Introduction
Although prostate cancer (PC) remains one of the
most diagnosed malignant diseases and is the second-leading cause
of tumor-associated mortality in males in the Western hemisphere
(1), progression mechanisms of PC
are only marginally understood. Phenotypically, it differs from a
localized hormone-naive state to an advanced, castration-resistant
and metastatic state that is predominantly attributed to cellular
proliferation in a low-level steroid hormonal environment. The
first line drug docetaxel is an option for castration-resistant PC
treatment; however, efficacy varies between patients and
therapeutic outcome is often unsatisfying (2). Heat shock protein (HSP) 27 has been
identified as controlling anti-therapeutic mechanisms by specific
alterations in proliferation, cell cycle regulation, apoptosis and
general stress response in cancer (3–5). In
PC, it has been demonstrated that critical events in tumor
progression, such as epithelial-to-mesenchymal transition,
metastasis, androgen receptor (AR) signaling and treatment
resistance, are promoted by HSP27 activities. Consequently,
attenuation of HSP27 by newly developed inhibitors is a promising
approach in the treatment of advanced PC (6). Regulation of cell physiology as well
as dysregulation in malignant cells frequently depends on protein
phosphorylation controlled by a coordinated network of specific
kinase and phosphatase activities. HSP27 protein contains three
regulatory phosphorylation sites located at the positions
serine-15, -78 and -82 which are phosphorylated in the presence of
diverse cellular signals, e.g. in response to oxidative and
pro-inflammatory stress (7,8). In PC cells, HSP27 is predominantly
phosphorylated by the mitogen-activated protein kinase p38 (MAPK
p38), mitogen-activated protein kinase-activated protein kinase 2
(MAPKAPK-2) pathway and by protein kinase D1 (PKD1) (9–11).
Although HSP27 is estimated to orchestrate pivotal functions in PC
progression and therapy, only little is known about HSP27 activity
modulated by protein phosphorylation. Hassan et al described
HSP27 phospho-status at serine-82 leads to suppress AR signaling in
PC cells (9); however, further
HSP27 functions regulated by protein phosphorylation remain
unclear. Our previous studies of HSP functionality in PC
progression showed HSP27 to have potent effects on AR activity
(12,13). Since HSP27 has been suggested to
play crucial roles in the initiation and development of
chemoresistance, we hypothesized that HSP27 functionality may be
associated with specific protein phosphorylation/dephosphorylation.
In the present study, we established a PC cell model system
containing PC-3 and PC-3 cells stably overexpressing HSP27 to
examine the input of HSP27 phosphorylation to HSP27-driven
cytoprotective properties of docetaxel-induced chemoresistance.
Materials and methods
Tumor cell lines and chemicals
Human epithelial PC cancer cell line PC-3 (Cell
Lines Service; CLS, Eppelheim, Germany) was grown in RPMI-1640
media with 10% fetal bovine serum and 1% penicillin/streptomycin
(PAN-Biotech, Aidenbach, Germany) in a 5% CO2 atmosphere
at 37°C. PC-3-HSP27 cells stably overexpressing HSP27 were selected
with 400 μg/ml G418 (Carl Roth, Karlsruhe, Germany) as previously
described (12). For incubation
experiments, docetaxel was purchased from Sigma-Aldrich (Munich,
Germany), bryostatin-1 (3×10−8 M) from Tocris Bioscience
(Minneapolis, MN, USA), CID755673 (5×10−5 M) from Axon
Medchem (Groningen, Netherlands), SB203580 (10−5 M) from
Selleckchem (Munich, Germany) and sorbitol (3×10−1 M)
was acquired from Carl Roth. Incubation with sorbitol was limited
to 30 min/day since continuous incubation revealed cell damaging
effects (14). Before the
hyperosmolar shock with sorbitol, culture supernatant was removed,
collected and re-added after sorbitol treatment. Drug treatment was
generally initiated in adherent cells seeded 24 h before.
Transfection experiments
One day before transfection, cells were plated into
6-well (150,000 cells/well) or 24-well (30,000 cells/well) culture
plates. For overexpression experiments, cells were transfected with
1 μg DNA (24-well) and 3 μg (6-well) per well, using Lipofectamine
2000 (Invitrogen, Darmstadt, Germany). The vectors pHSP27 wt,
pHSP27-3A and pHSP27-3D were kindly provided by C. Kubisch (Munich,
Germany) (15). pcDNA3.1
(Invitrogen) was used as empty control vector.
Western blotting
Cells were lysed in RIPA buffer [50 mM Tris (pH
7.5), 150 mM NaCl, 10 mM K2HPO4, 5 mM EDTA,
10% glycerol, 1% Triton X-100, 0.05% sodium dodecyl sulfate, 1 mM
Na3VO4, 20 mM NaF, 0.1 mM
phenylmethylsulfonyl fluoride, 20 mM 2-phosphoglycerate and
complete protease inhibitor cocktail from Roche Applied Science;
Mannheim, Germany]. Determination of protein concentration was
carried out utilizing Bradford reagent (Bio-Rad) and equal amounts
of protein were separated by SDS-PAGE and transferred onto a
nitrocellulose membrane (Whatman, Dassel, Germany). Proteins of
interest were detected by specific primary antibodies directed
against HSP27, P-Ser78-HSP27, P-Ser82-HSP27,
glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Cell Signaling
Technology, Danvers, MA, USA) and P-Ser15-HSP27 (Thermo
Scientific, Waltham, MA, USA) incubated overnight and
peroxidase-coupled secondary antibodies directed against mouse and
rabbit (Cell Signaling Technology) incubated for 1 h. Proteins were
visualized by using SuperSignal West Dura Chemiluminescent
Substrate (Thermo Scientific) and a ChemiDoc system (Bio-Rad).
Quantification of protein signals was carried out by Image Lab 3.0
software (Bio-Rad).
Cell viability assay
To verify effects on cell viability, we used the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. For MTT assays, cells were incubated with 0.1 μg/μl aqueous
MTT solution (Carl Roth) for 2 h at 37°C. Formazan complexes were
solubilized by the addition of 120 μl dimethyl sulfoxide lysis
buffer (DMSO; containing 10% SDS, 0.2% HCl) and dye formation was
measured in a plate reader Infinite 200 PRO (Tecan, Männedorf,
Switzerland) at 550 nm wavelength.
Cell proliferation assay
Cellular proliferation was examined by cell counting
by using a CASY Cell Counter and Analyzer Model TT (Roche Applied
Science). Therefore, adherent cells were detached by trypsin
treatment, suspended in CASYton (Roche Applied Science) as 1:100
dilution. Measurement of 400 μl cell suspension was performed in 3
cycles using a capillary of 150 μm in diameter and 7.20/15.45 nm as
gate settings to discriminate between living PC-3 and dead cells,
as well as cellular debris.
Statistical analysis
Statistical comparisons of at least three
independent measurements were performed using the unpaired
Student’s t-test with 95% confidence interval. For all statistical
analyses, results of p≤0.05 were considered statistically
significant. Data are provided as means ± SD.
Results
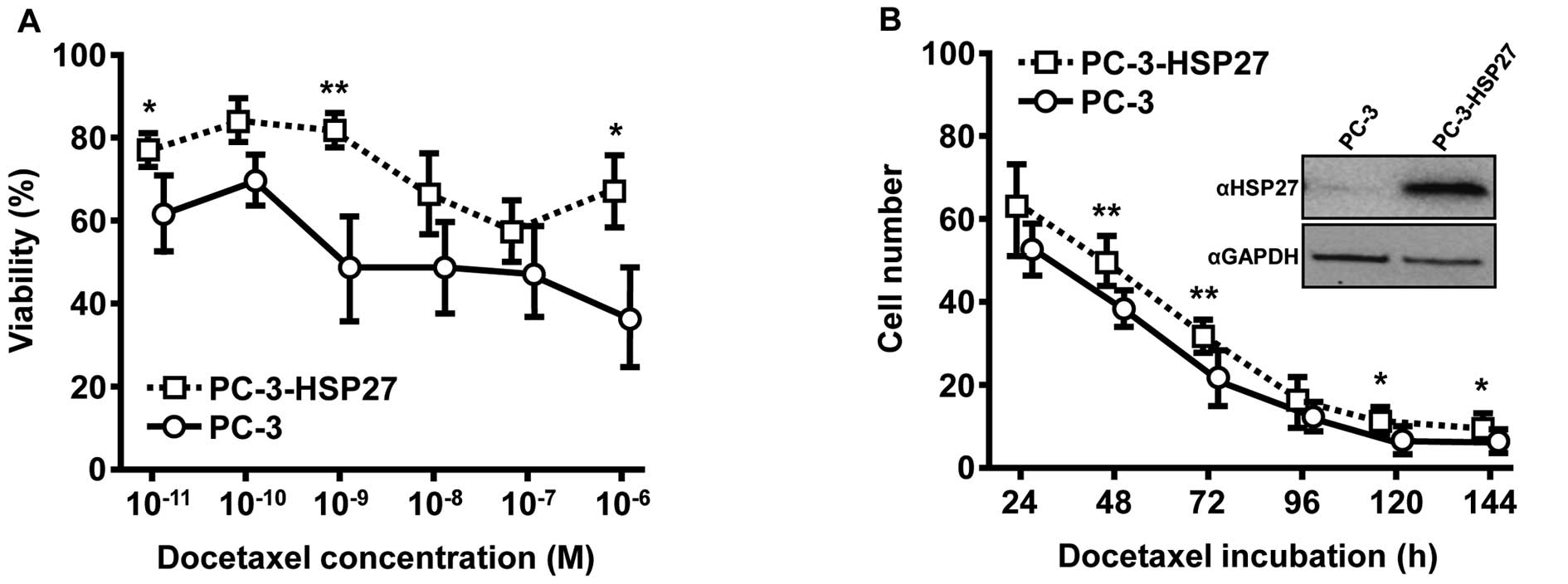
Establishment of an in vitro cell model
system for HSP27-driven chemoresistance in PC
High expression of HSP27 is known to induce
pro-oncogenic effects in PC (16).
We used an experimental system composed of the PC cell line PC-3
and PC-3 cells stably overexpressing HSP27 (PC-3-HSP27; inset
Fig. 1B) as a suitable model in
which to study drug-induced and HSP27-driven effects of acquired
chemoresistance. As shown by MTT assays, various concentrations of
docetaxel revealed a concentration-dependent cytostatic effect in
both cell lines after 72 h of incubation, although docetaxel
sensitivity was obviously lower in PC-3-HSP27 cells for all chosen
docetaxel concentrations (Fig. 1A).
Overexpression of HSP27 in PC-3-HSP27 cells led to significantly
increased survival rates in the presence of docetaxel at
concentrations of 10−6 M (1.9-fold, p=0.0258),
10−9 M (1.8-fold, p=0.0080) and 10−11 M
(1.3-fold, p=0.0170) compared to maternal PC-3 cells. These
findings were validated by growth kinetics utilizing a CASY Cell
Counter and Analyzer model TT for daily measurement over a period
of 144 h (Fig. 1B). Similarly,
elevated HSP27 levels resulted in enhanced cytoprotective effects
during drug treatment confirmed by significantly higher survival of
PC-3-HSP27 cells (48 h, 1.3-fold, p=0.0078; 72 h, 1.4-fold,
p=0.0022; 120 h, 1.5-fold, p=0.0392; 144 h, 1.4-fold,
p=0.0327).
Activation of MAPK p38 and PKD1 reduce PC
cell growth
MAPK p38 and PKD1 are known to be involved in
important cellular regulation pathways by phosphorylation of
diverse proteins, including HSP27 (9–11,17).
Therefore, we assessed changes in chemosensitivity of
docetaxel-treated PC-3-HSP27 cells co-incubated with modulators of
MAPK p38 and PKD1 activity. Specific protein phosphorylation was
activated by the use of 3×10−1 M sorbitol (sorb; MAPK
p38) and 3×10−8 M bryostatin-1 (bryo; PKD1) and
inhibited by the use of 10−5 M SB203580 (SB; MAPK p38)
and 5×10−5 M CID755673 (CID; PKD1), respectively.
Studies with PC-3-HSP27 cells treated with docetaxel and
exclusively incubated with a single kinase activator or inhibitor
exhibited no statistically significant differences for cellular
growth compared to controls (Fig.
2A). Thus, we compared combinations of both activators (sorb +
bryo) and both inhibitors (SB + CID). As a result, co-activated
MAPK p38 and PKD1 led to increased docetaxel sensitivity of cells
(Fig. 2B), measured by
significantly decreased cell numbers (24 h, 1.9-fold, p=0.0438; 48
h, 1.6-fold, p=0.0313; 72 h, 1.6-fold, p=0.1550; 96 h, 1.6-fold,
p=0.0271; 120 h, 2.0-fold, p=0.0117; 144 h, 2.0-fold, p=0.1383). In
addition to other putative regulatory pathways, our data indicated
kinase-dependent control of HSP27 functions in chemoresistance,
assuming higher phosphorylated HSP27 responsible for increasing
sensitivity to docetaxel treatment.
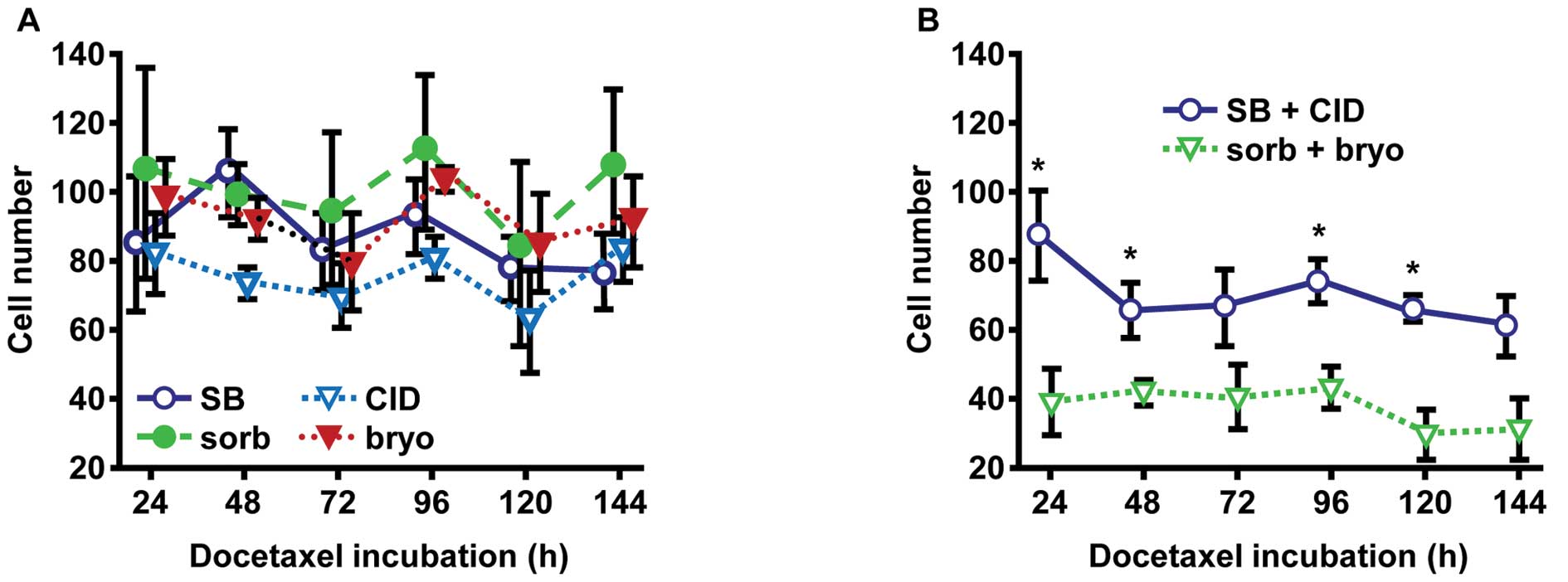 | Figure 2MAPK p38 and PKD1 suppress cell growth
in prostate cancer cells stably expressing HSP27 during docetaxel
treatment. (A) To study phosphorylation effects of the protein
kinases MAPK p38 and PKD1 on prostate cancer cells expressing HSP27
wt protein, PC-3-HSP27 cells were incubated over a period of 144 h
with 3×10−1 M MAPK p38 activator sorbitol (sorb),
3×10−8 M PKD1 activator bryostatin-1 (bryo),
10−5 M MAPK p38 inhibitor SB203580 (SB), and
5×10−5 M PKD1 inhibitor CID755673 (CID), respectively.
Cell numbers were measured at indicated time points using a CASY
Cell Counter and Analyzer Model TT. Results were standardized to
untreated control cells and illustrated as the means ± SD with
p-values determined by Student’s t-test. (B) Due to negligible
effects on cellular growth of cells treated with individual
activators or inhibitors, the experiments were replicated applying
combinations of both activators (sorb + bryo) and both inhibitors
(SB + CID). Given results are indicated as the means ± SD of cell
count and cell viability, respectively, and were compared to
control cells. *p≤0.05, as determined by Student’s
t-test. MAPK p38, mitogen-activated protein kinase p38; PKD1,
protein kinase D1; HSP, heat shock protein. |
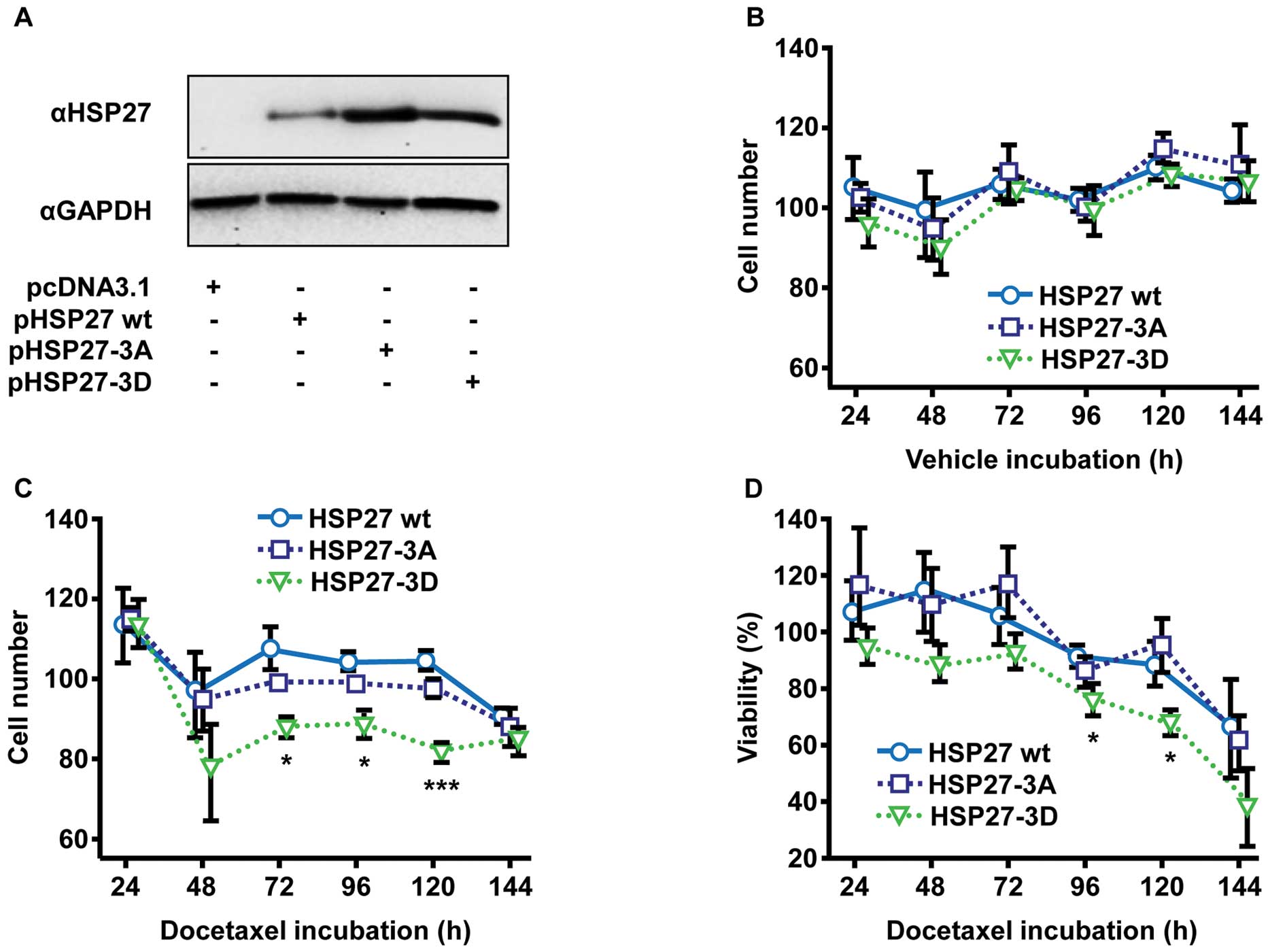
HSP27-driven chemoresistance is
diminished by overexpression of non-phosphorylatable HSP27-3D
mutant
Considering several cellular pathways potentially
being targeted by MAPK p38 and PKD1 activities and therefore being
responsible for the changes in chemosensitivity, we further
analyzed specific effects of HSP27 phosphorylation on
chemoresistance. Cellular functionality of HSP27 depends on
phosphorylation of the three regulatory phosphorylation sites,
namely serine-15, -78 and -82 (18). PC-3 cells were transiently
transfected with DNA plasmids encoding for HSP27 wild-type (HSP27
wt) and HSP27 phospho-mutants: i) the non-phosphorylatable triple
substitution mutant HSP27-3A (substitution of serine-15, -78 and
-82 with alanine-15, -78 and -82); and ii) the phosphorylation
mimicking triple mutant HSP27-3D (substitution of serine-15, -78
and -82 with aspartic acid-15, -78 and -82), imitating a triple
phosphorylated protein by charge and steric characteristics of the
aspartic acid residues.
Transient overexpression of HSP27 wt and both HSP27
mutants (Fig. 3A) in experiments
without docetaxel treatment were found to have no impact on
cellular growth (Fig. 3B). The
incubation with docetaxel, however, caused notable reductions in
proliferation of PC-3 cells overexpressing phosphorylation
mimicking HSP27-3D compared to cells overexpressing HSP27 wt and
non-phosphorylatable HSP27-3A (Fig.
3C). Compared to HSP27 wt and HSP27-3A, respectively,
HSP27-3D-expressing cells exhibited significantly reduced cell
proliferation (72 h, 1.2-fold reduction, p=0.0329; 96 h, 1.2-fold
reduction, p=0.0226; 120 h, 1.3-fold reduction, p=0.0005). As
expected, similar findings were noted by further analyses of
cellular viability using MTT assay (Fig. 3D). Again, HSP27-3D revealed poorer
cellular viability of transfected, docetaxel-treated cells, when
comparing overexpression of HSP27-3D with HSP27 wt and HSP27-3A (96
h, 1.2-fold reduction, p=0.0415; 120 h, 1.3-fold reduction,
p=0.0087). Analysis of cell counting (Fig. 3C) as well as cellular viability
(Fig. 3D) of HSP27 wt- and
HSP27-3A-transfected cells showed no differences.
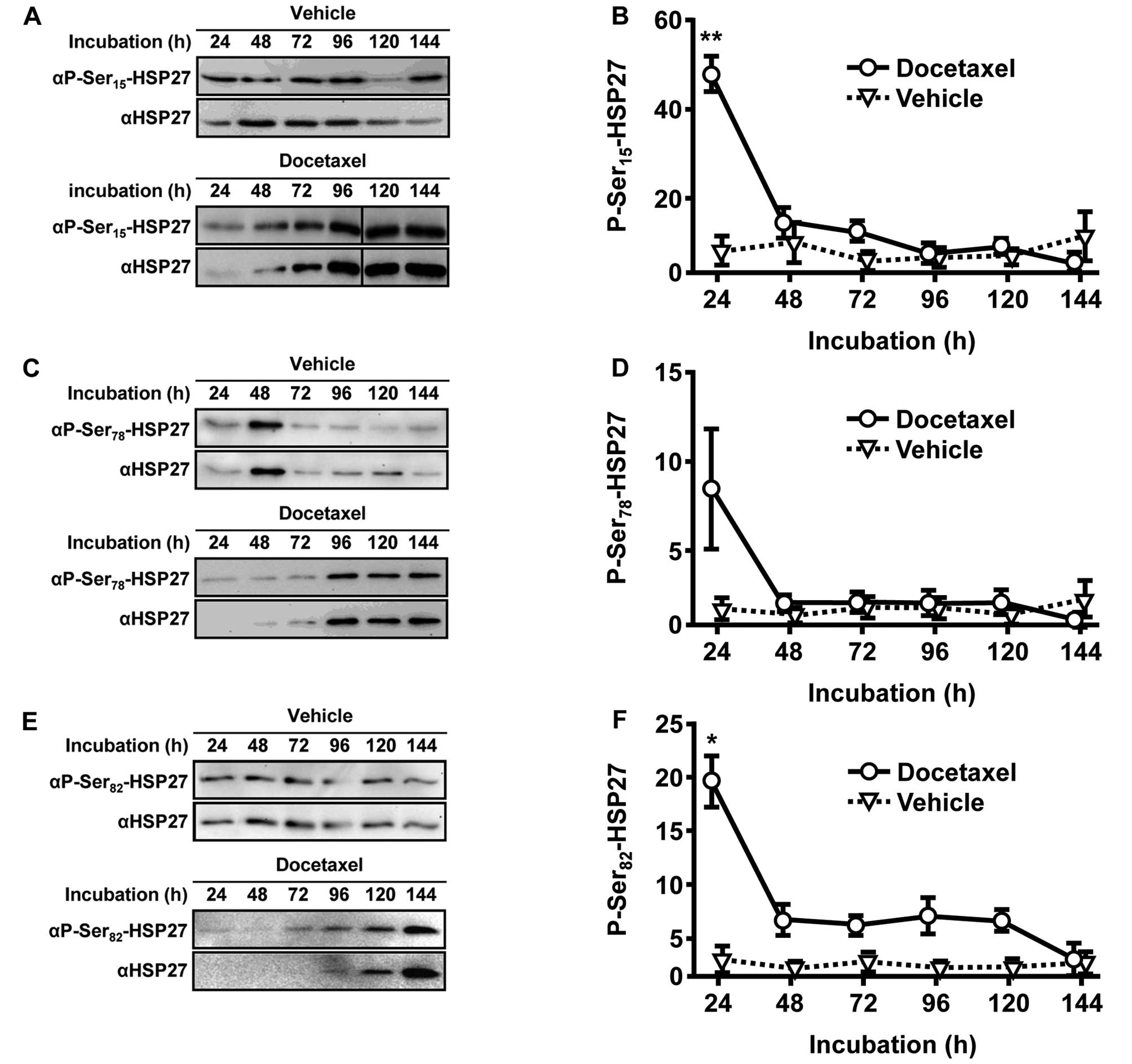
Docetaxel causes rapid and transient
phosphorylations of HSP27
To clarify how HSP27-driven chemoresistance is
activated and regulated throughout the treatment with docetaxel, we
analyzed changes in the phosphorylation status of HSP27 utilizing
phospho-specific HSP27 antibodies (Fig.
4A, C and E). Vehicle-treated control cells did not obviously
change their status of low-phosphorylated HSP27 (Fig. 4B, D and F). In comparison, protein
phosphorylation in the presence of docetaxel was significantly
increased after 24 h at HSP27 phosphorylation sites serine-15
(9.4-fold, p=0.0071) and serine-82 (12.6-fold, p=0.0150). Analysis
of serine-78 phosphorylation revealed similar results. However,
statistical assessment procedure failed to reach significance.
Notably, the strong induction of HSP27 phosphorylation was largely
reversed after 48 h of docetaxel incubation and remained constantly
low for 144 h of observation. Periods of incubation >24 h did
not differ in HSP27 phosphorylation between vehicle- and
docetaxel-treated groups (Fig. 4B, D
and F).
Discussion
In the present study, we demonstrated that treatment
of PC cells with docetaxel, a first-line therapy drug for
castration-resistant PC, induces phosphorylation of HSP27, which is
a cytoprotective factor counter-acting antiproliferative cancer
therapies (18). Markedly, we
detected a rapid phosphorylation within the first day of docetaxel
incubation at all of the three regulatory phosphorylation sites
immediately followed by dephosphorylation. Furthermore, permanently
phosphorylated HSP27 was associated with attenuated cytoprotective
properties, strongly supporting the theory that rapid but
short-termed phosphorylation of HSP27 is an important step in
HSP27-driven initiation of chemoresistance.
HSP27 induction has been detected in various solid
tumors. Hence, HSP27 may be regarded as a pivotal factor for
progression and treatment resistance. In PC cells, HSP27 has a
crucial role in tumor-specific and therapy-induced cytoprotection
enabling tumor growth in the setting of hormonal ablation and
cytostatic therapy. It is well recognized that HSP27 properties
control activities of various signaling molecules, such as
β-catenin, E-cadherin, interleukins, transforming growth factor β
and AR, with all these effector proteins frequently dysregulated in
PC and shown to exert oncogenic transformation (12,19–22).
As shown in Fig. 1, stable
overexpression of HSP27 in PC-3 cells revealed significantly
enhanced resistance to docetaxel treatment in a concentration- and
time-dependent manner. These findings, in accordance with previous
studies (14), reflect HSP27-driven
cytoprotectivity in PC.
Biological functions of HSP27 are mainly regulated
by protein phosphorylation, suggesting that HSP27 phosphorylation
may concur with chemoresistance mechanisms in PC cells. Therefore,
it is reasonable to assume that temporal and spatial
phosphorylation/dephosphorylation of HSP27 by kinase and
phosphatase activities are an essential part of HSP27 in the
regulation of treatment-resistant processes.
It has been reported that kinase pathways of MAPK
p38 and PKD1 are major regulators of HSP27 properties in PC
(9,23). Since we showed that the
pharmacological activation of MAPK p38 and PKD1 kinases led to
elevated docetaxel sensitivity (Fig.
2B), it is conceivable that this sensitizing effect, at least
to some extent, is mediated through HSP27 phosphorylation.
Accordingly, HSP27 phosphorylation by MAPK p38 and PKD1 was already
found in PC and other cancer entities (9–11,16).
However, due to both kinases operating in numerous signaling
pathways, we cannot exclude the possibility that both kinases
affect additional cellular targets whose modulation may contribute
to enhanced antiproliferative effects in the presence of docetaxel.
To examine this possibility, we focused on transfection experiments
utilizing DNA plasmids encoding for the well-approved HSP27 mutants
HSP27-3A and HSP27-3D representing an unphosphorylatable and a
phospho-mimicking type of HSP27, respectively. Expression of
HSP27-3A obtained no alteration on docetaxel sensitivity whereas
expression of HSP27-3D resulted in significantly increased
sensitivity to docetaxel (Fig. 3C and
D). These observations were supported by western blot analysis
using an experimental chemotherapy model which exhibited rapid and
transient phosphorylations of HSP27, a mechanism similar to
HSP27-driven drug resistance described in breast and pancreatic
cancer cells (24,25). In particular, we detected
docetaxel-induced phosphorylation of HSP27 amino acids serine-15,
-78 and -82 after 24 h of incubation (Fig. 4). Experiments in high time
resolution (1–24 h) exhibited a rapid but transient HSP27
phosphorylation within the first 8 h (data not shown).
Although our experiments clearly indicate that
permanently phosphorylated HSP27 increases chemosensitivity, our
current understanding of HSP27 phospho-regulation is still limited.
Nakashima et al showed enhanced chemosensitivity in
gemcitabine-treated pancreatic cancer cells with phosphorylated
HSP27 protein (25), confirming our
own observations. In contrast, Taba et al reported converse
effects of phosphorylated HSP27 in pancreatic cancer cells in the
presence of gemcitabine, which was confirmed in a cell model system
of 5-fluorouracil-resistant colon cancer (26,27).
Furthermore, some studies provide examples for growth regulatory
properties of phosphor-HSP27 in the absence of chemotherapeutics
(28,29). Collectively, regulation of HSP27
appears to be part of a complex regulatory network controlled by
kinases and phosphatases and may be induced in a drug- and cell
type-specific manner.
Protein phosphorylation at multiple sites is often
catalyzed in a fixed order. However, human HSP27 phosphorylation
does not occur in an obligatory sequence (11). Although HSP27 phosphorylation has
been shown in response to various stimuli, such as cytokines,
receptor ligands, glucose and metals (18), to our knowledge this study is the
first to report HSP27 phosphorylation initiated by drug treatment
of PC cells. Due to the transient character of the HSP27
phosphorylation which lasts several hours, strongly orchestrated
activities of kinases and phosphatases are required. According to
our observations and the data from previous studies, most probably
MAPK p38 and PKD1 pathways exert HSP27 phosphorylation (18). Since several kinases were described
using HSP27 as substrate for protein phosphorylation, e.g. various
MAPK-activated kinases and protein kinase C (30–32),
it cannot be excluded that further kinase pathways contribute to
HSP27 phosphorylation. Despite the increase in molecular insights
into HSP27 phosphorylation in cancer mechanisms, functions of HSP27
dephosphorylation are for the most part unknown. Several studies
have shown that inhibition of protein phosphatase 2A (PP2A) affects
the phospho-status of human HSP27 (33,34).
Notably, Liu et al recently described that dual specificity
protein phosphatase 1 (DUSP1) controls treatment resistance in
pancreatic cancer by affecting MAPK pathways (35), thus, it is consistent with our
observations indicating potential targets for further
examinations.
In conclusion, our findings enhance our
understanding of survival mechanisms in drug-resistant PC cells and
qualify HSP27 phosphorylation pathways as appropriate targets for
new anticancer strategies. Future research will clarify if kinase
activators as well as phosphatase inhibitors are a potent
opportunity to support existing HSP27-targeting anticancer
therapies.
Acknowledgements
This study was supported in part by a grant from the
Dr Robert Pfleger-Foundation, Bamberg, Germany. The authors thank
Anne Brandenburg and Katja Wittig for their technical
assistance.
References
|
1
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J,
Murray T and Thun MJ: Cancer statistics, 2008. CA Cancer J Clin.
58:71–96. 2008. View Article : Google Scholar
|
|
2
|
Hwang C: Overcoming docetaxel resistance
in prostate cancer: a perspective review. Ther Adv Med Oncol.
4:329–340. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Beere HM, Wolf BB, Cain K, Mosser DD,
Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM and Green DR:
Heat-shock protein 70 inhibits apoptosis by preventing recruitment
of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2:469–475.
2000. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Haslbeck M and Buchner J: Chaperone
function of sHsps. Prog Mol Subcell Biol. 28:37–59. 2002.
View Article : Google Scholar
|
|
5
|
Parcellier A, Brunet M, Schmitt E, Col E,
Didelot C, Hammann A, Nakayama K, Nakayama KI, Khochbin S, Solary E
and Garrido C: HSP27 favors ubiquitination and proteasomal
degradation of p27Kip1 and helps S-phase re-entry in
stressed cells. FASEB J. 20:1179–1181. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ischia J, Saad F and Gleave M: The promise
of heat shock protein inhibitors in the treatment of castration
resistant prostate cancer. Curr Opin Urol. 23:194–200. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu T, Guevara OE, Warburton RR, Hill NS,
Gaestel M and Kayyali US: Modulation of HSP27 alters
hypoxia-induced endothelial permeability and related signaling
pathways. J Cell Physiol. 220:600–610. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tsuji F, Oh-hashi K and Kiuchi K:
Differential effects of Akt pathway inhibitors on IL-1β-induced
protein phosphorylation in human fibroblast-like synoviocytes. J
Recept Signal Transduct Res. 32:22–28. 2012.
|
|
9
|
Hassan S, Biswas MH, Zhang C, Du C and
Balaji KC: Heat shock protein 27 mediates repression of androgen
receptor function by protein kinase D1 in prostate cancer cells.
Oncogene. 28:4386–4396. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Evans IM, Britton G and Zachary IC:
Vascular endothelial growth factor induces heat shock protein (HSP)
27 serine 82 phosphorylation and endothelial tubulogenesis via
protein kinase D and independent of p38 kinase. Cell Signal.
20:1375–1384. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Landry J, Lambert H, Zhou M, Lavoie JN,
Hickey E, Weber LA and Anderson CW: Human HSP27 is phosphorylated
at serines 78 and 82 by heat shock and mitogen-activated kinases
that recognize the same amino acid motif as S6 kinase II. J Biol
Chem. 267:794–803. 1992.PubMed/NCBI
|
|
12
|
Stope MB, Schubert T, Staar D, Rönnau C,
Streitbörger A, Kroeger N, Kubisch C, Zimmermann U, Walther R and
Burchardt M: Effect of the heat shock protein HSP27 on androgen
receptor expression and function in prostate cancer cells. World J
Urol. 30:327–331. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Stope MB, Bradl J, Peters S, Streitbörger
A, Weiss M, Zimmermann U, Walther R, Lillig CH and Burchardt M:
Shortened isoforms of the androgen receptor are regulated by the
cytoprotective heat-shock protein HSPB1 and the tumor-suppressive
microRNA miR-1 in prostate cancer cells. Anticancer Res.
33:4921–4926. 2013.PubMed/NCBI
|
|
14
|
Dokas LA, Malone AM, Williams FE, Nauli SM
and Messer WS Jr: Multiple protein kinases determine the
phosphorylated state of the small heat shock protein, HSP27, in
SH-SY5Y neuroblastoma cells. Neuropharmacology. 61:12–24. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kubisch C, Dimagno MJ, Tietz AB, Welsh MJ,
Ernst SA, Brandt-Nedelev B, Diebold J, Wagner AC, Göke B, Williams
JA and Schäfer C: Overexpression of heat shock protein Hsp27
protects against cerulein-induced pancreatitis. Gastroenterology.
127:275–286. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rocchi P, So A, Kojima S, Signaevsky M,
Beraldi E, Fazli L, Hurtado-Coll A, Yamanaka K and Gleave M: Heat
shock protein 27 increases after androgen ablation and plays a
cytoprotective role in hormone-refractory prostate cancer. Cancer
Res. 64:6595–6602. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cuenda A, Rouse J, Doza YN, Meier R, Cohen
P, Gallagher TF, Young PR and Lee JC: SB 203580 is a specific
inhibitor of a MAP kinase homologue which is stimulated by cellular
stresses and interleukin-1. FEBS Lett. 364:229–233. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kostenko S and Moens U: Heat shock protein
27 phosphorylation: kinases, phosphatases, functions and pathology.
Cell Mol Life Sci. 66:3289–3307. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jaggi M, Rao PS, Smith DJ, Hemstreet GP
and Balaji KC: Protein kinase C mu is down-regulated in
androgen-independent prostate cancer. Biochem Biophys Res Commun.
307:254–260. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mak P, Jaggi M, Syed V, Chauhan SC, Hassan
S, Biswas H and Balaji KC: Protein kinase D1 (PKD1) influences
androgen receptor (AR) function in prostate cancer cells. Biochem
Biophys Res Commun. 373:618–623. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Syed V, Mak P, Du C and Balaji KC:
β-catenin mediates alteration in cell proliferation, motility and
invasion of prostate cancer cells by differential expression of
E-cadherin and protein kinase D1. J Cell Biochem. 104:82–95.
2008.
|
|
22
|
Di K, Wong YC and Wang X: Id-1 promotes
TGF-β1-induced cell motility through HSP27 activation and
disassembly of adherens junction in prostate epithelial cells. Exp
Cell Res. 313:3983–3999. 2007.
|
|
23
|
Zoubeidi A, Zardan A, Beraldi E, Fazli L,
Sowery R, Rennie P, Nelson C and Gleave M: Cooperative interactions
between androgen receptor (AR) and heat-shock protein 27 facilitate
AR transcriptional activity. Cancer Res. 67:10455–10465. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xu Y, Diao Y, Qi S, Pan X, Wang Q, Xin Y,
Cao X, Ruan J, Zhao Z, Luo L, Liu C and Yin Z: Phosphorylated Hsp27
activates ATM-dependent p53 signaling and mediates the resistance
of MCF-7 cells to doxorubicin-induced apoptosis. Cell Signal.
25:1176–1185. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nakashima M, Adachi S, Yasuda I, Yamauchi
T, Kawaguchi J, Itani M, Yoshioka T, Matsushima-Nishiwaki R, Hirose
Y, Kozawa O and Moriwaki H: Phosphorylation status of heat shock
protein 27 plays a key role in gemcitabine-induced apoptosis of
pancreatic cancer cells. Cancer Lett. 313:218–225. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Taba K, Kuramitsu Y, Ryozawa S, Yoshida K,
Tanaka T, Maehara S, Maehara Y, Sakaida I and Nakamura K:
Heat-shock protein 27 is phosphorylated in gemcitabine-resistant
pancreatic cancer cells. Anticancer Res. 30:2539–2543.
2010.PubMed/NCBI
|
|
27
|
Sakai A, Otani M, Miyamoto A, Yoshida H,
Furuya E and Tanigawa N: Identification of phosphorylated serine-15
and -82 residues of HSPB1 in 5-fluorouracil-resistant colorectal
cancer cells by proteomics. J Proteomics. 75:806–818. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mouratidis PX, Colston KW, Bartlett JB,
Muller GW, Man HW, Stirling D and Dalgleish AG: Antiproliferative
effects of CC-8062 and CC-8075 in pancreatic cancer cells.
Pancreas. 38:78–84. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Garrido C, Ottavi P, Fromentin A, Hammann
A, Arrigo AP, Chauffert B and Mehlen P: HSP27 as a mediator of
confluence-dependent resistance to cell death induced by anticancer
drugs. Cancer Res. 57:2661–2667. 1997.PubMed/NCBI
|
|
30
|
Maizels ET, Peters CA, Kline M, Cutler RE
Jr, Shanmugam M and Hunzicker-Dunn M: Heat-schock protein-25/27
phosphorylation by the d isoform of protein kinase C. Biochem J.
332:703–712. 1998.PubMed/NCBI
|
|
31
|
Clifton AD, Young PR and Cohen P: A
comparison of the substrate specificity of MAPKAP kinase-2 and
MAPKAPkinase-3 and their activation by cytokines and cellular
stress. FEBS Lett. 392:209–214. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Guo K, Liu Y, Zhou H, Dai Z, Zhang J, Sun
R, Chen J, Sun Q, Lu W, Kang X and Chen P: Involvement of protein
kinase C β-extracellular signal-regulating kinase1/2/p38
mitogen-activated protein kinase-heat shock protein 27 activation
in hepatocellular carcinoma cell motility and invasion. Cancer Sci.
99:486–496. 2008.
|
|
33
|
Suga H, Nakajima K, Shu E, Kanno Y, Hirade
K, Ishisaki A, Matsuno H, Tanabe K, Takai S, Akamatsu S, Kato K,
Oiso Y and Kozawa O: Possible involvement of phosphatidylinositol
3-kinase/Akt signal pathway in vasopressin-induced HSP27
phosphorylation in aortic smooth muscle A10 cells. Arch Biochem
Biophys. 438:137–145. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yuan J and Rozengurt E: PKD, PKD2, and p38
MAPK mediate Hsp27 serine-82 phosphorylation induced by neurotensin
in pancreatic cancer PANC-1 cells. J Cell Biochem. 103:648–662.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu F, Gore AJ, Wilson JL and Korc M:
DUSP1 is a novel target for enhancing pancreatic cancer cell
sensitivity to gemcitabine. PLoS One. 9:e849822014. View Article : Google Scholar : PubMed/NCBI
|