Introduction
Chemokine C-C motif ligand 5 (CCL5), also called
regulated on activation normal T cell expressed and secreted
(RANTES), was originally identified as a product of activated T
cells and is capable of recruiting T cells to inflammatory sites
(1,2). Further study found that CCL5 can be
secreted by a variety of cells including endothelial cells,
mesenchymal stem cells, tissue resident stem cells and breast
cancer cells (3–6). Expression of CCL5 is upregulated in
breast cancer, and is correlated to disease progression (7,8).
Elevated CCL5 in tumours promotes cell motility, migration,
invasion and metastasis (9,10) and inhibition of tumour-derived CCL5
attenuates the metastasis of tumours (11).
While the role of CCL5 in tumour progression has
been recognized and investigated, the reason why CCL5 is
upregulated in tumours has not been clarified yet. Research
suggests that expression of CCL5 can be upregulated by signal
transducer and activator of transcription 3 (STAT3) with IL-6
stimulation (12,13). STAT proteins are one of several
different families of latent cytoplasmic transcription factors
(14). Seven members of the STAT
family are found in mammals: STAT1-6 and STAT5A. When activated,
STAT family members translocate to the cell nucleus acting as
transcription activators (15). Two
phosphorylation sites are found on STAT3 protein: Tyr705 and
Ser727. An increase in the concentration of endogenous
unphosphorylated STAT3 (U-STAT3) following long-term treatment with
IL-6 allows U-STAT3 to compete effectively with IκB for U-NFκB, to
form a novel transcription factor that induces CCL5 expression
(12,13). However, STAT3 regulation of CCL5
expression is related to the phosphorylation of Tyr705 site rather
than Ser727 (16).
Studies suggest that endoplasmic reticulum (ER)
stress affects the activation of STAT3, but controversy exists. In
hepatocarcinoma and retinal endothelial cells, ER stress activates
STAT3 (17,18), but in hepatocyte cells ER stress was
found to inhibit activation of STAT3 (19). In tumours, moderate ER stress
response functions as an anti-apoptotic mechanism protecting
intracellular homeostasis. However, if severe imbalances persist
which exceed the protective capacity of the ER stress response
system, a shift to a pro-apoptotic mode ensues to kill the faulty
cells for the benefit of the organism as a whole (20). Many types of cancer cells display
chronically elevated activity levels of protective components of ER
stress (21). One of the central
anti-apoptotic regulators of the ER stress response is
glucose-regulated protein 78 (GRP78/BiP) (22). In contrast, CCAAT/enhancer binding
protein homologous transcription factor (CHOP/GADD153) represents a
critical executioner of the pro-apoptotic arm of the ER stress
response (23).
The present study investigated the role of ER stress
induced by tunicamycin in regulating the expression of CCL5 and its
relationship with STAT3 expression and phosphorylation. The role of
ER stress and CCL5 on the transmigration of breast cancer MCF-7
cells was also analysed.
Materials and methods
Clinical samples and ethics
statement
The study protocol was approved by the Review
Committee for the Use of Human or Animal Subjects of Wuhan
University. Twelve pairs of breast cancer tissues and their related
normal counterparts were obtained from patients who underwent
surgery at Renmin Hospital of Wuhan University from March to
November 2009. All cases included were from female patients with
primary breast cancer who did not undergo chemotherapy and
radiotherapy before surgery. Signed consent for inclusion into the
study was obtained from all patients. The median age of the
patients was 53 years (range, 34 to 68); 17% (2/12) of the patients
had ductal carcinoma in situ (DCIS), 50% (6/12) had stage I
breast cancer, 25% (3/12) had stage II breast cancer, and 8% (1/12)
had stage III breast cancer.
Cell culture and lentiviral vector
infection
The human breast cancer cell line MCF-7 was
purchased from the American Type Culture Collection (ATCC,
Manassas, VA, USA) and maintained in RPMI-1640 medium (Hyclone,
Thermo Scientific, Logan, UT, USA), supplemented with 10% (v/v)
fetal calf serum (FCS; Hyclone) and 1% (v/v)
penicillin/streptomycin (Invitrogen, Grand Island, NY, USA) at 37°C
in a humidified incubator with 5% CO2.
Lentiviral vectors encoding non-target control
shRNA, CCL5-specific shRNA (5′-GAGACTCCGTCACAACAA CAA-3′) and the
infection kit were purchased from Genechem (Shanghai, China). MCF-7
cells were infected with lentiviral vectors using the kit provided
by the company. Cells were then cultured in antibiotic-free normal
growth medium supplemented with 5 μg/ml puromycin (Invitrogen) for
4 weeks to allow stable knockdown. Cells were then collected for
RT-PCR and western blotting to assess knockdown efficiency and
further study.
Western blot analysis
Subconfluent cells after treatment were harvested
and lysed with ice-cold RIPA lysis buffer (Sigma-Aldrich, St.
Louis, MO, USA) and 1% cocktail of protease inhibitors
(Sigma-Aldrich). Aliquots of 20 μg lysates were separated by
SDS-PAGE and transferred to a nitrocellulose membrane. After
blocking with 5% (w/v) BSA in Tris-buffered saline at 37°C for 1 h,
the nitrocellulose membrane was incubated with the primary antibody
at 4°C overnight. The following primary antibodies were used:
anti-tubulin, GAPDH, STAT3, phospho-STAT3 (Tyr705), (P-STAT3), CHOP
and CCL5 antibodies (Cell Signaling Technology, Danvers, MA, USA).
After extensive washing, the membrane was incubated with the
secondary antibody at room temperature for 1 h (Cell Signaling
Technology). Protein bands were visualized with ECL reagent (Thermo
Scientific, Rockford, IL, USA) and then were quantified by Quantity
One® software.
RNA isolation and real-time RT-PCR
Total RNAs were isolated from breast cancer cells or
specimens and reverse transcripted to cDNA as described previously
(6). Diluted cDNAs (1:10 dilution)
were used in a 25-μl real-time PCR reaction system [cDNA 2.5 μl,
SYBR-Green Mix (Toyobo, Japan) 12.5 μl, sense primer (5 μM) 1 μl,
antisense primer (5 μM) 1 μl, and DEPC-H2O 8 μl] in
triplicate for each gene. The primers used in the present study
were: CCL5 sense, 5′-CGTGCCCACATCAAGGAG-3′ and antisense, 5′-GGA
CAAGAGCAAGCAGAAAC-3′; GAPDH sense, 5′-CCATCA CCATCTTCCAGG-3′ and
antisense, 5′-ATGAGTCCTTCC ACGATAC-3′. Cycle parameters were 95°C
for 1 min hot start and 40 cycles of 95°C for 15 sec, 58°C for 15
sec and 72°C for 45 sec. Blank controls with no cDNA templates were
performed to rule out contamination. The specificity of the PCR
product was confirmed by melting curve analysis. At the extension
stage of each cycle, the value of the threshold cycle (Ct) was
recorded. A standard curve was generated by plotting the Ct. GAPDH
expression was assessed as a housekeeping gene to standardize the
expression level of the target genes. The comparative expression
level of the target gene = 2−ΔΔCt.
Enzyme-linked immunosorbent assay (ELISA)
to quantify the secretion of CCL5
Cells were treated with 10 μg/ml ER stress activator
tunicamycin (Sigma-Aldrich) in normal cell culture medium (cells
without drug treatment were set as the control). The supernatant
was collected after a 24-h treatment, and the secretion of CCL5 was
determined by commercial Human CCL5/RANTES DuoSet kit (R&D
Systems, Minneapolis, MN, USA) following the manufacturer’s
instructions.
Transmigration assay
The transmigration assay was carried out using
Transwell chambers (8.0 μm, Millipore, Billerica, MA, USA) with
polycarbonate membrane filters of 8-μm pore size which were used to
form dual compartments in a 24-well tissue culture plate. Cells
were harvested and resuspended in serum-free medium
(1×106 cells/ml). The cell suspension (100 μl) was added
into the upper chamber of the well. Media with 10% (v/v) serum or
with rhCCL5 (R&D Systems) at different concentrations (0, 0.1,
1, 10, 100 ng/ml) were loaded into the bottom well. After a 24-h
incubation, the Transwell chambers were fixed in 4% formaldehyde
and stained with crystal violet. The non-migrating cells were
carefully removed from the upper surface (inside) of the well.
Cells that had migrated to the bottom surface of the filter were
counted. Nine evenly spaced fields were counted in each well using
an inverted phase-contrast microscope at ×20 magnification.
Transmigration index = number of cells counted in the test
group/number of cells counted in the control group.
Statistical analysis
Data are expressed as mean ± SD and were analysed
using one-way analysis of variance (ANOVA), and the
Student-Newman-Keul’s test was used for individual comparisons. The
statistical program SPSS 19.0 for Windows was used for analysis. A
probability value P<0.05 was considered to indicate a
statistically significant result.
Results
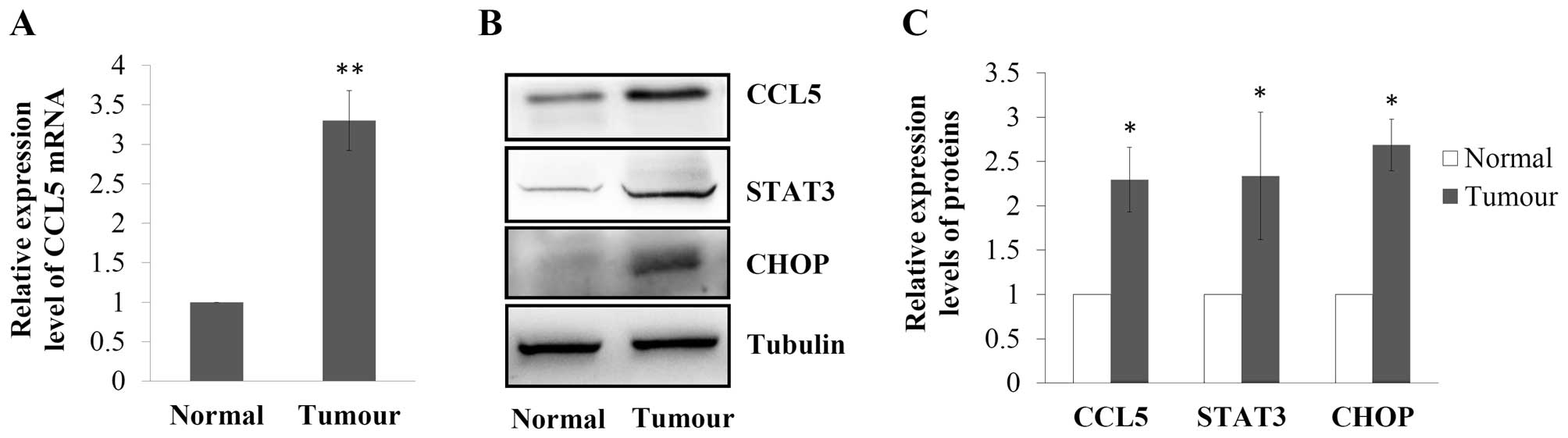
Expression of CCL5, U-STAT3 and CHOP are
upregulated in breast cancer
The CCL5 mRNA expression level in 12 pairs of breast
cancer tissues and their corresponding normal breast epithelial
tissues was assessed using real-time RT-PCR. As shown in Fig. 1A, CCL5 mRNA expression was
significantly elevated in the breast cancer tissues, which was
3.3-fold of that in the normal breast tissues (P<0.01).
Expression of CCL5 was also observed using western blotting, and
the results revealed that CCL5 expression at the protein level was
also upregulated in the tumour (P<0.05 vs. normal; Fig. 1B and C). In order to investigate
possible mechanisms for the upregulation of CCL5 in breast cancer,
the expression of STAT3, a transcriptional regulator of the CCL5
gene (12,13), and CHOP, an indicator of ER stress
(23), were further assessed.
Results showed that expression levels of STAT3 (2.3-fold of normal,
P<0.05) and CHOP (2.7-fold of normal, P<0.05) were higher in
breast cancer tissues than the levels in the corresponding normal
breast epithelial tissues (Fig. 1B and
C).
Tunicamycin elevates the expression of
CCL5, STAT3 and CHOP in a concentration- and time-dependent
manner
In order to investigate the role of ER stress in
regulating expression of CCL5 and STAT3 directly, a classical ER
stress activator tunicamycin was used. Breast cancer MCF-7 cells
were treated with different concentrations of tunicamycin (0, 2.5,
5, 10 and 20 μg/ml) for 24 h. CCL5 mRNA expression was markedly
elevated by tunicamycin (P<0.01, Fig. 2A). Similarly, expression of CCL5,
STAT3 and CHOP were also upregulated by tunicamycin in a
concentration-dependent manner (Fig.
2B). Tunicamycin (10 μg/ml) induced the highest CHOP and CCL5
expression levels (3.2- and 4.5-fold of the control, respectively,
P<0.01, Fig. 2C). Therefore, 10
μg/ml was chosen as the concentration of tunicamycin used in the
subsequent experiments. The effects of tunicamycin on expression of
CCL5, STAT3 and CHOP after different time periods of treatment were
also assessed. Western blotting showed that expression of CHOP and
STAT3 started to increase after a 6-h treatment, while CCL5
expression levels were not upregulated until 12 h of treatment
(Fig. 2D). After a 24-h treatment,
expression of CCL5, STAT3 and CHOP reached the highest levels
(Fig. 2E).
 | Figure 2Effect of tunicamycin on CCL5, STAT3
and CHOP protein as well as CCL5 mRNA expression in MCF-7 cells.
(A) Cultured MCF-7 cells were treated with tunicamycin at different
concentrations (2.5, 5, 10 and 20 μg/ml; 0 μg/ml was set as the
control) for 24 h. Real-time RT-PCR was conducted to detect the
expression of CCL5 mRNA. GAPDH was set as an internal control. The
CCL5 mRNA expression was presented as n-fold change of the control.
Data represent the mean value ± standard deviation of three
independent experiments. **P<0.01 vs. the control.
(B) Expression levels of CCL5, STAT3 and CHOP after treatment with
tunicamycin at different concentrations (as indicated) for 24 h
were assessed by western blotting. Tubulin was set as an internal
control. (C) Relative expression levels of the proteins were
quantified and presented as n-fold change of the control. Data
represent the mean value ± standard deviation of 3 independent
experiments. *P<0.05, **P<0.01 vs. the
control. (D) MCF-7 cells were treated with 10 μg/ml tunicamycin for
0, 6, 12, 18 and 24 h (0-h treatment was set as the control), and
expression levels of CCL5, STAT3 and CHOP were detected by western
blotting. Tubulin was set as an internal control. (E) Relative
expression levels of CCL5, STAT3 and CHOP were quantified. Data
represent the mean ± standard deviation of 3 independent
experiments. *P<0.05, **P<0.01 vs. the
control. |
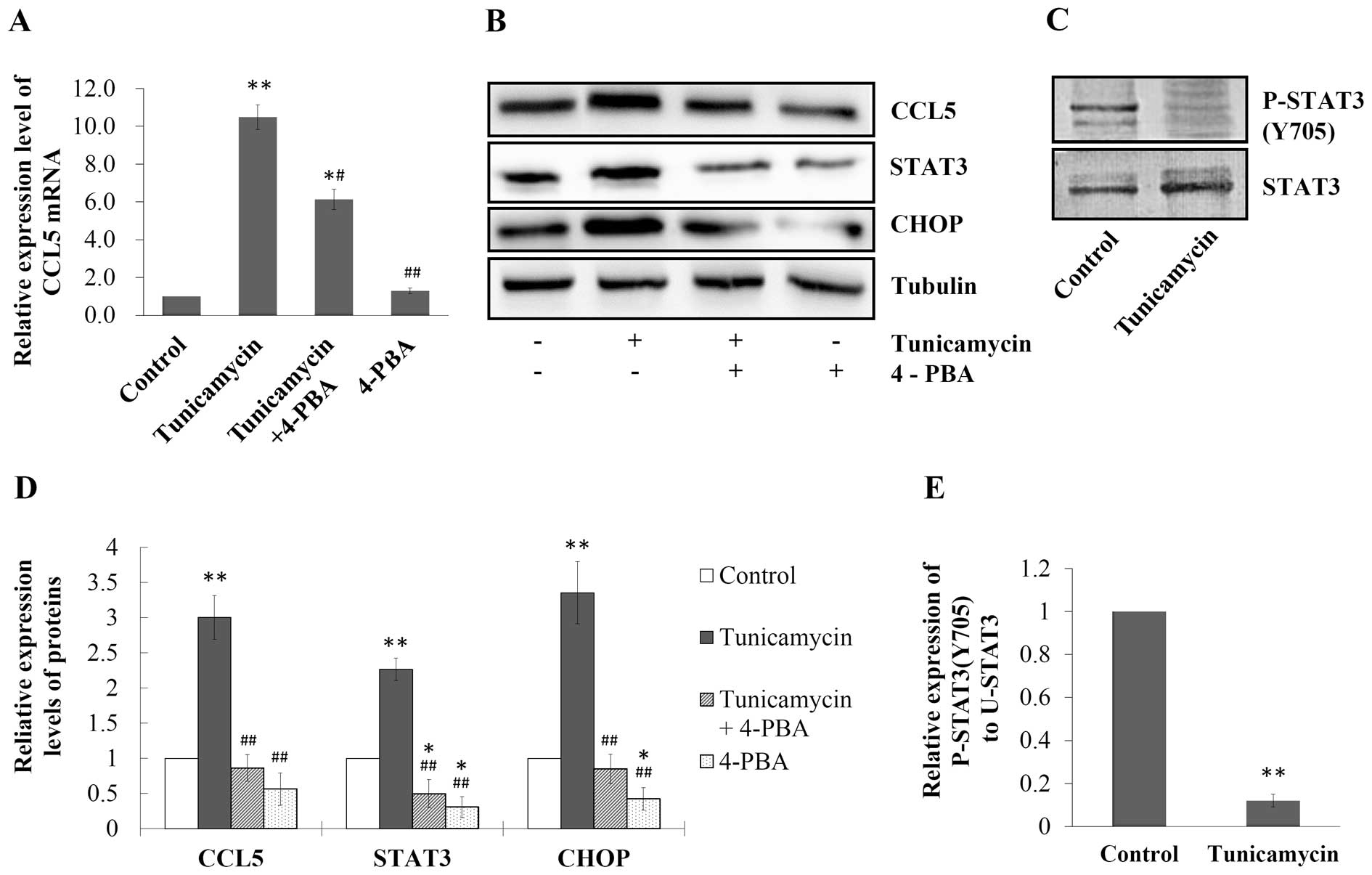
ER stress-induced CCL5 upregulation is
positively correlated to U-STAT3 expression but negatively related
to STAT3 phosphorylation
To confirm the role of ER stress in regulating CCL5
expression and to further investigate the mechanisms, we used ER
stress inhibitor 4-PBA (5 mM; Sigma-Aldrich) to block the ER stress
induced by tunicamycin (10 μg/ml) in the MCF-7 cells. As shown in
Fig. 3A, CCL5 mRNA expression in
the cells was induced by tunicamycin to 10.5-fold of the control
(P<0.01), which was blocked by 4-PBA as the expression of CCL5
mRNA decreased to 6.1-fold of the control when cells were treated
with both drugs (P<0.05, Fig.
3A). Western blotting revealed that tunicamycin increased the
expression of CCL5, STAT3 and CHOP in MCF-7 cells, compared with
the control (P<0.01, Fig. 3B and
D). In contrast, when cells were treated with both tunicamycin
and 4-PBA, the expression levels of these proteins were inhibited,
compared with the cells treated with tunicamycin alone (P<0.01,
Fig. 3B and D). However, while
STAT3 expression in breast cancer cells was upregulated by
tunicamycin, the expression level of P-STAT3 (Y705) was
dramatically downregulated (Fig.
3C), and the ratio of P-STAT3 (Y705) to U-STAT3 dropped to 12%
of the control (P<0.01, Fig.
3E).
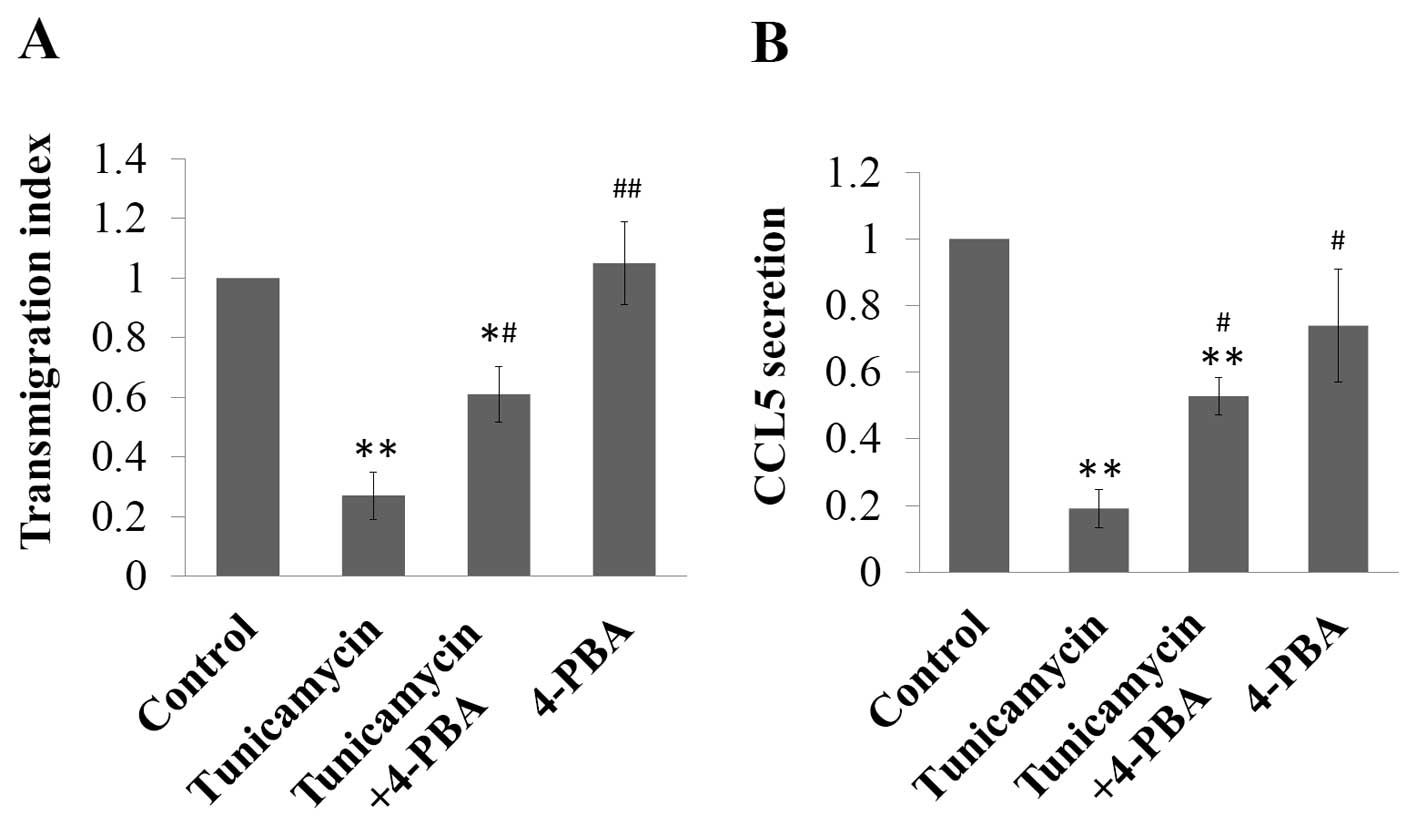
ER stress inhibits the transmigration and
CCL5 secretion of MCF-7 cells
Since CCL5 was previously found to be involved in
the migration and progression of breast cancer (6,11,24–26),
we further inestigated how ER stress-induced upregulation of CCL5
affects the transmigration of MCF-7 cells. Tunicamycin inhibited
the transmigration of MCF-7 to 27% of the control (P<0.01),
which was antagonised by 4-PBA treatment, while the transmigration
index of cells treated with both tunicamycin and 4-PBA was reverted
to 61% of the control (P<0.05), and 4-PBA treatment alone had no
effect on the transmigration of MCF-7 cells (Fig. 4A). Since these results were
contradictory to previous reports, which found that CCL5 induced
the migration and invasion of cancer (9,10), we
detected the CCL5 secreted in the cell culture medium by ELISA. We
found that CCL5 secretion was significantly attenuated by
tunicamycin treatment to 19% of the control (P<0.01), while in
the cells treated with both tunicamycin and 4-PBA, the secretion of
CCL5 was higher than that in the tunicamycin-treated cells, which
was 53% of the control (P<0.01). 4-PBA treatment alone did not
affect CCL5 secretion of MCF-7 cells (Fig. 4B).
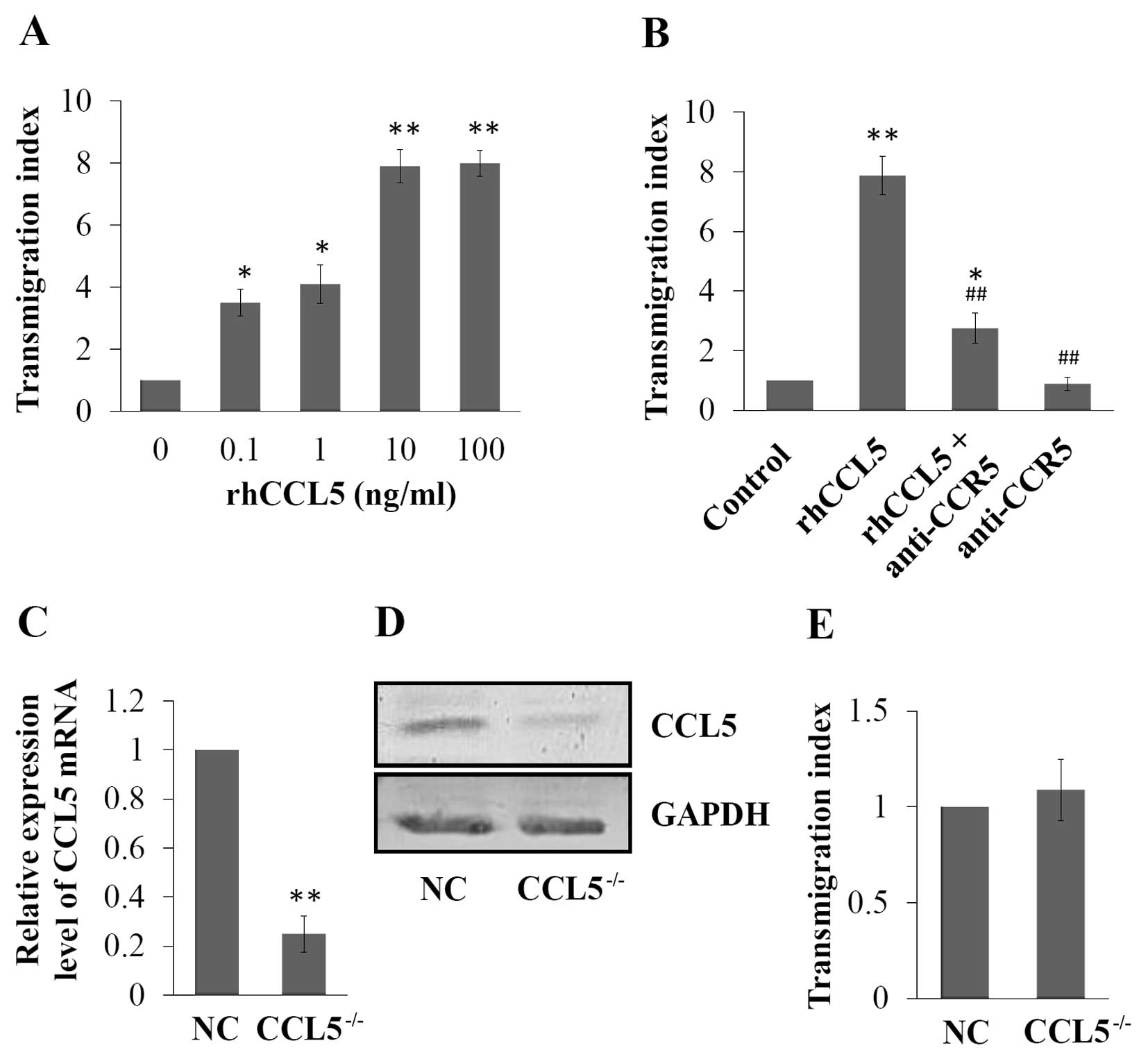
Extracellular CCL5 induces the
transmigration of MCF-7 cells, which was partially blocked by CCR5
monoclonal antibody
Effect of extracellular CCL5 on the transmigration
of MCF-7 was also observed by transmigration assay. The results
showed that extracellular rhCCL5 at different concentrations
induced the transmigration of MCF-7 cells, and 100 ng/ml of rhCCL5
induced the highest transmigration index in the MCF-7 cells, which
was 7.9-fold of the control (P<0.01, Fig. 5A). Furthermore, we investigated the
mechanism of the inductive effect of CCL5 on cell transmigration.
The results of the transmigration assay showed that rhCCL5
induction enhanced the transmigration ability of MCF-7 cells
(7.9-fold of the control, P<0.01), which was blocked by CCR5
monoclonal antibody treatment; the transmigration index decreased
to 35% of the index in the rhCCL5 only group (P<0.01, Fig. 5B). However, the transmigration index
of cells treated with both rhCCL5 and CCR5 monoclonal antibody was
still higher than that of the control (2.75-fold of the control,
P<0.05, Fig. 5B).
Knockdown of endogenous CCL5 expression
does not affect the transmigration of MCF-7 cells
To investigate the role of the endogenous expression
level of CCL5 on the transmigration of breast cancer MCF-7 cells, a
CCL5 knockdown (CCL5−/−) MCF-7 cell line was constructed
using lentiviral vectors encoding CCL5-shRNA. The expression of
CCL5 was assessed by RT-PCR and western blotting, and CCL5
expression at both the mRNA and protein levels were effectively
inhibited in the CCL5−/− cells compared with the NC
(Fig. 5C and D). Then the
transmigration abilities of NC and CCL5−/− MCF-7 cells
were assessed. No significant difference between these two cell
lines was noted (P>0.05, Fig.
5E).
Discussion
CCL5, a classical pro-inflammatory chemokine, can be
expressed and secreted by both breast cancer and non-malignant
stromal cells (3–6). Although the detailed mechanisms have
not yet been fully understood, the role of CCL5 in facilitating
cancer progression has been recognized. However, the reason why
CCL5 is upregulated in breast cancer is not well established.
Studies revealed that IL-6-induced U-STAT3 upregulation in
hTERT-HME1 cells led to an increase in CCL5 expression (12,13).
Moreover, since cancer cells display chronically elevated levels of
ER stress (21) and ER stress
regulates STAT3 activity (17–19),
we hypothesized that elevated expression of CCL5 in breast cancer
was attributed to ER stress-induced U-STAT3 upregulation.
In order to prove our hypothesis, we firstly
assessed the expression of CCL5, STAT3 and ER stress indicator CHOP
in tumour tissues from breast cancer patients and their
corresponding normal breast epithelial tissues. Elevated expression
of CCL5 was accompanied by upregulated expression of both CHOP and
STAT3 in breast tumour tissues when compared with levels in the
corresponding normal breast epithelial tissues. The results suggest
that a certain correlation may exist between the expression of
CCL5, STAT3 and ER stress responses of breast cancer cells. To
further investigate the effect of ER stress on the expression of
CCL5, we treated human breast cancer MCF-7 cells with a series of
concentrations of tunicamycin, an activator of ER stress (27). Expression of CCL5 was upregulated by
tunicamycin, in a concentration- and time-dependent manner.
Notably, changes in STAT3 and CHOP expression in MCF-7 cells were
parallel to the change in CCL5 expression after tunicamycin
treatment, which is consistent with the correlation pattern of the
expression of these proteins in breast cancer specimens. In order
to confirm the effect of ER stress on CCL5 expression, ER stress
inhibitor 4-PBA was used. Upregulation of CCL5 expression induced
by tunicamycin was inhibited by ER stress inhibitor 4-PBA, and
meanwhile, expression of STAT3 showed the same trend of change as
that of CCL5, which supports our hypothesis that ER stress elevates
endogenous CCL5 expression via STAT3 in breast cancer cells.
STAT3 regulation of CCL5 expression is related to
phosphorylation of STAT3 at Tyr705 site (16). Thus, the roles of
tunicamycin-induced ER stress in regulating STAT3 (Y705)
phosphorylation and U-STAT3 expression were further investigated.
Our results showed that the expression of P-STAT3 (Y705) was
markedly decreased by tunicamycin treatment while expression of
U-STAT3 was elevated. The results suggest that the upregulation of
endogenous CCL5 expression in human breast cancer MCF-7 cells
induced by ER stress is attributed to the elevated U-STAT3
expression, which is consistent with studies by Yang et al
that demonstrated that IL-6-induced U-STAT3 upregulation in
hTERT-HME1 cells led to an increase in CCL5 expression (12,13).
Since ER stress upregulated CCL5 expression in
breast cancer cells, and elevated CCL5 in tumours promotes cell
motility, migration, invasion and metastasis (9,10),
tunicamycin should enhance the transmigration of cells,
theoretically. Therefore, the action of tunicamycin on the
transmigration of MCF-7 cells was further investigated.
Importantly, our results showed that tunicamycin significantly
inhibited the transmigration of MCF-7 cells. To ascertain the
mechanism, we first detected CCL5 secretion (the concentration of
CCL5 in the cell culture supernatants) of MCF-7 cells after
tunicamycin treatment. The results revealed that although ER stress
upregulated intracellular CCL5 expression, it inhibited CCL5
secretion of MCF-7 cells. According to studies of Wang et al
(10) and Lin et al
(28), CCL5 promotes tumour cell
motility and migration by interacting with a specific receptor
chemokine C-C motif receptor 5 (CCR5). Thus, we assume that it is
the extracellular CCL5 in the microenvironment of tumour cells that
actually stimulates the migration, while the intracellular
expression level of CCL5 does not affect the migration ability of
cancer cells directly. The results confirmed the assumption
further, which indicated that, on one hand, extracellular CCL5
induced the transmigration of MCF-7 cells, which was partially
blocked by CCR5 monoclonal antibody; on the other hand, knockdown
of endogenous CCL5 expression by shRNA did not affect the
transmigration of MCF-7 cells.
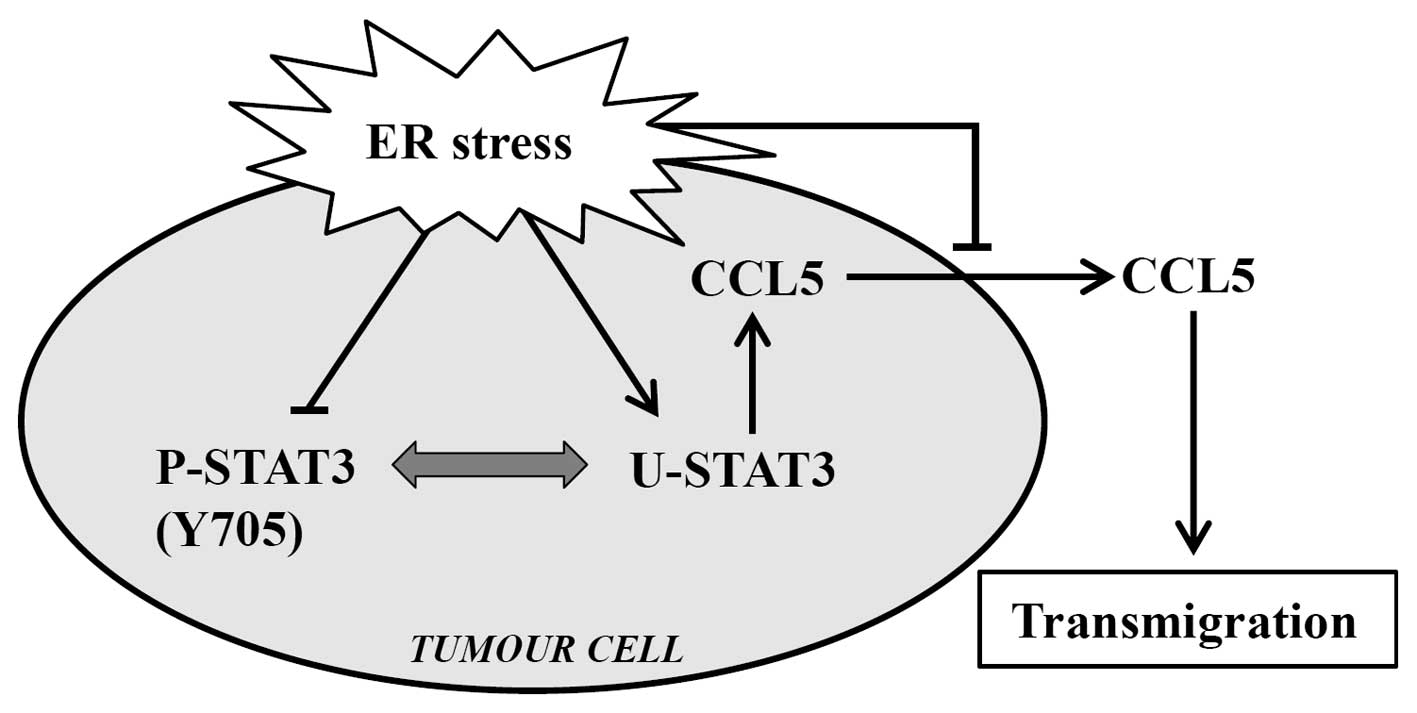
Taken together, as summarized in Fig. 6, the present study suggests that ER
stress induced endogenous expression of CCL5 via elevation of
U-STAT3 expression, and clarifies the mechanisms of CCL5
upregulation in breast cancer. Meanwhile, ER stress inhibited CCL5
secretion, which in turn, decreased the transmigration ability of
breast cancer MCF-7 cells. We determined that it was extracellular
CCL5 in the microenvironment of the tumour cells that stimulated
the transmigration, while the intracellular expression level of
CCL5 did not affect the transmigration ability of cancer cells
directly. If CCL5 is to be applied as a target to prevent the
migration and metastasis of tumours, in the clinic, it may be more
effective to block the CCL5-CCR5 interaction and/or to inhibit the
CCL5 concentration in the microenvironment of the tumour, than to
decrease the endogenous CCL5 expression in breast cancer cells.
However, our findings are based on in vitro experiments, and
more research, such as clinical data analysis and in vivo
experiments with animal models, is required before they can be
verified and applied in breast cancer treatment.
Acknowledgements
This project was supported by the Natural Science
Foundation of Hubei Province, China (2011CBD489).
References
|
1
|
Schall TJ, Jongstra J, Dyer BJ, et al: A
human T cell-specific molecule is a member of a new gene family. J
Immunol. 141:1018–1025. 1988.PubMed/NCBI
|
|
2
|
John AE, Berlin AA and Lukacs NW:
Respiratory syncytial virus-induced CCL5/RANTES contributes to
exacerbation of allergic airway inflammation. Eur J Immunol.
33:1677–1685. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hillyer P and Male D: Expression of
chemokines on the surface of different human endothelia. Immunol
Cell Biol. 83:375–382. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Karnoub AE, Dash AB, Vo AP, et al:
Mesenchymal stem cells within tumour stroma promote breast cancer
metastasis. Nature. 449:557–563. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pinilla S, Alt E, Abdul Khalek FJ, et al:
Tissue resident stem cells produce CCL5 under the influence of
cancer cells and thereby promote breast cancer cell invasion.
Cancer Lett. 284:80–85. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang Y, Yao F, Yao X, et al: Role of CCL5
in invasion, proliferation and proportion of
CD44+/CD24− phenotype of MCF-7 cells and
correlation of CCL5 and CCR5 expression with breast cancer
progression. Oncol Rep. 21:1113–1121. 2009.PubMed/NCBI
|
|
7
|
Luboshits G, Shina S, Kaplan O, et al:
Elevated expression of the CC chemokine regulated on activation,
normal T cell expressed and secreted (RANTES) in advanced breast
carcinoma. Cancer Res. 59:4681–4687. 1999.PubMed/NCBI
|
|
8
|
Niwa Y, Akamatsu H, Niwa H, Sumi H, Ozaki
Y and Abe A: Correlation of tissue and plasma RANTES levels with
disease course in patients with breast or cervical cancer. Clin
Cancer Res. 7:285–289. 2001.PubMed/NCBI
|
|
9
|
Long H, Xie R, Xiang T, et al: Autocrine
CCL5 signaling promotes invasion and migration of CD133(+) ovarian
cancer stem-like cells via NF-κB-mediated MMP-9 upregulation. Stem
Cells. 30:2309–2319. 2012.PubMed/NCBI
|
|
10
|
Wang SW, Wu HH, Liu SC, et al: CCL5 and
CCR5 interaction promotes cell motility in human osteosarcoma. PLoS
One. 7:e351012012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stormes KA, Lemken CA, Lepre JV, Marinucci
MN and Kurt RA: Inhibition of metastasis by inhibition of
tumor-derived CCL5. Breast Cancer Res Treat. 89:209–212. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang J, Chatterjee-Kishore M, Staugaitis
SM, et al: Novel roles of unphosphorylated STAT3 in oncogenesis and
transcriptional regulation. Cancer Res. 65:939–947. 2005.PubMed/NCBI
|
|
13
|
Yang J, Liao X, Agarwal MK, Barnes L,
Auron PE and Stark GR: Unphosphorylated STAT3 accumulates in
response to IL-6 and activates transcription by binding to
NFkappaB. Genes Dev. 21:1396–1408. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bandarian V, Pattridge KA, Lennon BW,
Huddler DP, Matthews RG and Ludwig ML: Domain alternation switches
B(12)-dependent methionine synthase to the activation conformation.
Nat Struct Biol. 9:53–56. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Levy DE and Darnell JE Jr: Stats:
transcriptional control and biological impact. Nat Rev Mol Cell
Biol. 3:651–662. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ng YP, Cheung ZH and Ip NY: STAT3 as a
downstream mediator of Trk signaling and functions. J Biol Chem.
281:15636–15644. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Waris G, Tardif KD and Siddiqui A:
Endoplasmic reticulum (ER) stress: hepatitis C virus induces an
ER-nucleus signal transduction pathway and activates NF-kappaB and
STAT-3. Biochem Pharmacol. 64:1425–1430. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen Y, Wang JJ, Li J, et al: Activating
transcription factor 4 mediates hyperglycaemia-induced endothelial
inflammation and retinal vascular leakage through activation of
STAT3 in a mouse model of type 1 diabetes. Diabetologia.
55:2533–2545. 2012. View Article : Google Scholar
|
|
19
|
Kimura K, Yamada T, Matsumoto M, et al:
Endoplasmic reticulum stress inhibits STAT3-dependent suppression
of hepatic gluconeogenesis via dephosphorylation and deacetylation.
Diabetes. 61:61–73. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cho HY, Thomas S, Golden EB, et al:
Enhanced killing of chemo-resistant breast cancer cells via
controlled aggravation of ER stress. Cancer Lett. 282:87–97. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schönthal AH: Endoplasmic reticulum stress
and autophagy as targets for cancer therapy. Cancer Lett.
275:163–169. 2009.PubMed/NCBI
|
|
22
|
Li J and Lee AS: Stress induction of
GRP78/BiP and its role in cancer. Curr Mol Med. 6:45–54. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
14:381–389. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Azenshtein E, Luboshits G, Shina S, et al:
The CC chemokine RANTES in breast carcinoma progression: regulation
of expression and potential mechanisms of promalignant activity.
Cancer Res. 62:1093–1102. 2002.PubMed/NCBI
|
|
25
|
Mañes S, Mira E, Colomer R, et al: CCR5
expression influences the progression of human breast cancer in a
p53-dependent manner. J Exp Med. 198:1381–1389. 2003.PubMed/NCBI
|
|
26
|
Yaal-Hahoshen N, Shina S, Leider-Trejo L,
et al: The chemokine CCL5 as a potential prognostic factor
predicting disease progression in stage II breast cancer patients.
Clin Cancer Res. 12:4474–4480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ozcan U, Cao Q, Yilmaz E, et al:
Endoplasmic reticulum stress links obesity, insulin action, and
type 2 diabetes. Science. 306:457–461. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lin S, Wan S, Sun L, et al: Chemokine C-C
motif receptor 5 and C-C motif ligand 5 promote cancer cell
migration under hypoxia. Cancer Sci. 103:904–912. 2012. View Article : Google Scholar : PubMed/NCBI
|