Introduction
DNA methylation determines spatial and temporal
silencing of gene expression (1),
and the aberrant methylation of CpG islands located in the promoter
regions of genes that regulate cell proliferation is a frequent
event in most types of cancers (2).
Methylation is often an early phenomenon during carcinogenesis
(3–5), and the methylation status of specific
genes has been proposed as an appropriated biomarker for early
detection of lung cancer (LC) (4,6,7). In
patients at risk for this disease, the assessment of the
methylation status of these genes in bronchial secretions, such as
sputum, bronchial washing and bronchial lavage may be useful for
the identification of subjects candidate for additional procedures
(6). Most of the studies requiring
respiratory samples have relied on sputum for the definition of
biomarkers with predictive ability (3,8).
Sputum samples are easily obtained, but contain a mixture of
inflammatory and oral cells, with an epithelial fraction that
comprises <3% of the sample. This cellular heterogeneity, which
varies across subjects, limits the possibilities for methylation
status assessment (3). Bronchial
washing (BW) samples can be obtained semi-invasively and contain a
higher proportion of epithelial cells (9). Therefore, BW may be more appropriate
for methylation studies of bronchial cells.
Methods based on methylation-sensitive PCR (MSP)
primers are used to identify low levels of DNA methylation and may
be useful for bronchial secretions (10–12).
With this procedure, two sets of primers allow the amplification of
the sequence of interest, with one pair recognizing the methylated
sequence and the other pair the unmethylated one. Subsequent
product analysis is performed in most cases by gel electrophoresis
(13). MSP does not require
specialized equipment and is sufficiently sensitive to identify
0.1% of the methylated template. However, the method is
non-quantitative and does not fully avoid false-positive results in
single sample analyses, since it is not a closed tube method
(14,15).
Methylation-sensitive high-resolution melting
(MS-HRM) analysis is a recently developed methodology with enormous
potential for the detection of DNA sequence changes (16). With this technique, sequence
differences between methylated and unmethylated DNA obtained after
bisulfite treatment can be analyzed by melting curve analysis. As
the PCR product originating from the methylated allele has a
different GC content from the PCR product obtained from the
unmethylated variant, the two products have distinct melting
temperatures (17). In this case,
the methylation level is estimated by comparing the melting
profiles of samples and standards of known ratios of methylated and
unmethylated DNA (15).
The goal of this study was to determine whether
methylation results in sputum samples parallel results obtained in
BW, a sample with a higher proportion of epithelial cells and the
effect of the used methodology on these results. To do so we
analyzed the methylation levels of three genes related to cancer
development in sputum and BW obtained from subjects at risk for LC
(7). Death-associated protein
kinase (DAPK), involved in apoptosis; cyclin-dependent kinase
inhibitor 2A gene (CDKN2A/p16), a tumor-suppressor gene which plays
a key role in cell cycle control; and ras effector homolog 1 gene
(RASSF1A), related to cell cycle progression at the G1-S
transition, were the assessed genes. Sputum and BW were analyzed,
and the methylation status after two different amplification
methods, MSP and MS-HRM, were compared to assess the impact of the
methodology on the identification of DNA methylation in these
bronchial samples.
Materials and methods
Ethics statement
The research protocol was approved by the ethics
committees of the affiliated hospitals, and written informed
consent was obtained from all subjects. Sputum and BW recoveries
were obtained in accordance with scheduled explorations for
standard care, which included diagnostic and follow-up
bronchoscopies for the enrolled individuals.
Design and population
This is a cross-sectional study in which bronchial
secretions of subjects at risk for LC were analyzed to detect the
methylation levels of three carcinogenesis-related genes. Included
subjects were smokers of >30 pack-years with no signs or
symptoms of LC and a normal chest X-ray when examined and not
treated for any cancer in the previous 10 years.
Sampling procedure
Induced sputum samples were obtained and processed
within 60 min at enrollment using standard methods (18,19).
Briefly, the subject was pretreated with an inhaled β2-agonist 10
min before the nebulization of isotonic saline (0.9%), followed by
increasing concentrations of hypertonic saline (3, 4 and 5%) for 7
min with each concentration. After each induction, the patient
attempted to provide a sputum sample by coughing, and the
nebulization procedure was discontinued when the sputum volume
collected was equal or higher than 1 ml. Sputum induction was
followed by a bronchoscopy performed under local anesthesia and
sedation, using a flexible video bronchoscope (BF180; Olympus
Optical Co., Tokyo, Japan). Local anesthesia and sedation were
achieved using topical lidocaine spray and intravenous midazolam
respectively, in accordance with standard recommendations (20,21).
BW was obtained aspirating bronchial secretions through the working
channel during the procedure.
Samples and DNA extraction
Sputum and BW samples were diluted with 5 ml of a
1:10 dilution of dithiothreitol (Sputasol; Oxoid, Basingstoke,
Hants, UK), centrifuged and the cellular fraction of the sample
separated and stored at −80°C until DNA extraction. The QIAamp DNA
Mini Kit and the QIAcube (both from Qiagen, Hilden, Germany) were
used for extraction, in accordance with the manufacturer’s
instructions, and DNA was quantified with a Nanodrop ND-1000
spectrophotometer (NanoDrop Technologies Inc., Wilmington, DE,
USA). Universal methylated DNA and unmethylated DNA (Chemicon,
Millipore, Billerica, MA, USA) were used as a fully methylated
positive control and unmethylated negative control,
respectively.
Bisulfite modification
One miligram of DNA was modified by treatment with
sodium bisulfite and then purified with the Wizard DNA purification
kit (Promega, Madison, WI, USA), desulfonated with NaOH,
precipitated with sodium acetate and ethanol, resuspended with 30
μl of PCR grade H2O and stored at −20°C for further
determinations (22). For the
MS-HRM study, DNA was modified using the EZ DNA Methylation-Gold
kit (Zymo Research Co., Irvine, CA, USA) according to
manufacturer’s instructions.
Primer design
Previously described primers were used for MSP
analysis (22,23) (Table
I). For HRM analysis, DAPK and RASSF1A primers were designed
with Primer Express software (Applied Biosystems, Foster City, CA,
USA), without any CpG dinucleotide in the sequence, determining the
annealing temperature experimentally to avoid the PCR bias
phenomenon. P16 primers for HRM analysis have been described
elsewhere (24) (Table II).
 | Table IMSP primers. |
Table I
MSP primers.
| Gene | Sequence | Annealing temperature
(°C) | Amplicon size
(bp) | Spanned region |
|---|
| p16 |
MF-ttattagagggtggggcggatcgcgtgc | 67 | 154 | ENSG000001478889 |
|
MR-acccgaccccgaaccgcgaccgtaa | | | 21,974,907- |
|
UF-ttattagagggtggggtggattgt | 65 | | 21,974,753 |
|
UR-caaccccaaaccacaaccataa | | | |
| DAPK |
MF-ggatagtcggatcgagttaacgtc | 60 | 108 | ENSG00000196730 |
|
MR-ccctcccaaacgccga | | | 90,112,771- |
|
UF-ggaggatagttggattgagttaatgtt | 60 | | 90,112,879 |
|
UR-caaatccctcccaaacaccaa | | | |
| RASSF1A |
MF-gggttttgcgagagcgcg | 62 | 170 | ENSG00000068028 |
|
MR-gccaagcgcaaacaatcg | | | 50,378,432- |
|
UF-ggttttgtgagagtgtgtttag | 62 | | 50,378,262 |
|
UR-cactaacaaacacaaaccaaac | | | |
| Table IIMS-HRM primers. |
Table II
MS-HRM primers.
| Gene | Sequence | Annealing
temperature (°C) | Amplicon size
(bp) | CpG between
primers | Spanned region |
|---|
| p16 |
F-gaagaaagaggaggggttggttggttatt | 68 | 84 | 6 |
ENSG000001478889 |
|
R-acctactctccccctctccgcaa | | | | 21,974,931-847 |
| DAPK |
F-gtttgtagggtttttattggt | 59 | 94 | 7 |
ENSG00000196730 |
|
R-actatcctcctcacactcc | | | | 90,112,712-806 |
| RASSF1A |
F-gtttagtttggattttggg | 60 | 139 | 12 |
ENSG00000068028 |
|
R-aactcaataaactcaaactcc | | | | 50,378,338-199 |
Methylation-specific PCR (MSP)
Two set of primers were used for specific
amplification of each region of interest with this technique. One
pair recognized the unmethylated sequence, whose cytosines were
changed to uracils by bisulfite modification and the other pair the
methylated sequence. Primer sequences and PCR conditions were
adjusted for each pair of primers and are described in Table I. Positive and negative methylation
controls were analyzed in each assay. PCRs were carried out in a
volume of 20 μl, which contained 1X reaction buffer, 1.5 mM of
MgCl2, 0.2 μM of each primer, 1 mM of dNTPs and 0.6
units of Taq polymerase (Qiagen). The procedure was
performed on a Thermal Cycler 2720 (Applied Biosystems). After
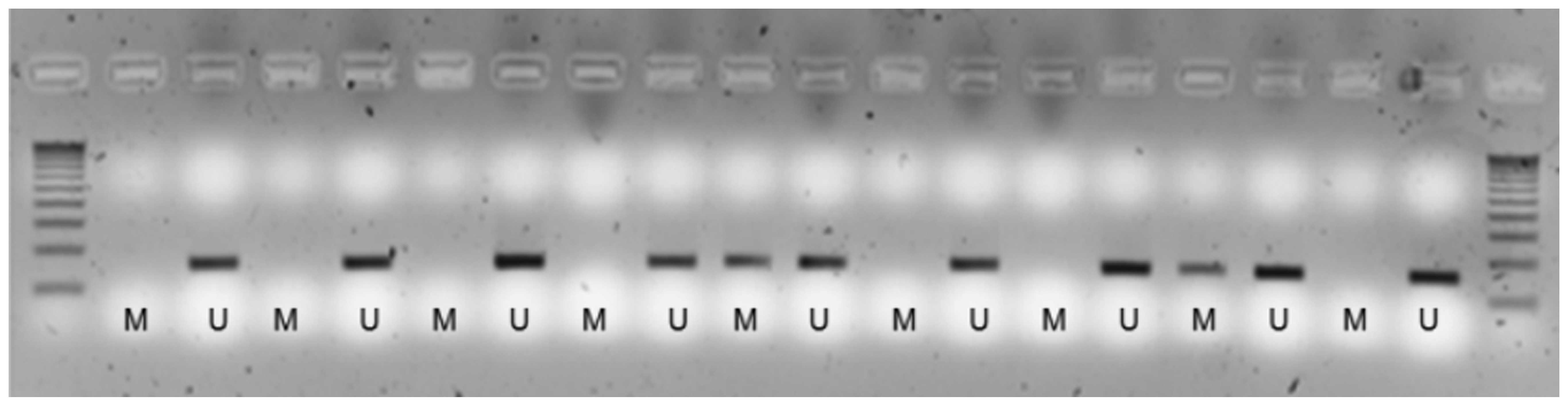
amplification, the products were visualized on 2% agarose gels with
ethidium bromide staining (Fig. 1).
Methylation assessment was conducting in duplicate, starting with
bisulfite modification. A positive result in 1 out of 2 was taken
as positive for methylation (3,25).
Methylation-sensitive high-resolution
melting (MS-HRM)
PCR cycling and HRM analysis were performed on a
LightCycler 480 II (Roche Applied Science, Mannheim, Germany).
Reactions were carried out in a total volume of 10 μl containing 2X
PCR Master Mix, 2.5–3 mM of MgCl2, 2–3 μM of each
primer, depending on the gene studied and 2 μl of the template. The
annealing temperatures were experimentally determined for each
assay to compensate for PCR bias, as shown in Table II.
Each assay was optimized so that no amplification
was observed in the unmodified control or in the non-template
control. Standard series of 100, 50, 30, 10 and 5% methylation
levels were prepared by diluting the fully methylated DNA into the
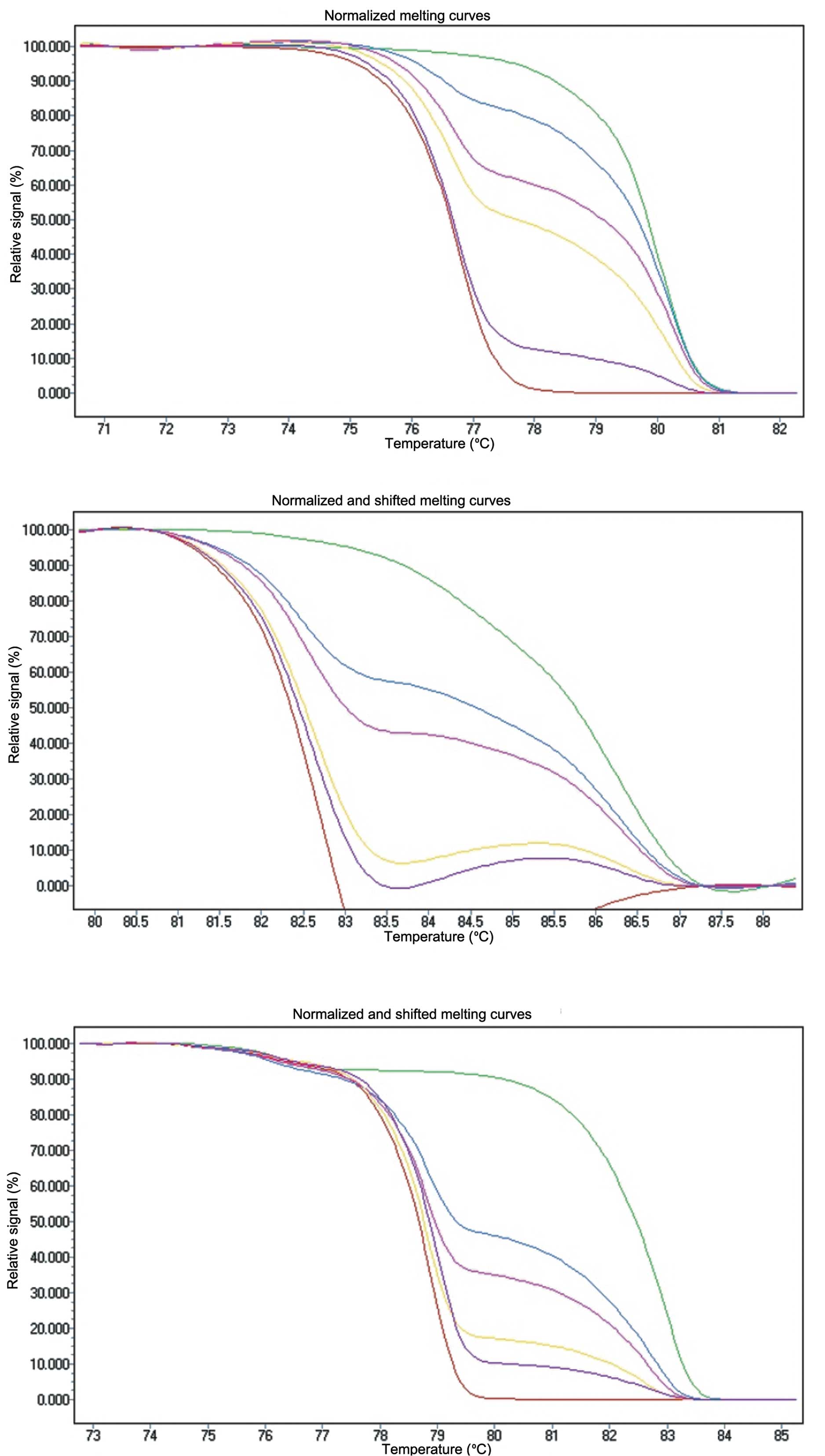
unmethylated DNA and were used as controls. The methylation level
of each sample was assessed by comparison of the PCR product
melting profile and standards with a known ratio of methylated and
unmethylated templates (Fig. 2).
The PCR bias of each assay was successfully corrected with the
annealing temperature and proportional amplifications of the
standard with 50% of methylated DNA were seen in a background of
unmethylated DNA. All samples were run in duplicate. Methylation
assessment was conducting in duplicate, starting with bisulfite
modification. A positive result in one out of two was taken as
positive for methylation.
Statistical analysis
Data were analyzed using SPSS Statistical Software
package version 18 (SPSS Inc., Chicago, IL, USA). Results for
categorical variables are expressed as absolute and relative
frequencies and results for continuous variables as means and
standard deviations (SD), or as medians and percentiles 25–75
(P25–P75) when the distribution was not normal.
First, clinical and functional variables of the
studied subjects were described, and the methylation results
obtained in sputum and in BW were compared. Considering BW as the
reference, the sensitivity and specificity of sputum were
calculated. Secondly, the results obtained after the two different
PCR methods were compared, to determine their specific advantages
in the study of bronchial secretions. All analyses were performed
using the Chi-square, Fisher’s exact, McNemar or Mann-Whitney U
test as required. Statistical tests were two-sided, and a P-value
of 0.05 or less was reported as statistically significant.
Results
Characteristics of the participants and
analysis of respiratory secretions
Bronchial secretions from 65 subjects with an
average age of 62 years (SD 9.4) were analyzed in this study.
Sixty-two of the enrolled subjects were men (95.4%) and their lung
function showed a mean post-bronchodilator forced expiratory volume
in 1 sec of 72% (SD 23) of the predicted value. All participants
were current or former smokers with a heavy cumulative smoking
history [median 48 (interquartile range 30–65) pack-years]. Ten
subjects had a history of LC (15%) but had been free from
neoplastic disease the 10 previous years.
First, sputum and BW were manually modified and the
methylation status of the DAPK, p16 and RASSF1A genes was
determined by MSP (Fig. 3).
Differences between these two samples were not statistically
significant for any of the studied genes (DAPK, P=0.690; p16,
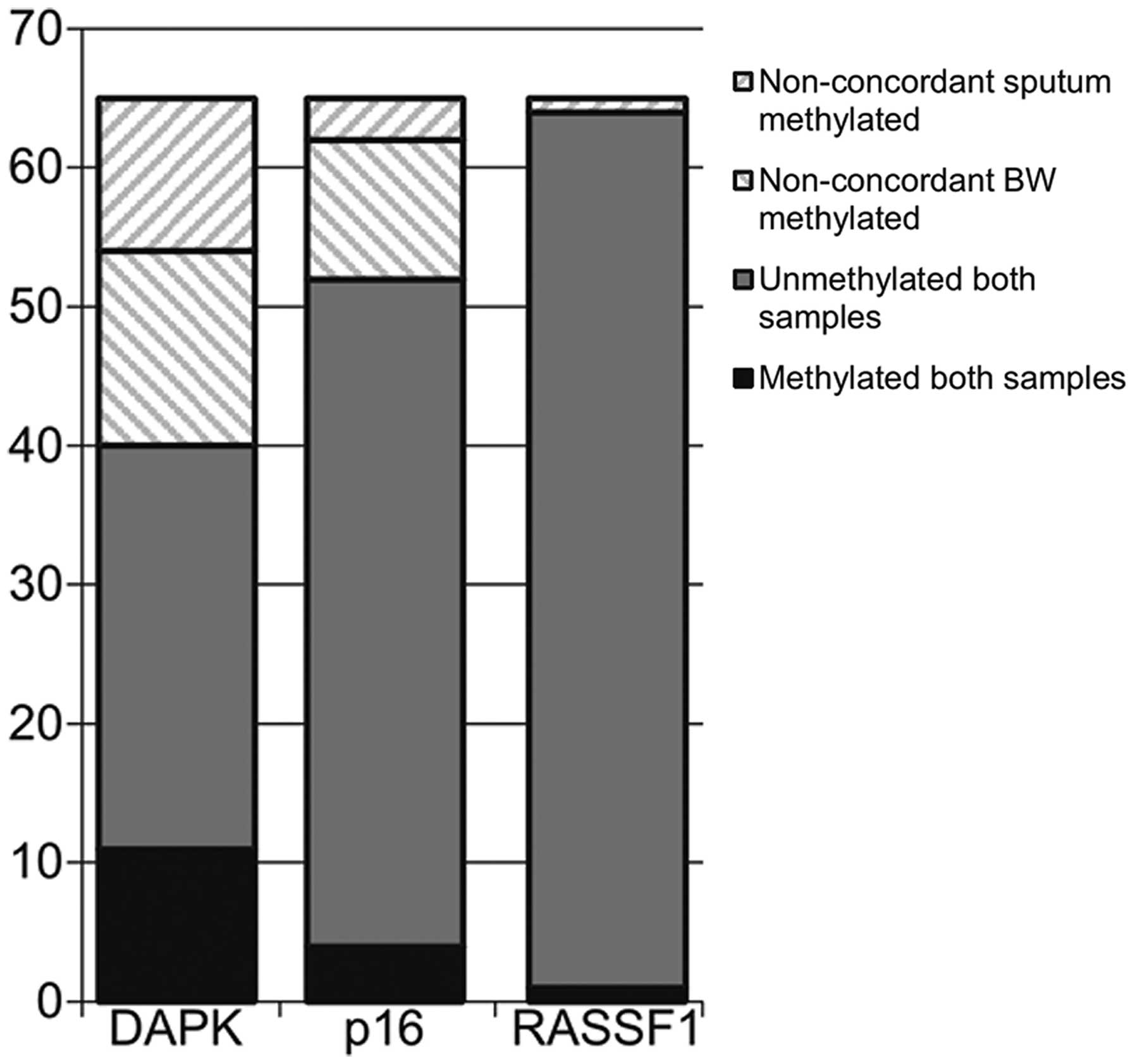
P=0.092; RASSF1, P=1.00; McNemar test). Concordant results between
sputum and BW were found in 40 patients for DAPK (61%), in 52
patients for p16 (80%) and in 63 patients for RASSF1 (97%)
(Fig. 4). More methylated samples
were found in BW, however, and considering this sample as the
reference, sputum sensitivity and specificity for the
identification of methylation status were calculated. Sensitivities
and specificities were 44 and 72% for the DAPK gene, 21 and 94% for
p16 and 100 and 98% for RASSF1A, respectively.
Comparison of MSP and MS-HRM
techniques
Forty samples of bronchial secretions were available
for a second testing by MS-HRM, for the comparison of MSP and
MS-HRM techniques. Twenty-three methylated samples for the DAPK
gene and 6 for p16 after MSP emerged to be unmethylated with the
MS-HRM technique, with a limit of detection of 5%. For RASSF1A, the
sample that appeared methylated by MSP showed the same result with
MS-HRM.
Discussion
In the present study, although the methylation
status of DAPK, p16 and RASSF1A genes was similar in single samples
of sputum and BW, higher levels of positive results were obtained
with BW for DAPK and p16 genes, a result attributable to the higher
proportion of epithelial cells in this sample (6,9). The
lack of statistically significant differences between both samples
of bronchial secretions confirms that sputum may be considered
useful as a source of DNA for the identification of epigenetic
changes in bronchial secretions, and the fact that this sample is
obtained non-invasively offers a significant advantage for
screening. Our results, however, suggest that single sputum samples
may underestimate the methylation status of bronchial cells and
support the use of repeated sputum samples, or its combination with
BW, to guarantee an accurate assessment of the methylation status
of the bronchial tree (26).
The methylation status was assessed again in a
subgroup of samples using MS-HRM to compare the obtained results
with two different techniques. This is a sensitive technique that
analyzes DNA methylation in a semi-quantitative manner, using
methylation-independent PCR primers which identify genes with
methylation rates over predetermined cutoffs (10,17,27,28).
The efficiency of the amplification of the methylated and
unmethylated templates with this procedure may differ (12,29), a
bias that is overcome by increasing the annealing temperature
(29). For DAPK and p16 genes, all
samples analyzed with the MS-HRM technique turned out to be
unmethylated, while the sample that appeared methylated for RASSF1A
gene when analyzed with MSP, showed the same methylation status
when tested by MS-HRM. The discrepancy in the obtained results
between MSP and MS-HRM is probably attributable to a low prevalence
of methylation in the studied sample, as MS-HRM was less sensitive
than MSP and only identifies concentrations over 5%. However,
false-positive results of MSP have been reported (10), and the confirmation of MSP-positive
results with another technique should be recommended. The use of
MS-HRM for this confirmation in bronchial secretions needs to take
into account the low proportion of methylation and accordingly,
adjust its detection limit. An alternative option to approach this
situation would be the reassessment of methylated samples by
pyrosequencing (27), a sensitive
and specific approach but more expensive in its performance. Then,
regardless of the technique chosen for methylation analysis,
analyzing sputum at least twice in independent samples and
confirming positive results in bronchial secretions that appear
repeatedly methylated may be recommended for the identification of
true positive results.
In conclusion, although DNA methylation has been
proposed as a biomarker for early detection of LC, techniques for
the measure of methylation status have not been standardized, and
the genes that need to be included in the analyses are not defined.
Our results confirmed that sputum and BW samples are equally valid
for the recognition of methylation of DAPK, p16 and RASSF1A genes
in bronchial secretions. Sputum may be used as a preferential
sample for the examination of methylation status as it is a sample
obtained non-invasively, although the analysis of independent
samples to increase the sensitivity of the test may be recommended.
Due to the variability in the results according to the technique
used, however, the use of combined techniques for the confirmation
of positive results may increase the reliability of the methylation
analysis of DAPK, p16 and RASSF1A genes.
Acknowledgements
We thank Michael Maudsley for providing an outline
for this manuscript and for his support in editing and journal
styling. This study was funded by Fondo de Investigación Sanitaria
(http://www.isciii.es/) (PI08/1042 and
PI12/02040), Fundació Parc Taulí (http://www.tauli.cat), Fundació Catalana de
Pneumologia (FUCAP) (http://www.fucap.org), PII Oncologia SEPAR (http://www.separ.es/) and CIBER de Enfermedades
Respiratorias - CIBERES (http://www.ciberes.org). CIBERES is an initiative of
Instituto de Salud Carlos III.
References
|
1
|
Adcock IM, Tsaprouni L, Bhavsar P and Ito
K: Epigenetic regulation of airway inflammation. Curr Opin Immunol.
19:694–700. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Barlesi F, Giaccone G, Gallegos-Ruiz MI,
Loundou A, Span SW, Lefesvre P, et al: Global histone modifications
predict prognosis of resected non small-cell lung cancer. J Clin
Oncol. 25:4358–4364. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Leng S, Do K, Yingling CM, Picchi MA, Wolf
HJ, Kennedy TC, et al: Defining a gene promoter methylation
signature in sputum for lung cancer risk assessment. Clin Cancer
Res. 18:3387–3395. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Malentacchi F, Forni G, Vinci S and
Orlando C: Quantitative evaluation of DNA methylation by
optimization of a differential-high resolution melt analysis
protocol. Nucleic Acids Res. 37:e862009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mikeska T, Bock C, Do H and Dobrovic A:
DNA methylation biomarkers in cancer: progress towards clinical
implementation. Expert Rev Mol Diagn. 12:473–487. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Anglim PP, Alonzo TA and Laird-Offringa
IA: DNA methylation-based biomarkers for early detection of
non-small cell lung cancer: an update. Mol Cancer. 7:812008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim H, Kwon YM, Kim JS, Lee H, Park JH,
Shim YM, et al: Tumor-specific methylation in bronchial lavage for
the early detection of non-small-cell lung cancer. J Clin Oncol.
22:2363–2370. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hubers AJ, van der Drift MA, Prinsen CF,
Witte BI, Wang Y, Shivapurkar N, et al: Methylation analysis in
spontaneous sputum for lung cancer diagnosis. Lung Cancer.
84:127–133. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fahy JV, Wong H, Liu J and Boushey HA:
Comparison of samples collected by sputum induction and
bronchoscopy from asthmatic and healthy subjects. Am J Respir Crit
Care Med. 152:53–58. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kristensen LS and Hansen LL: PCR-based
methods for detecting single-locus DNA methylation biomarkers in
cancer diagnostics, prognostics, and response to treatment. Clin
Chem. 55:1471–1483. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mikeska T, Candiloro IL and Dobrovic A:
The implications of heterogeneous DNA methylation for the accurate
quantification of methylation. Epigenomics. 2:561–573. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Warnecke PM, Stirzaker C, Song J, Grunau
C, Melki JR and Clark SJ: Identification and resolution of
artifacts in bisulfite sequencing. Methods. 27:101–107. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lorente A, Mueller W, Urdangarin E, Lazcoz
P, von Deimling A and Castresana JS: Detection of methylation in
promoter sequences by melting curve analysis-based semiquantitative
real time PCR. BMC Cancer. 8:612008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kristensen LS, Mikeska T, Krypuy M and
Dobrovic A: Sensitive melting analysis after real time-methylation
specific PCR (SMART-MSP): high-throughput and probe-free
quantitative DNA methylation detection. Nucleic Acids Res.
36:e422008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kristensen LS, Wojdacz TK, Thestrup BB,
Wiuf C, Hager H and Hansen LL: Quality assessment of DNA derived
from up to 30 years old formalin fixed paraffin embedded (FFPE)
tissue for PCR-based methylation analysis using SMART-MSP and
MS-HRM. BMC Cancer. 9:4532009.PubMed/NCBI
|
|
16
|
Do H, Krypuy M, Mitchell PL, Fox SB and
Dobrovic A: High resolution melting analysis for rapid and
sensitive EGFR and KRAS mutation detection in formalin fixed
paraffin embedded biopsies. BMC Cancer. 8:1422008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wojdacz TK, Dobrovic A and Hansen LL:
Methylation-sensitive high-resolution melting. Nat Protoc.
3:1903–1908. 2008. View Article : Google Scholar
|
|
18
|
Pin I, Gibson PG, Kolendowicz R,
Girgis-Gabardo A, Denburg JA, Hargreave FE and Dolovich J: Use of
induced sputum cell counts to investigate airway inflammation in
asthma. Thorax. 47:25–29. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pizzichini E, Pizzichini MM, Efthimiadis
A, Evans S, Morris MM, Squillace D, et al: Indices of airway
inflammation in induced sputum: reproducibility and validity of
cell and fluid-phase measurements. Am J Respir Crit Care Med.
154:308–317. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Du Rand IA, Blaikley J, Booton R,
Chaudhuri N, Gupta V, Khalid S, et al: British Thoracic Society
guideline for diagnostic flexible bronchoscopy in adults. Thorax.
68:i1–i44. 2013.
|
|
21
|
Reed AP: Preparation of the patient for
awake flexible fiberoptic bronchoscopy. Chest. 101:244–253. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Herman JG, Graff JR, Myohanen S, Nelkin BD
and Baylin SB: Methylation-specific PCR: a novel PCR assay for
methylation status of CpG islands. Proc Natl Acad Sci USA.
93:9821–9826. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Belinsky SA, Palmisano WA, Gilliland FD,
Crooks LA, Divine KK, Winters SA, et al: Aberrant promoter
methylation in bronchial epithelium and sputum from current and
former smokers. Cancer Res. 62:2370–2377. 2002.PubMed/NCBI
|
|
24
|
Smith E, Jones ME and Drew PA:
Quantitation of DNA methylation by melt curve analysis. BMC Cancer.
9:1232009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Belinsky SA, Liechty KC, Gentry FD, Wolf
HJ, Rogers J, Vu K, et al: Promoter hypermethylation of multiple
genes in sputum precedes lung cancer incidence in a high-risk
cohort. Cancer Res. 66:3338–3344. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Belinsky SA, Grimes MJ, Casas E, Stidley
CA, Franklin WA, Bocklage TJ, et al: Predicting gene promoter
methylation in non-small-cell lung cancer by evaluating sputum and
serum. Br J Cancer. 96:1278–1283. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Candiloro IL, Mikeska T and Dobrovic A:
Assessing combined methylation-sensitive high resolution melting
and pyrosequencing for the analysis of heterogeneous DNA
methylation. Epigenetics. 6:500–507. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wojdacz TK and Dobrovic A:
Methylation-sensitive high resolution melting (MS-HRM): a new
approach for sensitive and high-throughput assessment of
methylation. Nucleic Acids Res. 35:e412007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shen L, Guo Y, Chen X, Ahmed S and Issa
JP: Optimizing annealing temperature overcomes bias in bisulfite
PCR methylation analysis. Biotechniques. 42:48–58. 2007. View Article : Google Scholar : PubMed/NCBI
|