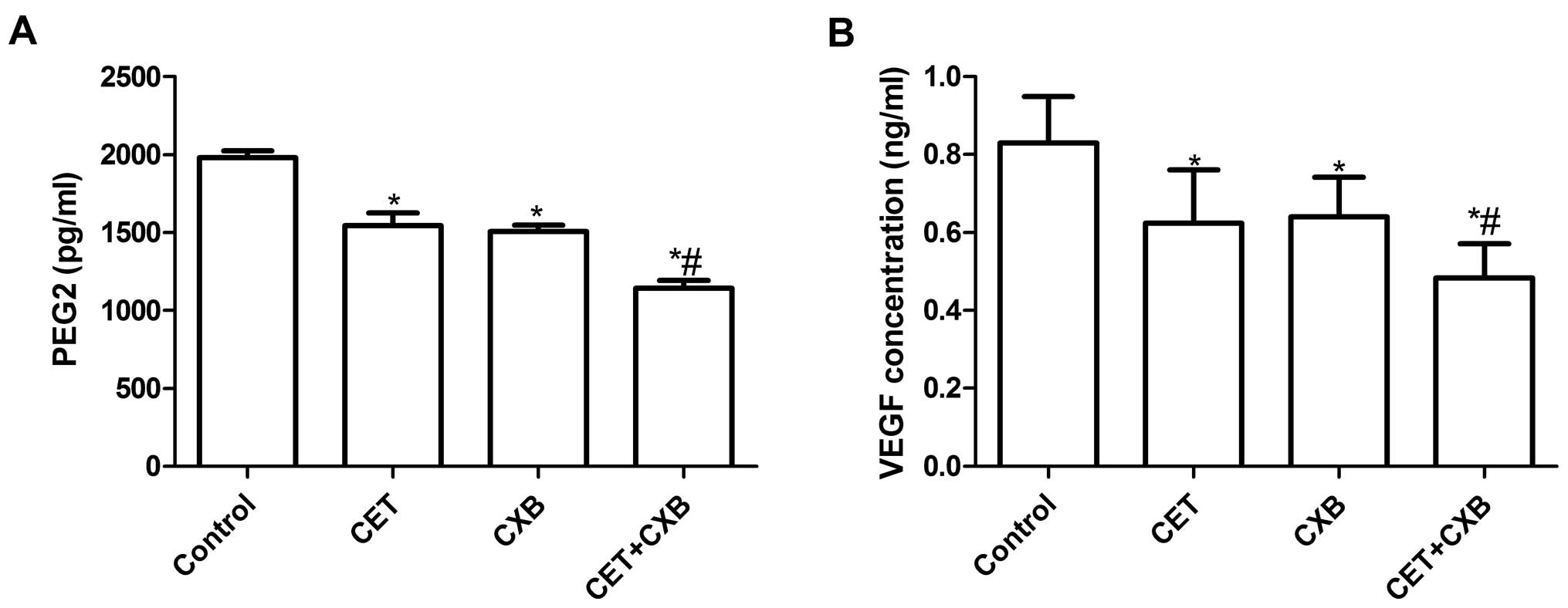
To examine the effect of CET in combination with CXB
on PGE2 production in HSC3 cells, ELISA was performed. As shown in
Fig. 4A, all treatment groups
inhibited PGE2 production when compared with the control group. CXB
in combination with CET significantly inhibited PGE2 production
compared to the single drug groups (P<0.05).
We also determined VEGF protein expression by ELISA
to evaluate the effect of CET in combination with CXB on VEGF
expression. As shown in Fig. 4B,
ELISA analysis revealed that VEGF excretion in the supernatant from
all treatment groups was significantly decreased compared to the
control group (P<0.05). Compared to single drug treatments, CET
in combination with CXB significantly inhibited VEGF expression
(P<0.05).
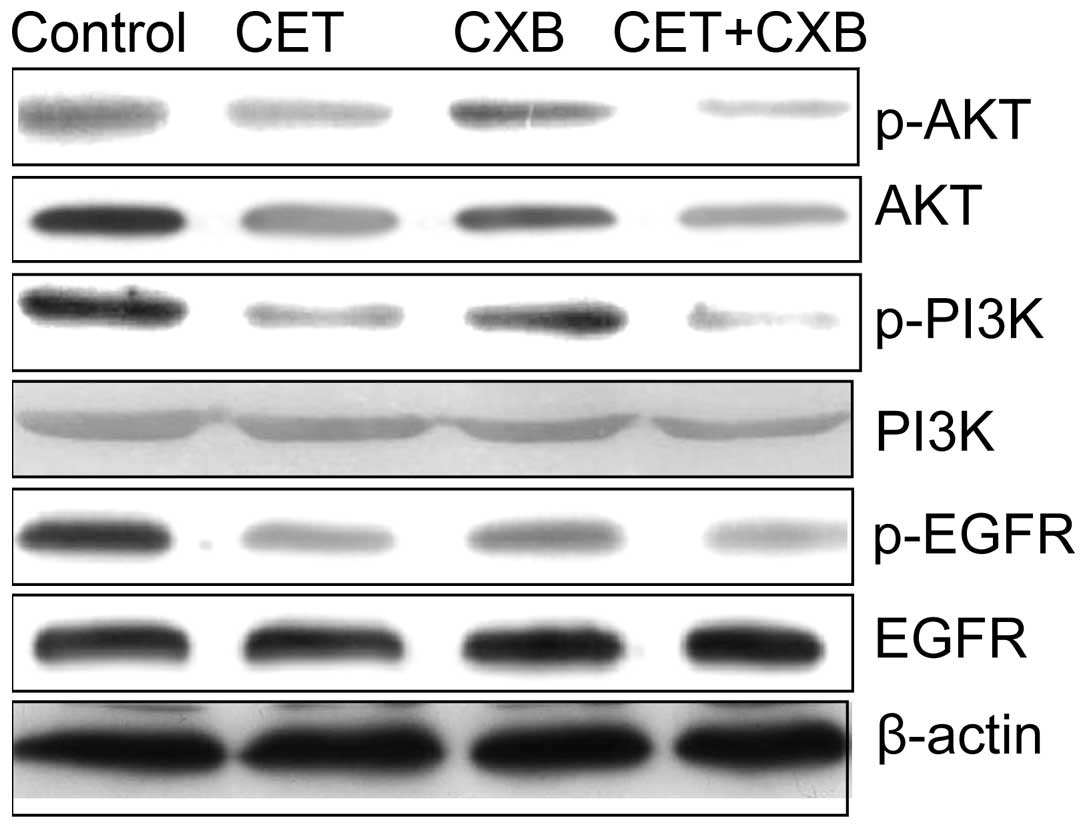
Effect of CET in combination with CXB on
EGFR signaling
The EGFR signaling pathway plays a crucial role in
cell proliferation and survival in various types of cancer; thus,
we evaluated the effect of CET or CXB alone and in combination on
several key downstream molecules involved in the EGFR signaling
pathway by western blotting. CET and CXB alone and the combination
inhibited the phosphorylation of PI3K, p-AKT and p-EGFR (Fig. 5). In addition, treatment of cells
with CET combined with CXB further reduced the expression levels of
the activated forms of EGFR (p-EGFR), p-PI3K and p-Akt, in the HSC3
cells.
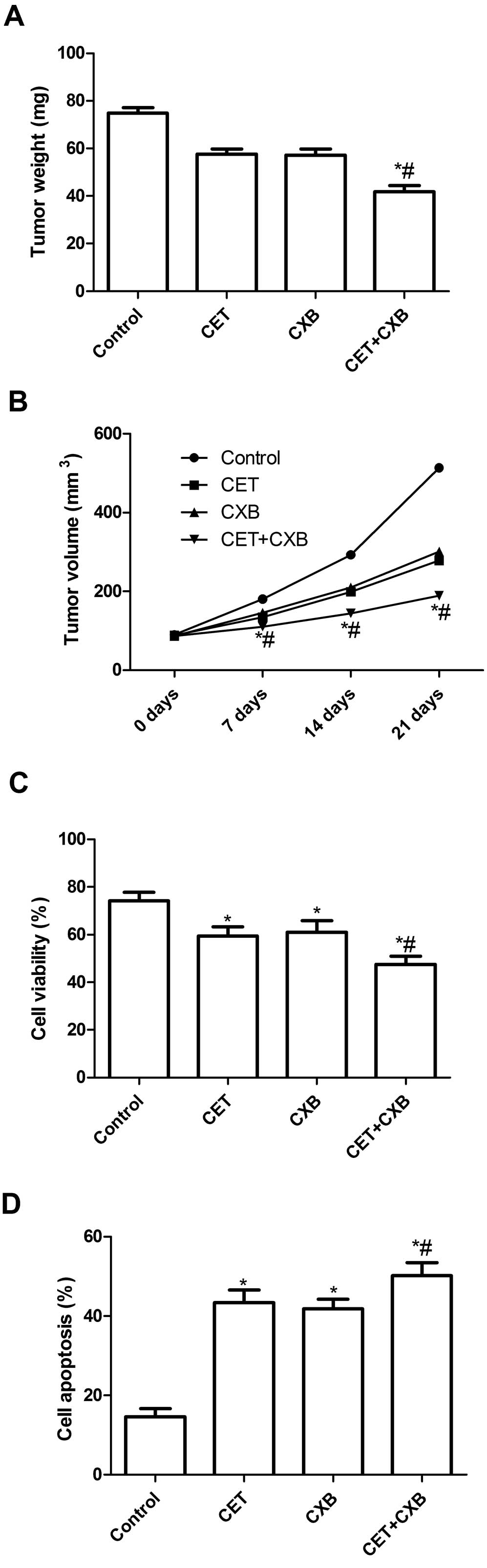
Effect of CET in combination with CXB on
tumor growth
We assessed the in vivo therapeutic efficacy
of CET and CXB in female BALB mice bearing HSC3 tumor cells. Mice
were sacrificed and tumor tissues were excised at the end of 7
days. Tumor weight of the animals was then measured. It was found
that the tumor weight of all treatment groups was lower than that
of the untreated group. Compared to the single drug groups, the
tumor weight of the combination group was the lowest (P<0.05,
Fig. 6A). In addition, we also
found that the tumor volume in the combination group was
significantly lower compared with that in the single drug groups
and the untreated group (P<0.05, Fig. 6B).
To assess the efficacy of CET and CXB in modulating
splenocyte proliferation, MTT assay was performed. As shown in
Fig. 6C, the inhibitory rate of the
CET combination treatment group was higher than that of the control
group and single drug groups (P<0.05). In addition, we also
determined tumor tissue cell apoptosis in vivo by TUNEL. The
cell apoptosis ratios of all treatment groups in vivo were
significantly higher than the ratio of the control group
(P<0.05, Fig. 6D). Compared to
the single drug groups, the cell apoptosis ratio in the combination
groups was markedly increased. Taken together, these results
demonstrated that the CET and CXB combination suppressed tumor
growth of oral squamous cell carcinoma in vivo.
Discussion
In the present study, we provide convincing evidence
that the combined tumor treatment with cetuximab (CET) and
celecoxib (CXB) significantly reduced oral squamous cell carcinoma
cell proliferation, migration and invasion and induced cell
apoptosis in vitro and inhibited OSCC tumor growth in
vivo, whereas the single treatments did not significantly
improve the anticancer effect. Although both compounds have each
been extensively studied, to our knowledge, this study is the first
to show that combining a clinically applied therapy with CET and
CXB inhibits OSCC tumor growth in vivo and in
vitro.
The epidermal growth factor receptor (EGFR) pathway
plays key roles in cell proliferation, adhesion, invasion, survival
and angiogenesis. Tumors in which the EGFR pathway is activated
include lung cancer, OSCC, pancreatic cancer, breast cancer and
head and neck cancer (24).
Antibodies and small-molecule inhibitors acting on different parts
of the EGFR pathway have shown effectiveness as antineoplastic
agents in these cancers (25),
while monoclonal antibodies have shown low efficacy in oral
squamous cell carcinoma due to intrinsic and acquired resistance
(18), which require a combination
of drugs. For example, Park et al showed that a combination
of CET and genistein significantly inhibited tumor growth and
caused a substantial growth delay in in vivo models of both
cell lines while each exposure to a single-agent caused no delay of
tumor growth of OSCC (26). Maseki
et al found that gemcitabine and CET are effective drugs
against HNSCC, and an enhanced antitumor effect may be expected
when using gemcitabine in combination with CET (27). These studies revealed that CET in
combination with other anticancer drugs contributes to the
treatment of cancer. In the present study, we selected the
combination of CET and CXB (a COX-2 inhibitor) for the treatment of
OSCC as COX-2 overexpression leads to EGFR expression, which
enhances tumor growth (28). In
addition, preclinical data showed that upregulation of COX-2 in
tumor cells resulted in increased PGE2 production and increased
tumor invasiveness (29). Increased
PGE2 production contributes to active EGFR and results in the
activation of Ras and the mitogen-activated protein kinase (MAPK)
pathway (30). Upregulation of
COX-2 in tumor cells also promoted proangiogenic factors such as
VEGF, bFGF, PDGF and TGF-β1 (31),
all of which favor tumor growth and dissemination. Importantly, Xia
et al showed that CET in combination with CXB
synergistically inhibited the growth of A549 cells and
downregulated the expression of KDR and AQP1 in A549 cells
(32). Jalili et al reported
that tumors regressed partially and the patients Karnofsky index
improved when the combination of CET and CXB was used to treat an
88-year-old man presenting a recurrent, locoregionally metastatic
squamous cell carcinoma (SCC) of the right parietal region, which
was resistant to radiotherapy (33). In the present study, our results
revealed that the combination of low concentrations of CET and CXB
significantly suppressed the proliferation, migration and invasion
of HSC3 cells in vitro, and decreased PEG2 production and
VEGF expression in vitro and inhibited tumor growth in
vivo compared to the actions of either agent alone. These
results are in agreement with previous results (32).
Chemotherapeutic drugs are most effective when
administered in combination (combined chemotherapy) since
chemotherapy drugs with different mechanisms of action are used,
which contribute to a decreased possibility of drug resistance of
cancer cells. In addition, drugs are combined having different
effects, and each drug can be used at its optimal dose, without
intolerable side effects (34).
Currently, growing evidence suggests that the anticancer activity
of standard chemotherapeutic agents can be enhanced by using CXB
(35). For instance, Zhang et
al showed that the combination of low concentrations of
sorafenib (SOR) and CXB in A549 tumor cells significantly
suppressed the proliferation in vitro and suppressed tumor
growth in vivo compared to the actions of either agent alone
(23). Morisaki et al showed
that the combination of low concentrations of SOR (<5 μM) and
CXB (<20 μM) resulted in enhanced inhibition of cell growth and
AKT activation in hepatocellular carcinoma (HCC), and increased the
induction of apoptosis of HCC cells when compared to the actions of
either agent alone, and that the growth inhibitory effect was
synergistic by combination index (CI) analysis (36). Jeon et al demonstrated that a
combination of CXB and luteolin could provide superior inhibition
of breast cancer cell growth than either CXB or luteolin treatment
alone (37). Consistent with these
results, our data showed that CET in combination with CXB
significantly inhibited cell proliferation and induced cell
apoptosis compared to each single drug. These results further
confirm that CXB in combination with anticancer drugs can improve
their antitumor effects.
In conclusion, the present study showed that CET in
combination with CXB enhanced the anti-proliferative and
pro-apoptotic effects on oral squamous cell carcinoma in
vitro and inhibited OSCC tumor growth in a nude mouse model
allowing for the use of lower doses of CET and CXB than those
currently used. Therefore, it may be worthwhile to consider their
combination for the treatment of oral squamous cell carcinoma and
this combination further warrants evaluation in clinical
trials.
Acknowledgements
This research was supported by the Youth Science
Fund of the Innovation Team of the Department of Science and
Technology of Jilin Province (20130522036JH).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar
|
|
2
|
Scully C and Bagan J: Oral squamous cell
carcinoma overview. Oral Oncol. 45:301–308. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Saraswathi TR, Kavitha B and Vijayashree
Priyadharsini J: Gene therapy for oral squamous cell carcinoma: an
overview. Indian J Dent Res. 18:120–123. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shurin MR, Umansky V, Malyguine A, et al:
Cellular and molecular pathways in the tumor immunoenvironment: 3rd
Cancer Immunotherapy and Immunomonitoring (CITIM) meeting, 22–25
April 2013, Krakow, Poland. Cancer Immunol Immunother. 63:73–80.
2014.PubMed/NCBI
|
|
5
|
Sihver W, Pietzsch J, Krause M, Baumann M,
Steinbach J and Pietzsch HJ: Radiolabeled cetuximab conjugates for
EGFR targeted cancer diagnostics and therapy. Pharmaceuticals.
7:311–338. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Olayioye MA, Neve RM, Lane HA and Hynes
NE: The ErbB signaling network: receptor heterodimerization in
development and cancer. EMBO J. 19:3159–3167. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
de Bono JS and Rowinsky EK: The ErbB
receptor family: a therapeutic target for cancer. Trends Mol Med.
8:S19–S26. 2002.PubMed/NCBI
|
|
8
|
Carpenter G: Receptors for epidermal
growth factor and other polypeptide mitogens. Annu Rev Biochem.
56:881–914. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Salomon DS, Brandt R, Ciardiello F and
Normanno N: Epidermal growth factor-related peptides and their
receptors in human malignancies. Crit Rev Oncol Hematol.
19:183–232. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wells A: EGF receptor. Int J Biochem Cell
Biol. 31:637–643. 1999. View Article : Google Scholar
|
|
11
|
Yarden Y: The EGFR family and its ligands
in human cancer: signalling mechanisms and therapeutic
opportunities. Eur J Cancer. 37:S3–S8. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wikstrand CJ, Hale LP, Batra SK, et al:
Monoclonal antibodies against EGFRvIII are tumor specific and react
with breast and lung carcinomas and malignant gliomas. Cancer Res.
55:3140–3148. 1995.PubMed/NCBI
|
|
13
|
Herbst RS and Langer CJ: Epidermal growth
factor receptors as a target for cancer treatment: the emerging
role of IMC-C225 in the treatment of lung and head and neck
cancers. Semin Oncol. 29:27–36. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sridhar SS, Seymour L and Shepherd FA:
Inhibitors of epidermal-growth-factor receptors: a review of
clinical research with a focus on non-small-cell lung cancer.
Lancet Oncol. 4:397–406. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Humblet Y: Cetuximab: an IgG(1) monoclonal
antibody for the treatment of epidermal growth factor
receptor-expressing tumours. Expert Opin Pharmacother. 5:1621–1633.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Harding J and Burtness B: Cetuximab: an
epidermal growth factor receptor chemeric human-murine monoclonal
antibody. Drugs Today. 41:107–127. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Park SJ, Kim MJ, Kim YK, Kim SM, Park JY
and Myoung H: Combined cetuximab and genistein treatment shows
additive anti-cancer effect on oral squamous cell carcinoma. Cancer
Lett. 292:54–63. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tol J, Koopman M, Cats A, et al:
Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal
cancer. N Engl J Med. 360:563–572. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hattar K, Savai R, Subtil FS, et al:
Endotoxin induces proliferation of NSCLC in vitro and in vivo: role
of COX-2 and EGFR activation. Cancer Immunol Immunother.
62:309–320. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wei D, Wang L, He Y, Xiong HQ, Abbruzzese
JL and Xie K: Celecoxib inhibits vascular endothelial growth factor
expression in and reduces angiogenesis and metastasis of human
pancreatic cancer via suppression of Sp1 transcription factor
activity. Cancer Res. 64:2030–2038. 2004. View Article : Google Scholar
|
|
21
|
Jeon YW and Suh YJ: Synergistic apoptotic
effect of celecoxib and luteolin on breast cancer cells. Oncol Rep.
29:819–825. 2013.PubMed/NCBI
|
|
22
|
Kao J, Sikora AT and Fu S: Dual EGFR and
COX-2 inhibition as a novel approach to targeting head and neck
squamous cell carcinoma. Curr Cancer Drug Targets. 9:931–937. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang H, Li Z and Wang K: Combining
sorafenib with celecoxib synergistically inhibits tumor growth of
non-small cell lung cancer cells in vitro and in
vivo. Oncol Rep. 31:1954–1960. 2014.PubMed/NCBI
|
|
24
|
Ratti M and Tomasello G: Emerging
combination therapies to overcome resistance in EGFR-driven tumors.
Anticancer Drugs. 25:127–139. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pal SK and Pegram M: Epidermal growth
factor receptor and signal transduction: potential targets for
anti-cancer therapy. Anticancer Drugs. 16:483–494. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Park SJ, Kim MJ, Kim YK, Kim SM, Park JY
and Myoung H: Combined cetuximab and genistein treatment shows
additive anti-cancer effect on oral squamous cell carcinoma. Cancer
Lett. 292:54–63. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Maseki S, Ijichi K, Nakanishi H, Hasegawa
Y, Ogawa T and Murakami S: Efficacy of gemcitabine and cetuximab
combination treatment in head and neck squamous cell carcinoma. Mol
Clin Oncol. 1:918–924. 2013.PubMed/NCBI
|
|
28
|
Kinoshita T, Takahashi Y, Sakashita T,
Inoue H, Tanabe T and Yoshimoto T: Growth stimulation and induction
of epidermal growth factor receptor by overexpression of
cyclooxygenases 1 and 2 in human colon carcinoma cells. Biochim
Biophys Acta. 1438:120–130. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tsujii M, Kawano S and DuBois RN:
Cyclooxygenase-2 expression in human colon cancer cells increases
metastatic potential. Proc Natl Acad Sci USA. 94:3336–3340. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sheng H, Shao J, Dixon DA, Williams CS,
Prescott SM, DuBois RN and Beauchamp RD: Transforming growth factor
beta1 enhances Ha-ras-induced expression of cyclooxygenase-2 in
intestinal epithelial cells via stabilization of mRNA. J Biol Chem.
275:6628–6635. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tsujii M, Kawano S, Tsuji S, Sawaoka H,
Hori M and DuBois RN: Cyclooxygenase regulates angiogenesis induced
by colon cancer cells. Cell. 93:705–716. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xia H, Ye J, Bai H and Wang C: Effects of
cetuximab combined with celecoxib on apoptosis and KDR and AQP1
expression in lung cancer. Zhongguo Fei Ai Za Zhi. 16:625–631.
2013.(In Chinese).
|
|
33
|
Jalili A, Pinc A, Pieczkowski F, Karlhofer
FM, Stingl G and Wagner SN: Combination of an EGFR blocker and a
COX-2 inhibitor for the treatment of advanced cutaneous squamous
cell carcinoma. J Dtsch Dermatol Ges. 6:1066–1069. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ullah MF: Cancer multidrug resistance
(MDR): a major impediment to effective chemotherapy. Asian Pac J
Cancer Prev. 9:1–6. 2008.PubMed/NCBI
|
|
35
|
Irie T, Tsujii M, Tsuji S, et al:
Synergistic antitumor effects of celecoxib with 5-fluorouracil
depend on IFN-gamma. Int J Cancer. 121:878–883. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Morisaki T, Umebayashi M, Kiyota A, et al:
Combining celecoxib with sorafenib synergistically inhibits
hepatocellular carcinoma cells in vitro. Anticancer Res.
33:1387–1395. 2013.PubMed/NCBI
|
|
37
|
Jeon YW and Suh YJ: Synergistic apoptotic
effect of celecoxib and luteolin on breast cancer cells. Oncol Rep.
29:819–825. 2013.PubMed/NCBI
|