Introduction
During cancer metastasis, tumor cells detach from
their neighbors, invade the surrounding stroma and undergo
intravasation. They survive in the circulation and arrest in the
vessels of the target organ where they undergo extravasation,
invade the matrix, and proliferate within the organ parenchyma to
complete the metastatic process (1–3).
However, reaching another location in the body does not guarantee
that a metastatic tumor will form, and successful colonization and
proliferation of tumor cells depends on the interaction of the
cells with the microenvironment at the site of metastasis.
Previous studies found that the suitability of a
site for metastatic colonization was determined not only by the
characteristics of cancer cells, but also by the local
microenvironment (1,4–10).
Hematopoietic progenitor cells and myeloid cells were reportedly
recruited to the lung by primary tumors to form the
microenvironment. Furthermore, emerging evidence has revealed that
bone marrow-derived cells (BMDCs) are a major player in the
formation of pre-metastatic niches (1,8).
The lung is a common target site for cancer cell
metastasis. Recent reports showed that the lung microenvironment
was modulated by distant primary tumors before the metastatic
cancer cells arrived. This process occurred through changes in the
lung in response to factors secreted by the primary tumors.
Considering the key role of the microenvironment in
tumor metastasis, there has been speculation regarding the possible
molecular processes whereby the primary tumor modulates the
pre-metastatic niche in the lung prior to the formation of
metastasis. Although several molecules have been implicated
(11,12), such as matrix metalloproteinase-9,
S100A8 and S100A9, the mechanisms underlying lung
microenvironmental changes in the pre-metastatic phase remain
largely unknown.
In the present study, we investigated the effects of
the primary tumor on the lung microenvironment, and explored the
role of tumor-derived vascular endothelial growth factor (VEGF) as
a key initiator of pre-metastatic modulation, and its relationship
with prostaglandin E2 (PGE2) production.
Materials and methods
Cells and reagents
4T1 cells were cultured in RPMI-1640 with 10% fetal
calf serum medium. Six-week-old female Balb/c mice were acclimated
and caged in groups of 6 or less. Celecoxib was manufactured by
Pharmacia (Peapack, NJ, USA), rVEGF was purchased from Roche
Applied Science (Basel, Switzerland) and a rat anti-mouse CD31
monoclonal antibody was from BD-Pharmingen. Celecoxib was fed on
diet after 14 days of tumor cells implanted on the back of the
mice, rVEGF was intraperitoneally injected daily at a dose of 5
mg/kg.
Bone marrow transplantation
All animal studies were reviewed and approved by the
Animal Care and Use Committee of the Shandong Provincial Animal
Board (Jinan, China). Mice (6–8 weeks old) labeled with enhanced
green fluorescent protein (EGFP) were used as donors of bone marrow
(BM). BM cells (BMCs) were prepared according to procedures
previously described (1). BMDCs
were flushed out with RPMI-1640 medium from femurs and tibiae using
21-gauge needles. Then cells were harvested by centrifugation,
resuspended with phosphate-buffered saline (PBS), and intravenously
injected at a total number of 1×106 cells per 100 μl PBS
into recipient syngenic Balb/c mice that had received whole body
irradiation of 900 rad immediately before the transplantation.
RNA extraction and RT-qPCR
Total RNA was extracted from PBS perfused lungs with
the RNeasy kit (Qiagen) according to the manufacturer’s protocol.
First-strand cDNA was synthesized from 1 μg total tissue RNA using
the RevertAid™ First Strand cDNA synthesis kit (Thermo Fisher
Scientific, Waltham, MA, USA) with random primers. Then cDNA was
amplified for quantitative real-time PCR. The specific primers used
were: Cox-2, forward, 5′-ATCAGGTCATTGGTGGAG-3′ and reverse,
5′-ACACTCTGTTGTGCTCCC-3′; β-actin, forward,
5′-CTGTCCCTGTATGCCTCTG-3′ and reverse, 5′-ATGTCACGCACGATTTCC-3′.
Real-time PCR reactions were performed at: 95°C, 10 sec
(denaturation); 55°C, 30 sec (annealing); 72°C, 30 sec (extension)
for 35 cycles. Real-time PCR reactions were performed on the ABI
7000 Fast real-time PCR system with SYBR-Premix Ex Taq™ according
to the manufacturer’s instructions.
Microarray analysis of the lungs
Balb/c mice were injected with 4T1 cells; control
mice received PBS only. Twelve days after tumor-cell inoculation,
mice were euthanized and lungs were perfused with PBS to completely
remove blood from the lung vasculature. Cleaned lung lobes were
immersed in RNAlater™ to stabilize RNA. Total RNA was isolated and
used for subsequent microarray analysis. Affymetrix Gene Chip mouse
genome 430 2.0 array was applied to analyze the expression profile
of tissue samples. The microarray chip consisting of 27,326
different human cDNAs (Angilent, Wilmington, DE, USA) was employed
to the analysis of the MPVECs treated with lipopolysacharide (LPS)
and VEGF.
Cancer cell labeling and in vivo homing
assay
4T1 cells were labeled with DiI, then
2×104 labeled cells were infused i.v. In mice with
primary tumors, the labeled cells were infused when the tumor
reached 6 mm in diameter. In mice without tumors, labeled cells
were infused i.v. 6–15 min after VEGF treatment. Five and 24 h
after cancer cell infusion, the lungs were perfused with PBS under
physical pressure to remove any circulating tumor cells and were
then excised. Three lung tissue fragments (3 mm in diameter) were
randomly selected and two 50-μM sections per fragment were examined
with a confocal microscope. The number of labeled cancer cells was
normalized by total tissue surface area.
Metastasis assay in vivo
4T1 cells (1×105) labeled with DiI were
intravenously injected at 2 weeks after the subcutaneous
implantation of tumor cells (2×106) into the mice. Two
weeks after injection, animals were sacrificed and lungs were
resected. Surface metastasis was examined with light microscopy and
further identification was performed with scanning for DiI-positive
metastasis and processed for histological analysis. The metastasis
was quantified as the total number of metastatic foci per reference
area.
Immunofluorescence of lung tissue and
MPVECs
The method of MPVEC isolation was described
previously (2). MPVECs were
cultured in a 24-well plate with various concentrations of VEGF for
4 h and then fixed by 4% paraformaldehyde. After washing with PBS,
cells were incubated with 3% bovine serum albumin at room
temperature for 30 min, followed by incubation of celecoxib at a
dilution of 1:100 overnight at 4°C. For the immunofluorescence of
lung tissue, the procedures of the first day were described above.
Similarly, an FITC-conjugated fluorescent secondary antibody was
added on the next day, and cells (or tissue sections) were detected
under an inverted fluorescent microscope.
Tumor cell-MPVEC adhesion assay
MPVECs were cultured in 24-well plates with various
concentrations of VEGF for 6 h. 4T1 cells were suspended at a
concentration of 1×106 cells/ml in DMEM and labeled with
2′,7′-bis-5-carboxyfluorescetin acetoxymethyl ester (BCECF, AM) at
37°C for 15 min. Then, the labeled 4T1 cells were added to the
MPVEC 24-well plates, and incubated at 37°C for 30 min. After
washing 3 times with PBS to remove unattached cells, the number of
adhered AM-labeled 4T1 cells was counted using an inverted
fluorescent microscope.
Measurement of circulating cytokines
The RayBio Custom mouse cytokines antibody array kit
was purchased from RayBiotech and used according to the
manufacturer’s instructions. Briefly, after blocking with 1X
blocking buffer, membranes were incubated for 1.5 h with the
experimental serum. The membranes were washed and incubated with
biotin-conjugated antibodies for 1.5 h. The membranes were washed
again and incubated with streptavidin-conjugated horseradish
peroxidase for 2 h, washed, and developed using an enhanced
chemiluminescent substrate for horseradish peroxidase.
Chemiluminescence was detected using EpiChemi3® Darkroom
imaging system and LabWorks® densitometry software. Data
was corrected for background signal and normalized to positive
controls using RayBio® Analysis Tool software (UVP
Bioimaging, Upland, CA, USA).
Statistical analyses
The data were analyzed using JMP ver. 6. The groups
were compared using Chi-square test and Fisher’s exact test. The
analyses yielded 95% confidence intervals and P-values. Values of
P<0.05 were considered to indicate a statistically significant
difference.
Results
Primary breast cancer tumor cells
stimulate inflammation and BMDC recruitment in pre-metastatic
lungs
We examined the effects of breast cancer-cell
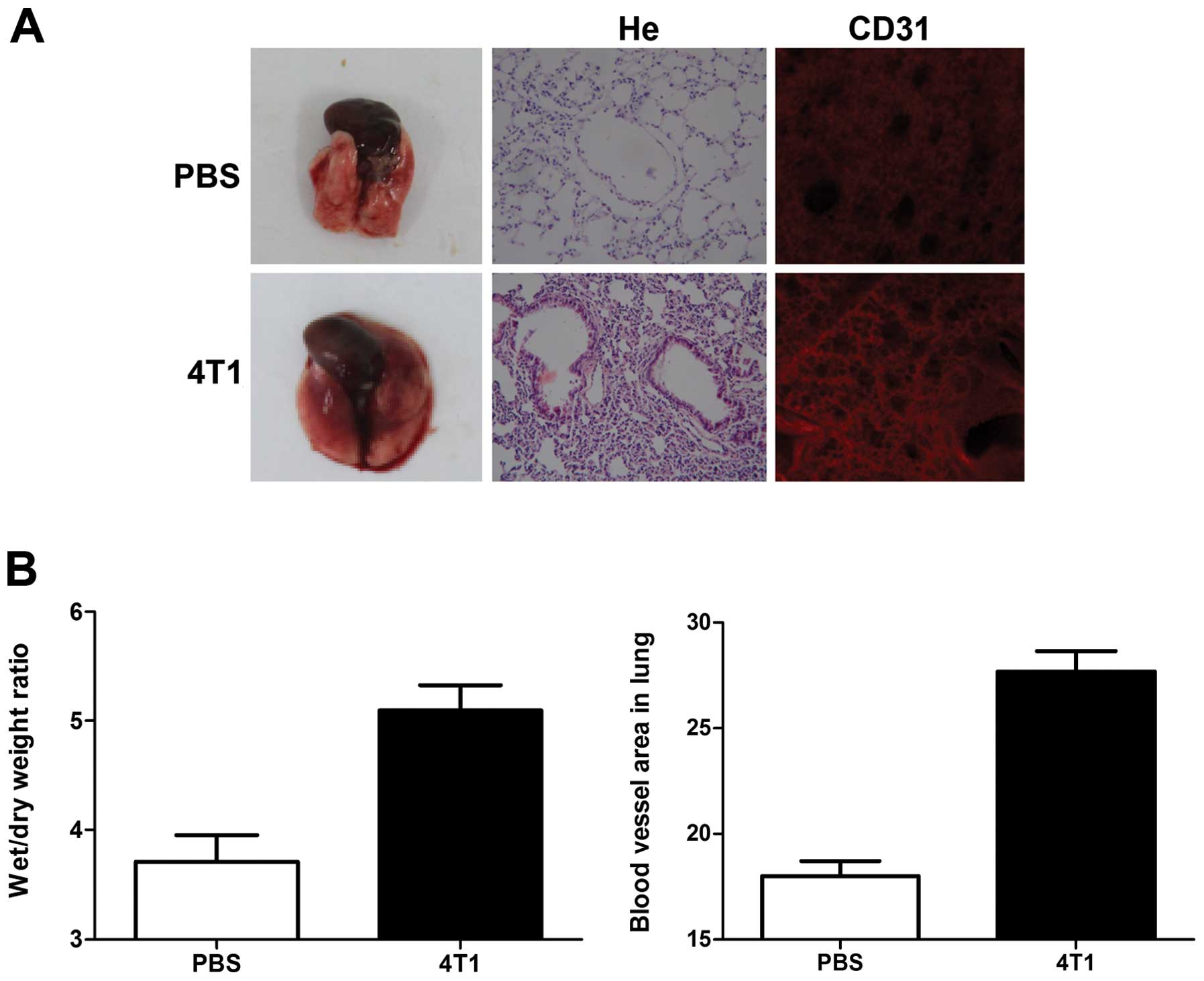
induction during the pre-metastatic phase on the morphology of lung
tissues excised from 4T1 tumor-bearing mice. Edema and lung
wet/dry-weight ratios were significantly increased by breast cancer
cell induction, compared with controls (Fig. 1). Hematoxylin and eosin (H&E)
staining also showed a higher level of inflammatory cell
infiltration in the lungs of tumor-bearing mice, compared with
non-tumor-bearing mice (Fig.
1A).
Immunohistochemical analysis with CD31 antibody
showed that blood vessels in the lungs of 4T1-tumor-bearing mice
appeared as primitive and dilated sinusoidal vascular structures,
consisting of disorganized, tortuous, and interconnected vascular
plexuses. Total blood vessel density was markedly increased in the
lungs of tumor-bearing mice compared with non-tumor-bearing mice
(Fig. 1A and B).
Metastatic breast cancer cells
preferentially home to BMDC recruitment sites in pre-metastatic
mouse lungs
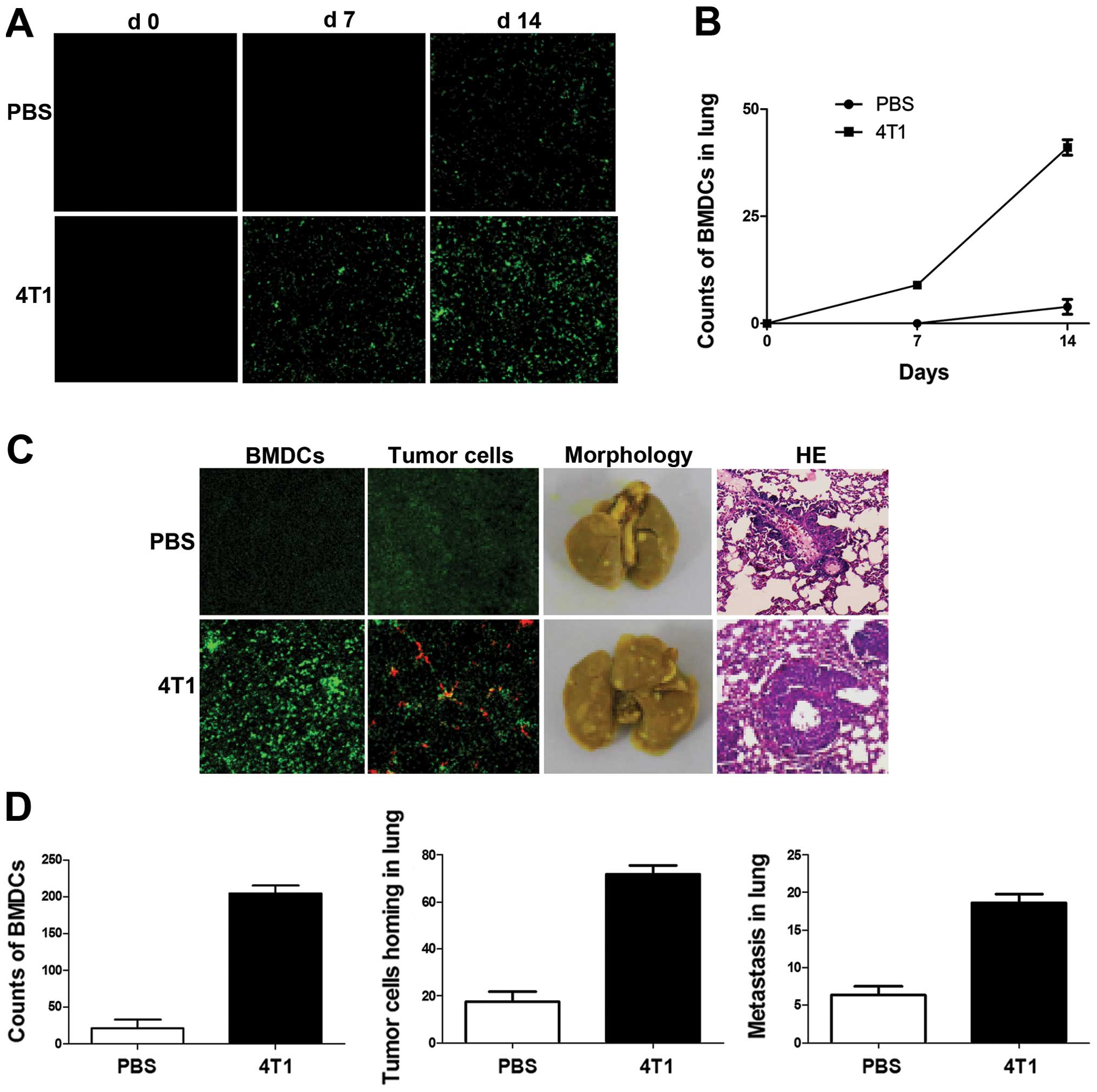
During tumor progression, BMDCs have been shown to
promote inappropriate growth, invasion and ultimately metastasis.
In the present study, BMDC mobilization and recruitment changed in
response to breast cancer cell inoculation. Green fluorescent
protein (GFP) was used to label BMDCs and pre-metastatic lung
fluorescence was detected by confocal microscopy following
4T1-tumor-cell injection in mice. Mice inoculated with PBS were
used as controls. No GFP+ BMDCs were observed in lungs
without tumor cell injection (Fig.
2A), but GFP+ BMDCs were found scattered in lung
tissue by day 7 after tumor implantation in tumor-bearing mice. No
BMDCs were observed in control lungs at the same stage. The
difference increased at day 14, when clusters of GFP+
BMDCs were detected in lung tissues in tumor-bearing mice and near
terminal bronchioles, while minimal GFP+ BMDCs were
observed in lung tissues in a small number of control mice
(Fig. 2A and B).
The homing of tumor cells and their subsequent
survival are critical for metastasis. To investigate the effects of
BMDC recruitment on breast cancer cell homing in mouse lungs,
DiI-labeled 4T1 cells were injected into mice 2 weeks after tumor
inoculation. Clusters of GFP+ BMDCs were detected near
terminal bronchioles in 4T1-tumor-bearing mice (Fig. 2C). A subsequent metastasis assay
revealed that the number of metastatic foci in lung lesions in
4T1-tumor-bearing mice was significantly higher than that in the
lungs of non-tumor-bearing mice (Fig.
2C and D).
Primary breast cancer cells trigger BMDC
recruitment and inflammation in lungs through VEGF
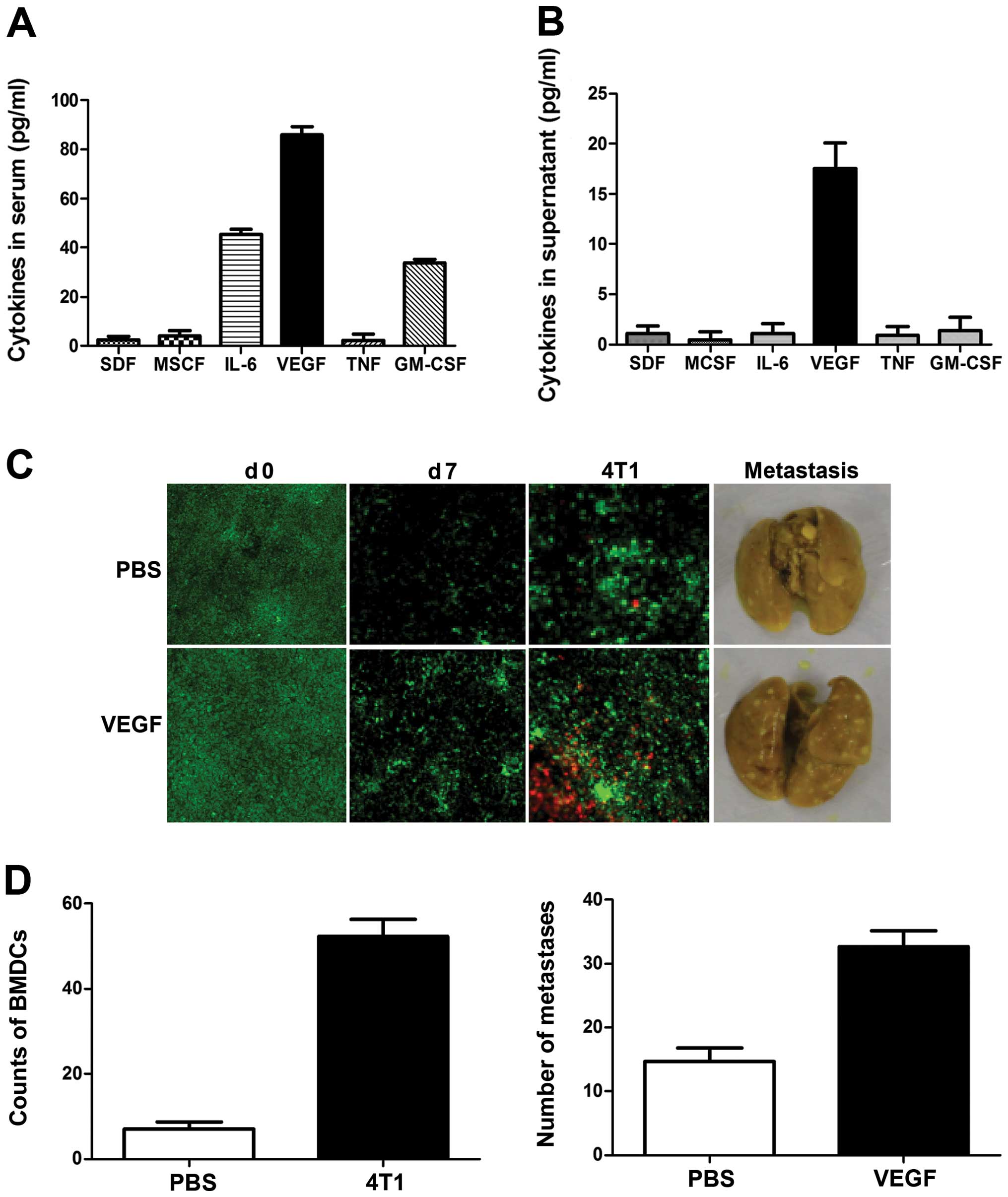
We investigated the factor secreted by breast cancer
cells responsible for inducing the pre-metastatic niche and BMDC
recruitment. Mouse sera and 4T1 cell culture media were assayed
using a RayBio Custom mouse cytokine antibody array kit. As shown
in Fig. 3A, levels of VEGF,
granulocyte-macrophage colony-stimulating factor (GM-CSF) and
interleukin-6 (IL-6) were significantly increased in sera from
4T1-tumor-bearing mice. However, only VEGF was detected in culture
media from 4T1 cells, at levels as high as 20 pg/ml (Fig. 3B); levels of GM-CSF, IL-6 and tumor
necrosis factor-α (TNFα) were too low for detection, suggesting
that 4T1 cell-derived VEGF may be the critical triggering molecule
responsible for initiating the formation of the pre-metastatic
niche.
To investigate the role of VEGF in the initiation of
the pre-metastatic niche, mice were treated daily with recombinant
VEGF for 7 consecutive days and BMDC recruitment in the lung was
tested. As expected, the frequency of BMDCs within the lungs, as
well as angiogenesis, increased significantly in response to VEGF
(Fig. 3C). To determine the effect
of VEGF on the metastatic potential of breast cancer cells, mice
were treated with VEGF for 7 consecutive days, and 4T1 cells were
then injected through the tail vein. The number of visible
metastatic foci in lungs revealed increased metastases (Fig. 3C and D), and examination of the
lungs showed enlarged lung masses in VEGF-treated mice, compared
with controls. These results suggest that VEGF is a crucial
triggering molecule initiating the formation of the pre-metastatic
niche.
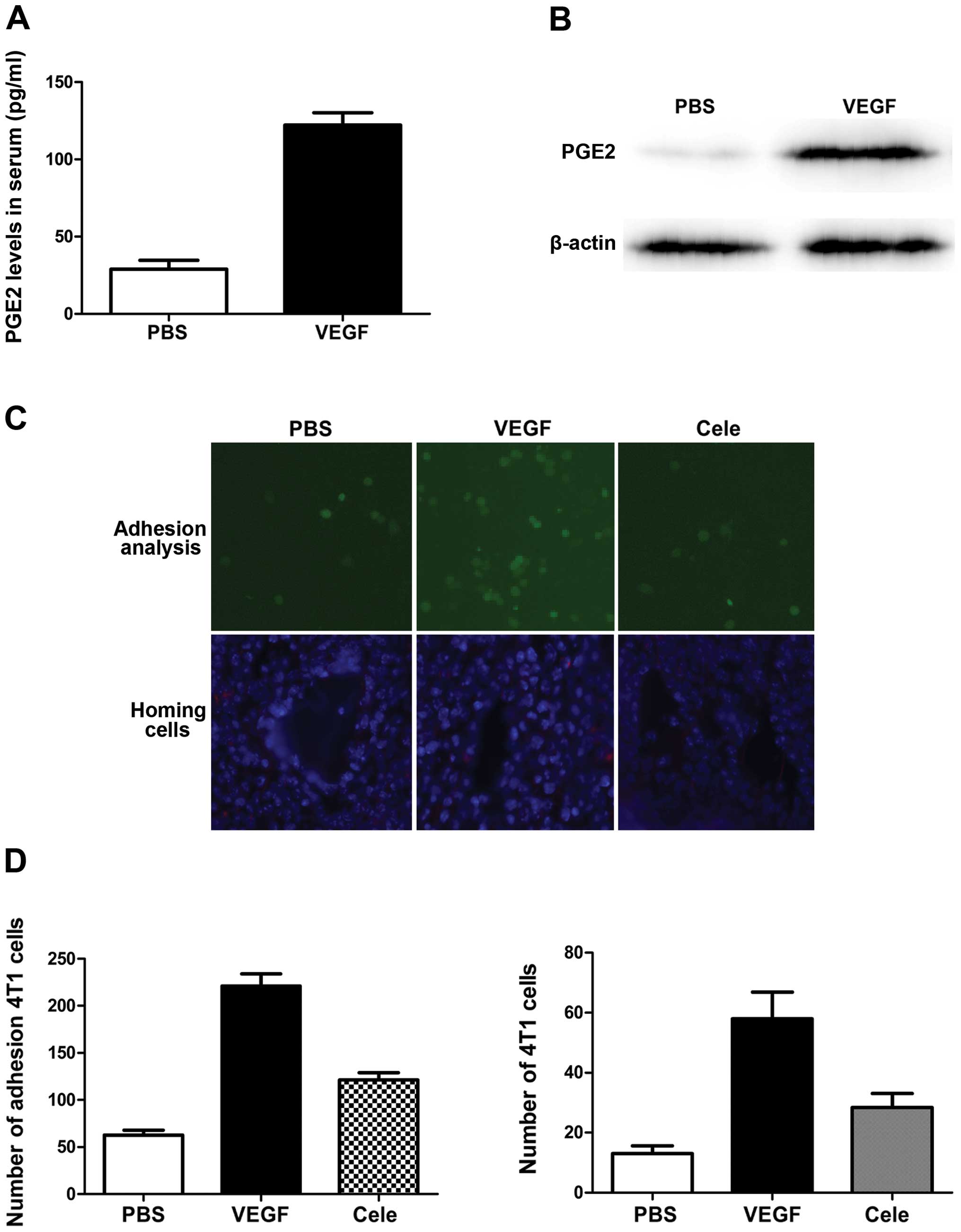
Primary tumors induce PGE2 production and
inflammatory response in pre-metastatic lungs
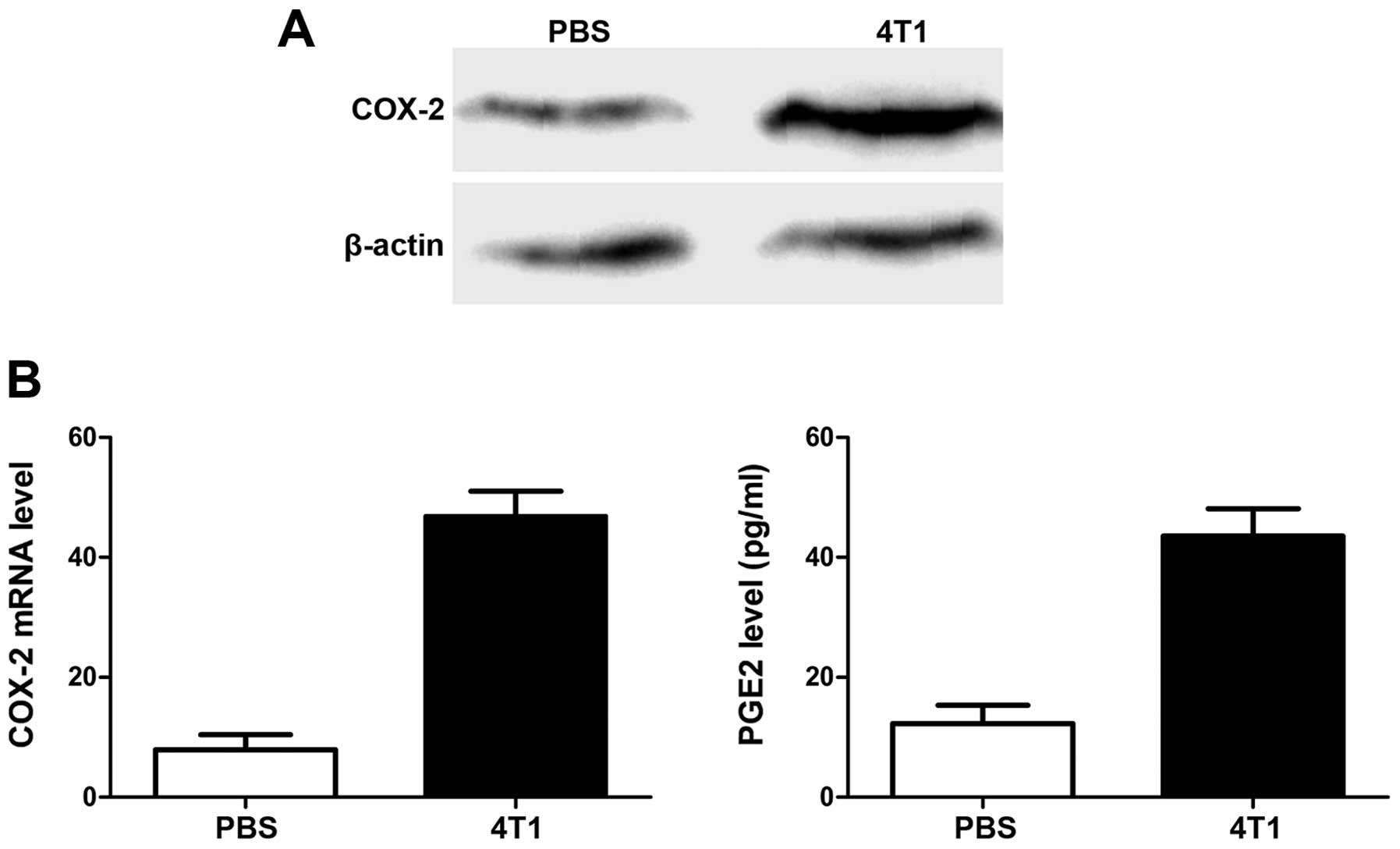
The important role of PGE2 in tumor angiogenesis and
metastasis suggests that its expression may be correlated with
VEGF. We therefore compared PGE2 levels, and levels of its
synthesis regulator cyclooxygenase-2 (COX-2) gene, in
pre-metastatic lungs in mice with and without 4T1 tumors. As shown
in Fig. 4, COX-2 expression in lung
tissues was upregulated at both mRNA and protein levels by
inoculation of 4T1 cells, and the increase in PGE2 production
confirmed the COX-2-expression results.
Notably, the expression profiles in MPVECs induced
by VEGF and PGE2 were similar. Among the genes with >2-fold
changes in expression in response to stimulation, 48 genes were
upregulated and 35 were downregulated in response to VEGF, and 65
and 33, respectively, in response to PGE2. Notably, 33 upregulated
genes and 14 downregulated genes were common to both responses,
suggesting that they used the same downstream pathways.
VEGF plays key roles in inducing
pre-metastatic recruitment of BMDCs in lungs and its function is
suppressed by COX-2 inhibition
PGE2 is a secreted protein previously characterized
as a proinflammatory factor. We defined the role of PGE2 in
VEGF-induced BMDC recruitment and lung metastasis in primary mouse
lung endothelial cells exposed to VEGF. As expected, VEGF
stimulation significantly increased COX-2 expression and PGE2
levels in endothelial cells (Fig. 5A
and B). We also evaluated the role of PGE2 induction by VEGF in
endothelial cell adhesion to metastatic cancer cells in an in
vitro cancer cell adhesion assay. VEGF stimulation increased
the number of fluorescently-labeled metastatic cancer cells
attached to lung endothelial cells (Fig. 5C and D). The addition of celecoxib
abolished tumor cell adhesion to the endothelial cells (Fig. 5C and D). PGE2 overexpression
resulted in a significant increase in the homing of infused
metastatic cancer cells to the lungs of mice treated with VEGF
(Fig. 5C and D), measured 48 h
after metastatic cancer cell infusion (Fig. 5C and D). Collectively, these data
show that blocking lung endothelial PGE2 production reduced
VEGF-induced breast cancer metastasis to the lungs.
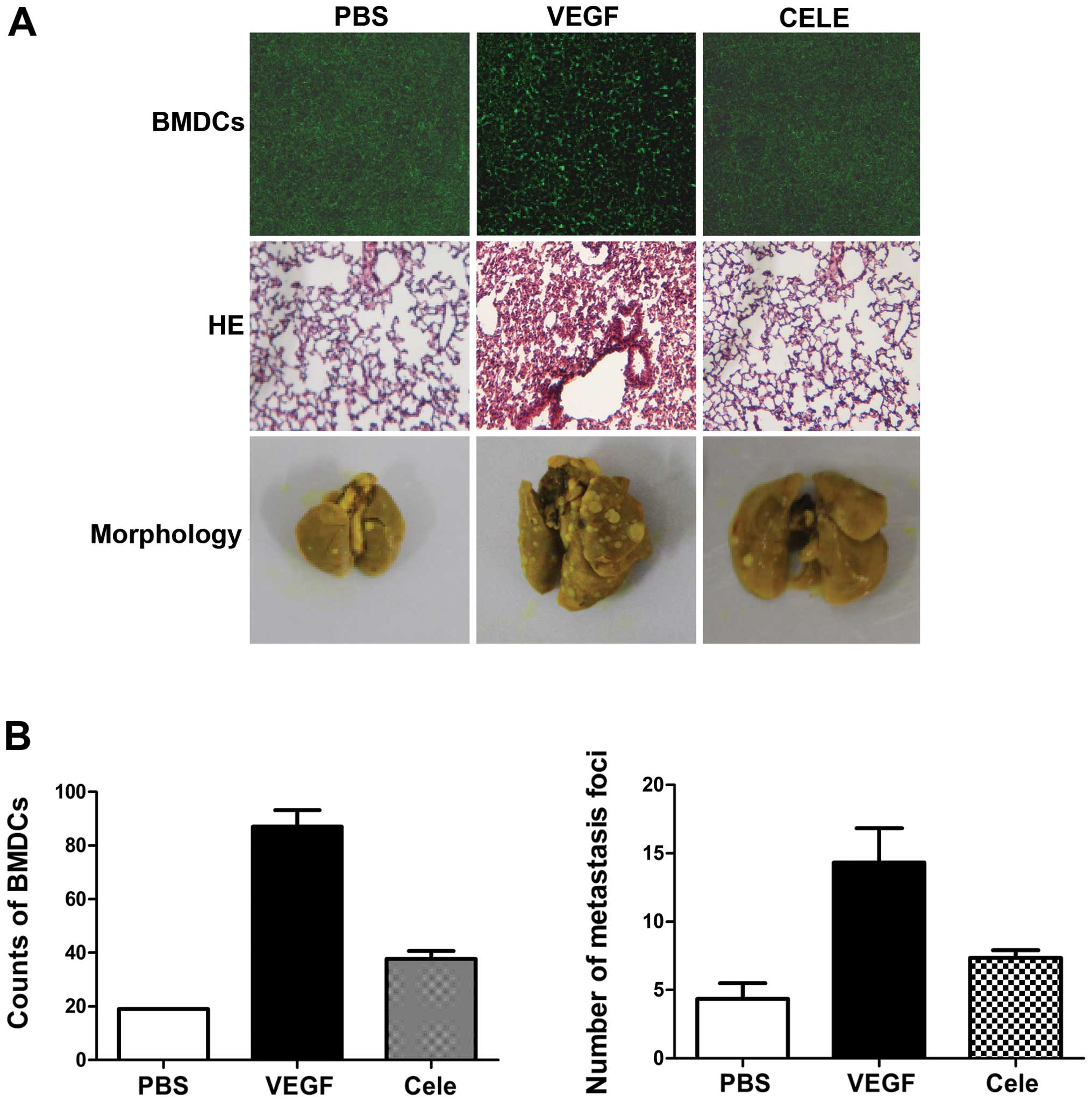
VEGF mediates 4T1 cell homing to the
lungs via COX-2
The role of COX-2 in VEGF-induced BMDC recruitment
and lung metastasis was defined by treating mice with VEGF and the
COX-2 inhibitor, celecoxib. Celecoxib treatment reduced the
recruitment of BMDCs in lungs (Fig. 6A
and B), and lung inflammation was also reduced, as shown by
H&E staining (Fig. 6A). 4T1
cells were then injected into the tail vein of mice pretreated with
VEGF in the presence or absence of celecoxib. Celecoxib treatment
significantly reduced VEGF-induced lung metastases of 4T1 cells
(Fig. 6A and B). These results
indicate that PGE2 is involved in VEGF-induced pre-metastatic
microenvironmental changes.
Discussion
Some researchers have suggested that therapy for
metastasis should be targeted not only against tumor cells, but
also against the host microenvironment that contributes to and
supports the progressive growth and survival of metastatic cancer
cells (13–18). The results of this study showed that
BMDC recruitment and inflammatory responses could be induced in
pre-metastatic lungs by distant 4T1 breast cancer in mice, and that
pre-metastatic changes contributed to the homing of metastatic
cells in lungs. The process of cancer metastasis is complex and
consists of a series of inter-related steps. Extravasation and
homing of metastatic tumor cells in specific organs is a checkpoint
in the production of clinically-relevant lesions. Previous studies
have suggested that primary tumors are able to modify the distant
microenvironment before the arrival of metastatic tumor cells, to
create what is known as the pre-metastatic niche (19–23).
This ability of tumors to affect distant tissues is expected to
enable cancer cells to target specific organs, and BMDCs are
thought to be major players in these processes. In accordance with
these reports, our results showed that clusters of GFP+
BMDCs and an inflammatory response were detected in lung tissues by
day 14 after tumor implantation.
To determine which factor(s) secreted by tumor cells
might be responsible for mobilizing BMDCs, we measured plasma and
tumor levels of several factors previously implicated in BMDC
mobilization. However, only VEGF correlated with the ability of
tumors to metastasize, and pretreatment with VEGF was sufficient to
mimic the pre-metastatic environment initiated by primary tumors.
Previous studies have shown that multiple secreted factors
overexpressed in primary tumors, including VEGF, transforming
growth factor-β, TNF-α, and angiopoietin-2, can induce lung
microenvironmental changes and enhance the metastatic properties of
several tumors. VEGF released from lung-metastasizing cancer cells
can activate the Src-FAK complex in lung endothelial cells and
promote vascular hyperpermeability, upregulation of endothelial
adhesion molecules, and cancer-cell homing. We demonstrated that
primary tumor-derived VEGF was involved in induction of the
inflammatory response in pre-metastatic lungs.
Our data indicated that tumor-secreted VEGF induced
PGE2 expression in pre-metastatic lungs. Treatment with celecoxib
significantly reduced the number of VEGF-induced metastases,
emphasizing the important role of BMDC homing in the lung. Our
results suggest that anti-inflammatory agents may modulate the
microenvironment in target organs through suppression of the
inflammatory response, leading to marked inhibition of lung
metastasis, which may be an alternative mechanism of action for
these agents.
The mechanism by which celecoxib modulates the
microenviroment in the lung remains to be elucidated. PGE2
expression is rapidly induced in response to inflammatory stimuli,
such as TNF-α or lipopolysaccharide (24,25).
VEGF overexpression has been reported to lead to PGE2 production
(26,27), while exposure of cultured
endothelial cells to tumor-secreted factors increased PGE2
expression (28–30), and VEGF directly induced PGE2
expression in endothelial cells (31). We showed that PGE2 can function as a
chemoattractant to enhance the mobilization of BMDCs and facilitate
their homing into the lung, before the arrival of tumor cells. Lung
regions thus provide discrete, fertile fields of pre-metastatic
‘soil’ where increased tumor-cell-homing is facilitated by the
secretion of factors by endothelial cells.
In summary, our results provide direct evidence for
the ability of primary tumor-derived VEGF to trigger a host
inflammatory response in the lung microenvironment through PGE2
production, leading circulating tumor cells to localize
preferentially in regions of high BMDC recruitment. These results
are in agreement with those of other studies that have implicated
the proinflammatory host response in promoting the process of lung
metastasis.
Acknowledgements
We gratefully acknowledge the financial support from
the Ph.D. Programs Foundation of Ministry of Education of China
(20120131120046), the Natural Science Foundation (General programs
81272351), the Natural Science Foundation of Shandong Province
(ZR2012HM020, BS2010YY038), and the Key Development Program for
Basic Research of Shandong Province (2012G0021826).
References
|
1
|
Nguyen DX, Bos PD and Massagué J:
Metastasis: from dissemination to organ-specific colonization. Nat
Rev Cancer. 9:274–284. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fidler IJ: The organ microenvironment and
cancer metastasis. Differentiation. 70:498–505. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Das Roy L, Pathangey LB, Tinder TL,
Schettini JL, Gruber HE and Mukherjee P: Breast-cancer-associated
metastasis is significantly increased in a model of autoimmune
arthritis. Breast Cancer Res. 11:Jul 30–2009.(Epub ahead of print).
View Article : Google Scholar
|
|
4
|
Hiratsuka S, Watanabe A, Aburatani H and
Maru Y: Tumour-mediated upregulation of chemoattractants and
recruitment of myeloid cells predetermines lung metastasis. Nat
Cell Biol. 8:1369–1375. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kaplan RN, Riba RD, Zacharoulis S, et al:
VEGFR1-positive haematopoietic bone marrow progenitors initiate the
pre-metastatic niche. Nature. 438:820–827. 2005. View Article : Google Scholar
|
|
6
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar
|
|
7
|
Chen SF, Fei X and Li SH: A new simple
method for isolation of microvascular endothelial cells avoiding
both chemical and mechanical injuries. Microvasc Res. 50:119–128.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lu H, Ouyang W and Huang C: Inflammation,
a key event in cancer development. Mol Cancer Res. 4:221–233. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fidler IJ: The pathogenesis of cancer
metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev
Cancer. 3:453–458. 2003.
|
|
10
|
Murdoch C, Muthana M, Coffelt SB and Lewis
CE: The role of myeloid cells in the promotion of tumour
angiogenesis. Nat Rev Cancer. 8:618–631. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim S, Takahashi H, Lin WW, et al:
Carcinoma-produced factors activate myeloid cells through TLR2 to
stimulate metastasis. Nature. 457:102–106. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Carmeliet P: VEGF as a key mediator of
angiogenesis in cancer. Oncology. 69:4–10. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mantovani A, Allavena P, Sica A and
Balkwill F: Cancer-related inflammation. Nature. 454:436–444. 2008.
View Article : Google Scholar
|
|
14
|
Padua D, Zhang XH and Wang Q: TGFbeta
primes breast tumors for lung metastasis seeding through
angiopoietin-like 4. Cell. 133:66–77. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Serafini P, Borrello I and Bronte V:
Myeloid suppressor cells in cancer: recruitment, phenotype,
properties, and mechanisms of immune suppression. Semin Cancer
Biol. 16:53–65. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang L, Huang J, Ren X, et al: Abrogation
of TGFbeta signaling in mammary carcinomas recruits
Gr-1+CD11b+ myeloid cells that promote
metastasis. Cancer Cell. 13:23–35. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Olkhanud PB, Baatar D, Bodogai M, et al:
Breast cancer lung metastasis requires expression of chemokine
receptor CCR4 and regulatory T cells. Cancer Res. 69:5996–6004.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang Y, Song N, Ding Y, et al: Pulmonary
vascular destabilization in the premetastatic phase facilitates
lung metastasis. Cancer Res. 69:7529–7537. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kitamura T, Kometani K, Hashida H, et al:
SMAD4-deficient intestinal tumors recruit CCR1+ myeloid
cells that promote invasion. Nat Genet. 39:467–475. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dawson MR, Duda DG, Fukumura D and Jain
RK: VEGFR1-activity independent metastasis formation. Nature.
461:E4–E5. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Erler JT, Bennewith KL, Cox TR, et al:
Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow
cell recruitment to form the premetastatic niche. Cancer Cell.
15:35–44. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Duda DG, Cohen KS, Kozin SV, et al:
Evidence for incorporation of bone marrow-derived endothelial cells
into perfused blood vessels in tumors. Blood. 107:2774–2776. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dawson MR, Duda DG, Chae SS, Fukumura D
and Jain RK: VEGFR1 activity modulates myeloid cell infiltration in
growing lung metastases but is not required for spontaneous
metastasis formation. PLoS One. 4:e65252009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bos CL, Richel DJ, Ritsema T, et al:
Prostanoids and prostanoid receptors in signal transduction. Int J
Biochem Cell Biol. 36:1187–1205. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ma X, Kundu N, Ioffe OB, et al:
Prostaglandin E receptor EP1 suppresses breast cancer metastasis
and is linked to survival differences and cancer disparities. Mol
Cancer Res. 8:1310–1318. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Narumiya S, Sugimoto Y and Ushikubi F:
Prostanoid receptors: structures, properties, and functions.
Physiol Rev. 79:1193–1226. 1999.PubMed/NCBI
|
|
27
|
Yang T and Du Y: Distinct roles of central
and peripheral prostaglandin E2 and EP subtypes in blood pressure
regulation. Am J Hypertens. 25:1042–1049. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gupta GP, Nguyen DX, Chiang AC, et al:
Mediators of vascular remodeling co-opted for sequential steps in
lung metastasis. Nature. 446:765–770. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nozawa H, Chiu C and Hanahan D:
Infiltrating neutrophils mediate the initial angiogenic switch in a
mouse model of multistage carcinogenesis. Proc Natl Acad Sci USA.
103:12493–12498. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fridlender ZG, Sun J, Kim S, Kapoor V,
Cheng G, Ling L, et al: Polarization of tumor-associated neutrophil
(TAN) phenotype by TGF-beta: ‘N1’ versus ‘N2’ TAN. Cancer Cell.
16:183–194. 2009.PubMed/NCBI
|
|
31
|
Chang SH, Liu CH, Conway R, et al: Role of
prostaglandin E2-dependent angiogenic switch in cyclooxygenase
2-induced breast cancer progression. Proc Natl Acad Sci USA.
101:591–596. 2004. View Article : Google Scholar : PubMed/NCBI
|