Introduction
Acute myeloid leukemia (AML) is characterized by the
accumulation of immature myeloid cells in the bone marrow and the
suppression of normal hematopoiesis. Increased understanding of the
pathogenesis of AML has fostered the development of targeted
therapies intended particularly for older patients. Successful
targeting of each of the numerous genotypic variants of AML is a
major challenge (1,2). AML blasts develop from normal blasts
affected by two types of genetic damage. The first (class 1)
results in constitutive activation of cell-surface receptors, such
as RAS, or receptor tyrosine kinases, such as FLT3 and c-KIT.
Through various downstream pathways, constitutive activation
confers a survival or proliferative advantage leading to clonal
expansion of the affected hematopoietic progenitors. In mouse
models, abnormalities in RAS, FLT3 or c-KIT by themselves produce
only a myeloproliferative disorder and not AML. The second type of
lesion (class 2) exemplified by overexpression of HOX genes or
formation of fusion genes, resulting from the t(8;21) or
inv(16) abnormalities block
myeloid differentiation but, similar to class 1 lesions, do not
cause leukemia in mouse models. AML may develop only when both
classes of lesions are present (2–7).
Current efforts in clinical research have focused on the assessment
of targeted therapies. Such new approaches may lead to an increase
in the cure rate. At present, the strategy of gene therapy may
offer new hope for expanded treatment options for patients with
AML.
To improve therapeutic effect, the gene therapy
treatment strategy has been suggested as a future direction. Gene
therapy has emerged as a powerful tool with which to regulate
biological functions in diseased tissues and to treat cancers.
Oncolytic viruses not only have the capacity to express therapeutic
genes in tumor cells, but also can be used as a direct
tumor-destruction medicament. For safety, oncolytic viral
replication must be controlled strictly within tumor cells
(8–11). Thus, different types of viruses have
been genetically modified, including adenovirus, adeno-associated
virus (AAV), herpes simplex virus type I, reovirus, lentivirus,
hepatitis B virus and Newcastle disease virus (12–16).
One of the common strategies used to design oncolytic adenoviruses
is to modify adenoviral E1A protein. The CR2 region of the
adenoviral E1A binds to retinoblastoma protein (RB) and the
RB-related proteins which regulate the E2F family of transcription
factors and induces quiescent cells to enter the S phase. Since
tumor cells often have dysfunctional RB and an uncontrolled cell
cycle, deletion of the CR2 region allows this engineered adenovirus
to selectively replicate in tumor cells but not in quiescent normal
cells. We previously constructed several conditionally replicative
adenovirus systems in which viral replication only occurred in
cancer cells with high expression of hTERT and an abnormal cell
cycle checkpoint. However, among these oncolytic adenoviruses,
therapeutic genes were controlled by exogenous constitutive
promoters. Thus, expression of therapeutic genes in normal tissue
may induce an undesired effect even if the virus does not replicate
(17–21). To overcome this limitation, we
developed the AdCN205 system in which therapeutic gene expression
is controlled by the adenovirus E3 endogenous promoter. We
confirmed that this vector expresses the therapeutic gene in a
predictable and safe manner (22).
The human adenovirus serotype 11 (Ad11), with a fiber different
from that of Ad5, can enter cells which have a capacity for
secreting complement regulatory protein CD46 (a specific membrane
protein) to the cytomembrane. The Ad11 adenoviral vector is an
alternative means for cancer therapy, particularly in leukemia.
Therefore, we developed the AdCN205–11 system to selectively
replicate in AML cell lines, and it exhibited marked antitumor
activity. Chimeric oncolytic adenoviruses, which are increasingly
being developed as oncolytic vectors, have recently emerged as a
novel approach for treating various neoplasms (23–25).
Because of their capability to replicate in transduced tumor cells
but not in normal tissue, they infect neighboring tumor cells after
selective viral propagation resulting in virus-mediated lysis of
tumor cells and tumor regression. Chimeric oncolytic adenoviruses
may have better antitumor efficacy than that of nonreplicating
adenoviruses.
We chose melanoma differentiation-associated
gene-7/inter-leukin-24 (MDA-7/IL-24), a novel member of the IL-10
family of cytokines, as inserted therapeutic genes to augment the
anti-cancer effects of the AdCN205-11 system since MDA-7/IL-24 is a
novel tumor-suppressor gene that plays an important role in
tumorigenesis. The overexpression of MDA-7/IL-24 using a
recombinant adenovirus causes in vitro selective growth
suppression and apoptosis of a variety of tumor cells, including
melanoma, glioblastoma multilforme, osteosarcoma and carcinomas of
the breast, colon, lung, cervix, kidney and prostate (26–31).
Studies by several laboratories have uncovered many of the unique
properties of MDA-7/IL-24. These include cancer-specific apoptosis
induction, cell cycle regulation, an ability to inhibit
angiogenesis, potent ‘bystander antitumor activity’ and a capacity
to enhance the sensitivity of tumor cells to radiation,
chemotherapy and monoclonal antibody therapy. Moreover, based on
its profound cancer tropism, substantiated by in vivo human
xenograft studies in nude mice, MDA-7/IL-24 (administered as
Ad.mda-7) was evaluated in a phase I clinical trial in patients
with melanomas and solid cancers. These studies document that
MDA-7/IL-24 is well tolerated and demonstrate the evidence of
significant clinical activity. In these contexts, MDA-7/IL-24
represents a unique cytokine gene with the potential for therapy of
human cancers. There are three unique properties of MDA-7/IL-24,
namely its potent ‘bystander antitumor activity’, ability to
sensitize tumor cells to radiation and its antiangiogenesis
properties. Additionally, the phase I clinical trial is provided.
These studies confirm that MDA-7/IL-24 has promise for the
management of diverse types of cancers (26–31).
In the present study, we confimed that the application of IL-24 in
the gene therapy system has a dramatic antitumor effect.
Materials and methods
Cell cultures
The normal human liver L02 cell line was purchased
from the Shanghai Cell Collection (Shanghai, China). The HEK293
cell line was purchased from Microbix Biosystems Inc. (Toronto, ON,
Canada). The human AML KG-1 cell line was purchased from the
American Type Culture Collection (ATCC; Rockville, MD, USA). L02
and HEK293 cells were maintained in Dulbecco’s modified Eagle’s
medium (DMEM; Gibco-BRL, Grand Island, NY, USA) supplemented with
10% fetal bovine serum (FBS; Gibco-BRL), 4 mM glutamine, 50 U/ml
penicillin and 50 mg/ml streptomycin. KG-1 cells were grown in
RPMI-1640 medium containing 10% FBS. All cell lines were incubated
at 37°C in a humidified air atmosphere with 5% CO2.
Construction of the oncolytic adenoviral
vectors
The constructs including pCN205-11-EGFP and
pCN205-11-IL-24 were generated according to the standard molecular
cloning protocol. The homologous recombination between the
pCN205-11-EGFP and pCN205-11-IL-24 plasmids and the pCN103 plasmid
carrying the oncolytic adenoviral backbone was carried out in E.
coli strain BJ5183, to create pAdCN205-EGFP and pAdCN205-IL-24,
respectively. Viral particles were produced in HEK293 cells by
transfection with PacI-digested pAdCN205-EGFP and
pAdCN205-IL-24 to obtain recombinant AdCN205-11-EGFP and
AdCN205-11-IL-24.
Generation, purification and titration of
the recombinant adenoviruses
To obtain the viruses, the plasmids pCN205-11-EGFP
and pCN205-11-IL-24 were digested by PacI and transfected
into HEK293 cells using Effectene (Qiagen, Hilden, Germany). The
recombinant adenoviruses were amplified in HEK293 cells and
purified by cesium chloride gradient ultracentrifugation. The
titration of the recombinant adenovirus was carried out with Tissue
Culture Infectious Dose (TCID)50 assay in HEK293
cells.
Virus progeny assay
To determine the virus progeny, acute myeloid
leukemia or normal cells were infected with Ad-wt, AdCN205-EGFP,
AdCN205-IL-24, AdCN205-11-EGFP or AdCN205-11-IL-24 at an MOI of 10.
After 48 h, the supernatants and cells were collected separately.
The cells were resuspended in PBS, and the virus was released by
three freeze-thaw cycles. Virus production in the supernatants and
cell lysates was determined by TCID50 assay in HEK293
cells.
Cell viability assay
Cells were seeded in 96-well plates at a density of
1×104 per well. When cells grew to subconfluency, they
were infected with Ad-wt, AdCN205-EGFP, AdCN205-IL-24,
AdCN205-11-EGFP or AdCN205-11-IL-24 at a multiplicity of infection
(MOI) of 10. Fresh medium containing 0.5 mg/ml
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
(Sigma Chemical Co., St. Louis, MO, USA) solution was added to each
well at different times after infection. Cells were incubated at
37°C for 4 h and then 150 μl of dimethyl sulfoxide was added to
each well and mixed thoroughly for 10 min. Absorbance was read at
595 nm with a DNA Expert (Tecan, Switzerland).
In vitro transduction studies
For suspension of cells, the day before infection,
3×105 cells per well (24-well plate) were seeded. The
next day, the attached cells were counted and the viruses were
added at the multiplicities of infection (MOIs) indicated in the
figure legends in 1 ml of growth medium. Cells were incubated with
the virus for 24–48 h, and incubated at 37°C in a humidified
atmosphere with 5% CO2. Percentages of GFP-positive
cells were detected by fluorescence microscopy.
Hoechst 33342 staining and apoptotic cell
staining
Cells (4×105) were cultured in each well
of 6-well plates. Twelve hours later, the cells were infected with
Ad-wt, AdCN205-EGFP, AdCN205-IL-24, AdCN205-11-EGFP or
AdCN205-11-IL-24 at an MOI of 10. Forty-eight hours after
infection, the medium was replaced with PBS and then the cells were
stained with Hoechst 33342 (25 μg/ml). The percentage of apoptotic
cells was analyzed with and observed under a fluorescence
microscope.
Flow cytometric analysis
For apoptosis detection, the KG-1 cells were seeded
in 6-well culture plates and infected with Ad-wt, AdCN205-EGFP,
AdCN205-IL-24, AdCN205-11-EGFP or AdCN205-11-IL24 at an MOI of 10.
Cells infected with the adenovirus were trypsinized and washed once
with complete medium. Aliquots of cells (5×105) were
resuspended in 500 μl of binding buffer and stained with
fluorescein isothiocyanate-labeled Annexin V and propidium iodide
(PI; BioVision, Palo Alto, CA, USA) according to the manufacturer’s
instructions. A fluorescence-activated cell sorting (BD
Biosciences) assay was performed immediately after staining. To
determine transduction efficiency of the virus, KG-1 cells were
infected with Ad-wt, AdCN205-IL-24 or AdCN205-11-IL-24 at an MOI of
10 for 48 h and then subjected to flow cytometric analysis.
Western blot analysis
To determine the expression of various proteins,
western blot analysis was performed as described previously. The
cells were harvested by trypsinization and resuspended in lysis
buffer [62.5 mM Tris-HCl (pH 6.8), 2% sodium dodecylsulfate, 10 mM
glycerol and 1.55% dithiothreitol]. The total protein concentration
was determined by the BCA protein assay kit (Pierce Corporation,
Rockford, IL, USA). The protein samples were separated by 10%
sodium dodecylsulfate-polyacrylamide gel electrophoresis and
transferred to nitrocellulose membranes (Millipore Corporation,
Billerica, MA, USA). The membranes were blocked in a 5% bovine
serum albumin solution and incubated with the primary antibodies,
followed by secondary fluorescent antibodies. The primary
antibodies included rabbit monoclonal anti-mda-7/IL-24 (Abcam,
Cambridge, UK), rabbit polyclonal anti-cleaved caspase-3 (Cell
Signaling Technology, Danvers, MA, USA), poly(ADP-ribose)
polymerase-1/2 (H-250) rabbit polyclonal antibody (Santa Cruz
Biotechnology, Santa Cruz, CA, USA) and β-actin, rabbit monoclonal
(Epitomics, Burlingame, CA, USA). The membranes were then incubated
with anti-rabbit infrared dye 700. The fluorescent signal was
detected with an Odyssey Infrared Imaging System (LI-COR
Biosciences, Lincoln, NE, USA).
Statistical analysis
The data reported represent the means of three
independent experiments, and the bars indicate the standard
deviation. The Student’s t-test was used to calculate the
statistical significance of the experimental results. The level of
significance was set at P<0.05.
Results
The construction and characterization of
the Ad5/11 chimeric oncolytic adenoviruses
Previously, our group developed a double-controlled
oncolytic adenovirus system, AdCN205, in which the hTERT promoter
was used to control the expression of the CR2-deleted E1A region,
and the 6.7K/gp19K of E3 region were substituted by the exogenous
genes. This vector allows selective adenoviral replication in tumor
cells harboring overexpression of hTERT and dysfunction of RB. The
exogenous genes in the vector controlled by the adenovirus
endogenous E3 promoter are expressed in tumor cells following virus
replication. In the present study, we engineered the Ad5/11
chimeric oncolytic adenovirus AdCN205-11-EGFP or AdCN205-11-IL-24
by replacement of the Ad5 fiber from the AdCN205 system with the
Ad11 fiber. The structures of the Ad5/11 chimeric oncolytic
adenovirus AdCN205-11-GFP and AdCN205-11-IL-24 are shown in
Fig. 1A.
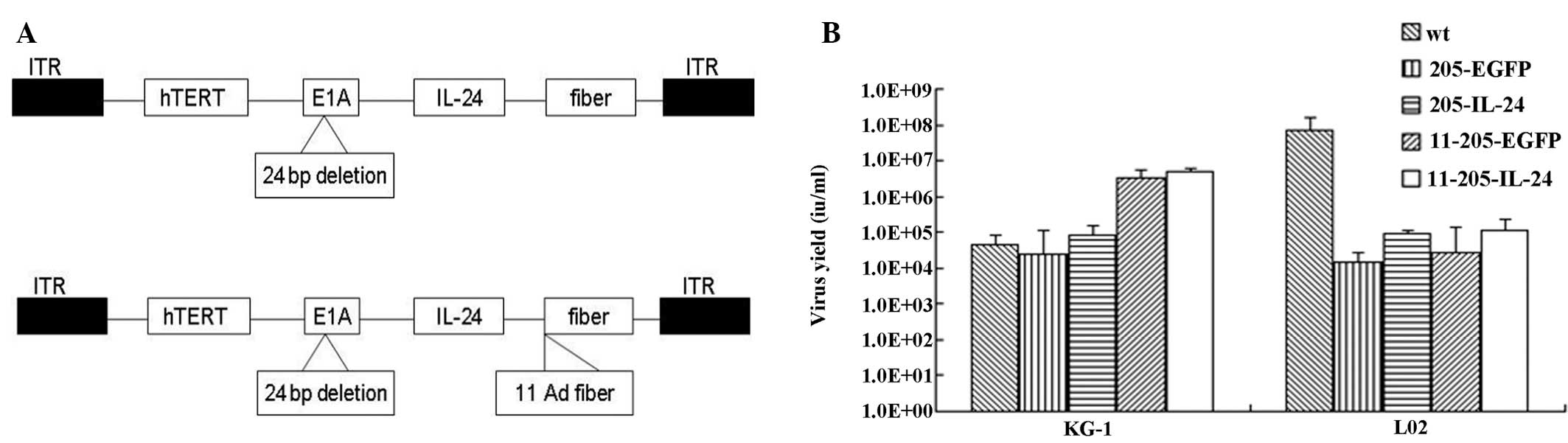 | Figure 1Construction and characterization of
the oncolytic adenoviral vectors. (A) Structures of the oncolytic
adenoviral vectors carrying EGFP and IL-24. Schematic description
of the structures of AdCN205, AdCN205-11-GFP and AdCN205-11-IL24.
In AdCN205, the E1A promoter was replaced by the hTERT promoter and
deletion of the adenoviral genome 923 to 946 nucleotides, which
enables viral replication within malignant cells with abnormal RB
function. The fibers on the human adenovirus type 5 (Ad5) were
replaced by fibers on the human adenovirus type 11 (Ad11). In
AdCN205-11-GFP and AdCN205-11-IL-24, E3 6.7K/gp19K genes were
substituted by the GFP reporter gene and the hIL-24 therapeutic
gene, respectively. (B) Selective replication of the recombinant
adenoviruses in vitro. The AML KG-1 and normal L02 cell
lines were infected with Ad-wt, AdCN205-EGFP, AdCN205-IL-24,
AdCN205-11-EGFP or AdCN205-11-IL-24 at an MOI of 10. After 48 h,
the supernatants and cells were collected separately. The cells
were resuspended in PBS and subjected to three freeze-thaw cycles.
The virus yield was measured in the supernatants and cell lysates.
*P<0.01 as compared with the AdCN205-EGFP or Ad-wt
group. IL-24, interleukin-24; AML, acute myeloid leukemia; MOI,
multiplicity of infection. |
Selective replication of the oncolytic
adenoviral vectors in vitro
To examine whether the transgenes could interfere
with the selective replication ability of the recombinant
adenoviruses, a progeny assay was conducted in the AML cells (KG-1)
and normal cells (L02) infected with Ad-wt, AdCN205-EGFP,
AdCN205-IL-24, AdCN205-11-EGFP or AdCN205-11-IL-24 at an MOI of 10.
As shown in Fig. 1B,
AdCN205-11-IL-24 and AdCN205-11-EGFP replicated at similar levels
in the AML cells; these levels were higher than the levels in AML
cells infected with Ad-wt, AdCN205-EGFP or AdCN205-IL-24. In
contrast, the replication capacity of these vectors was markedly
reduced in the normal cells. These data indicate that expression of
IL-24 and EGFP did not affect the selective replication ability of
the oncolytic vectors.
Furthermore, to confirm whether the exogenous gene
could be expressed properly in the AML cells infected with the
oncolytic adenoviruses, the KG-1 cell line was infected with
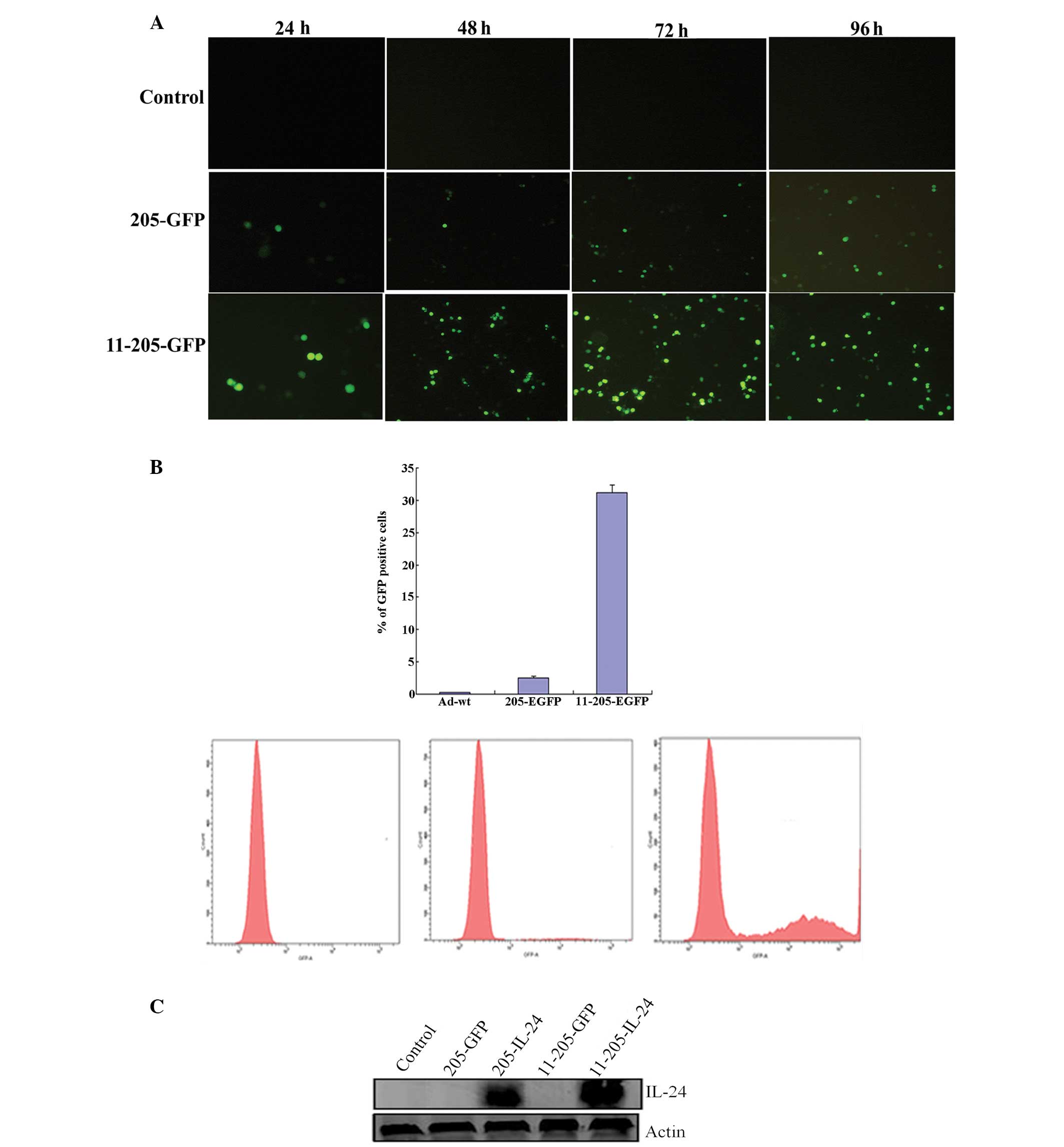
AdCN205-EGFP, AdCN205-11-EGFP or Ad-wt at an MOI of 10. Our results
showed that the KG-1 cells infected with AdCN205-11-EGFP expressed
a high level of EGFP protein as compared with the KG-1 cells
infected with AdCN205-EGFP and Ad-wt as observed under fluorescence
microscopy (Fig. 2A). To evaluted
the transduction efficiency of the recombinant oncolytic
adenoviral, KG-1 cells were infected with Ad-wt, AdCN205-EGFP or
AdCN205-11-EGFP at an MOI of 10 for 48 h and then subjected to flow
cytometric analysis. AdCN205-11-EGFP transduced KG-1 cells with
high efficiency (Fig. 2B).
Similarly, a high level of IL-24 was observed in the KG-1 cells
after infection with AdCN205-11-IL-24 (Fig. 2C). These data indicate that the
Ad5/11 chimeric oncolytic adenovirus could selectively replicate in
the AML cells and express a high level of the transgenes.
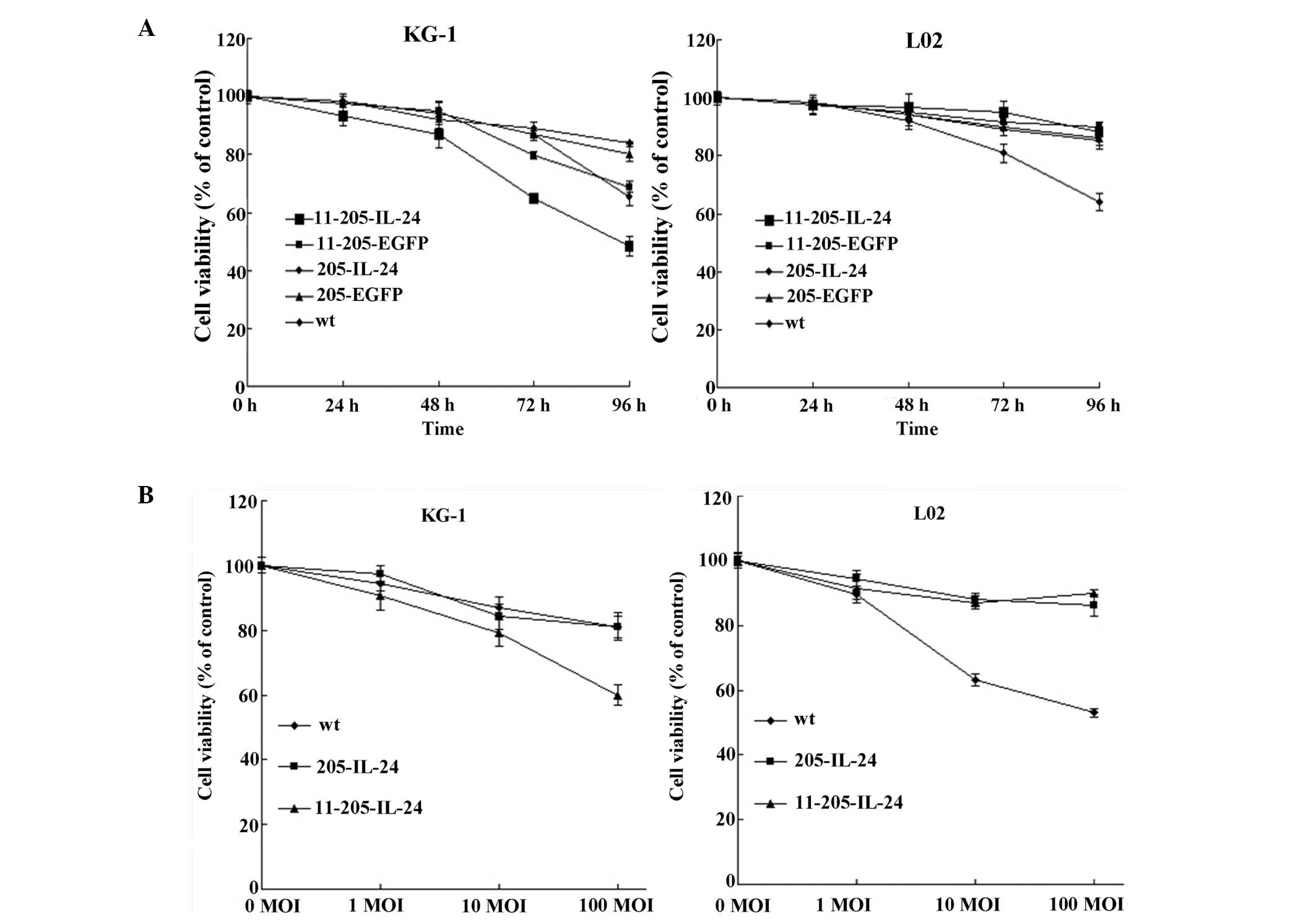
A cytotoxic effect is induced by the
oncolytic adenoviral vectors in AML cells in vitro
To analyze the antitumor efficiency of the
recombinant adenoviruses, the AML cell line (KG-1) and a normal
cell line (L02) were infected with Ad-wt, AdCN205-EGFP,
AdCN205-IL-24, AdCN205-11-EGFP or AdCN205-11-IL-24 at an MOI of 10,
and cell survival was determined by the MTT assay. As shown in
Fig. 3A, the AdCN205-EGFP,
AdCN205-IL-24 and Ad-wt induced a cytotoxic effect on AML cells at
a similar level in a time-dependent manner. Moreover,
AdCN205-11-IL-24 significantly reduced the viability of the AML
cells, as compared with that induced by Ad-wt, AdCN205-EGFP,
AdCN205-IL-24 or AdCN205-11-EGFP. In contrast, oncolytic
adenoviruses AdCN205-11-EGFP and AdCN205-11-IL-24 did not induce a
cytotoxic effect on the normal cell line L02. Furthermore, we
observed that AdCN205-11-IL-24 increased the cytotoxicity to AML
cells in a dose-dependent manner, as compared with that induced by
AdCN205-IL-24 and Ad-wt (Fig. 3B).
These data indicate that AdCN205-11-IL-24 can exert an obvious
cytotoxic effect on AML cells in vitro.
Induction of apoptosis in AML cells after
infection with the oncolytic adenoviral vectors in vitro
To examine whether IL-24 induces apoptosis in AML
cells, AML cells were infected with Ad-wt, AdCN205-EGFP,
AdCN205-IL-24, AdCN205-11-EGFP or AdCN205-11-IL-24 at an MOI of 10
for 48 h. Fig. 4A shows that
infection of KG-1 cells with AdCN205-11-IL-24 led to marked
morphological changes characteristic of apoptosis, such as
chromatin condensation, nuclear fragmentation and apoptotic body
formation in the KG-1 cells. Fluorescence-activated cell sorting
analysis was performed to examine apoptosis induced by
AdCN205-11-IL-24. Our results showed that the apoptotic rate
induced by AdCN205-11-IL-24 was significantly higher than that
induced by AdCN205-EGFP, AdCN205-IL-24 or Ad-wt (Fig. 4B). Furthermore, activation of the
caspase pathway was examined to explore the mechanism of AML cell
apoptosis. As shown in Fig. 4C, the
cleavage of caspase 3 and PARP was obviously increased in the KG-1
cells treated with AdCN205-11-IL-24, as compared with Ad-wt,
AdCN205-EGFP, AdCN205-IL-24, AdCN-EGFP or phosphate-buffered saline
(PBS). These results suggest that AdCN205-11-IL-24 markedly induced
KG-1 cell apoptosis by activation of extrinsic pathways.
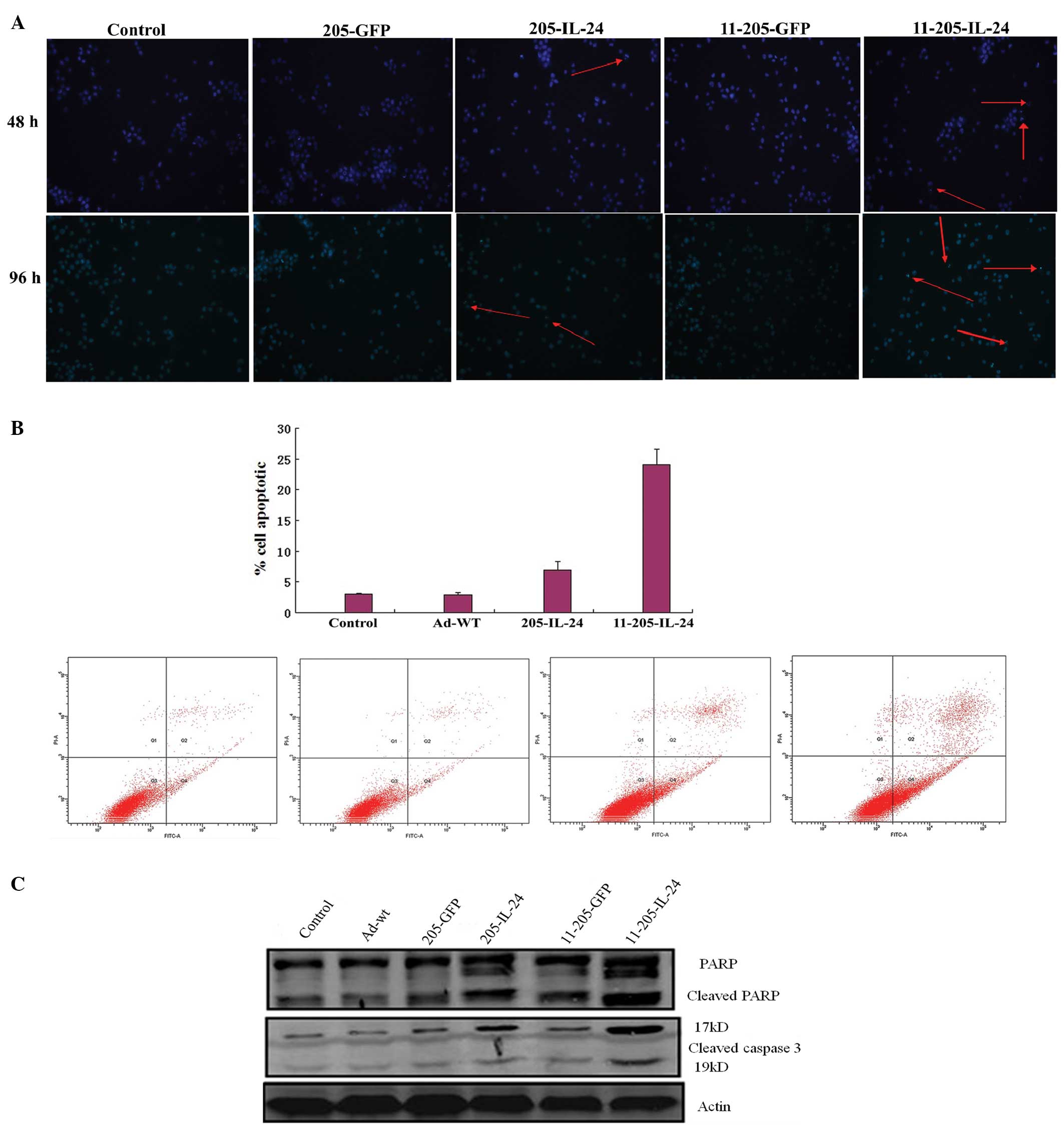 | Figure 4Induction of cell apoptosis in AML
cells after infection with AdCN205-11-IL-24 in vitro. (A)
AML cells were stained with Hoechst 33342 after infection with
Ad-wt, AdCN205-EGFP, AdCN205-IL-24, AdCN205-11-EGFP or
AdCN205-11-IL-24 at an MOI of 10 for 48 h. Arrows indicate positive
apoptotic cells. Original magnification, ×100. (B) Detection of
apoptosis by staining with anti-Annexin V in KG-1 cells after
infection with Ad-wt, AdCN205-IL-24 or AdCN205-11-IL-24 at an MOI
of 10, after 48 h by flow cytometric analysis. (C) Detection of
cleaved caspase 3 and PARP in KG-1 cells after infection with
Ad-wt, AdCN205-EGFP, AdCN205-IL-24, AdCN205-11-EGFP or
AdCN205-11-IL-24 at an MOI of 10. The total protein from the
treated cells after 48 h of infection was subjected to western blot
analysis with antibodies against total IL-24, cleaved caspase 3,
PARP and β-actin. AML, acute myeloid leukemia; MOI, multiplicity of
infection; IL-24, interleukin-24. |
Discussion
Human leukemias are characterized by acquired
recurring chromosomal translocations. Cloning of these
translocation breakpoints has provided important insight into the
pathogenesis of the disease as well as novel therapeutic
approaches. Acute myeloid leukemia (AML) is characterized by
uncontrolled proliferation, increased survival and impaired
differentiation of hematopoietic progenitor cells. Analysis of
primary human AML samples and data from animal models of leukemia
have demonstrated that these characteristics can be attributed to
deregulation of signal transduction pathways and disruption of
cellular differentiation programs that cooperate in AML
pathogenesis. These activated kinases are validated targets for
therapy with selective tyrosine kinase inhibitors, a paradigm that
may have broad applications in the treatment of hematologic
malignancies as well as solid tumors. This represents a critical
issue in the effort to design molecular therapies.
Activation of signal transduction in AML may occur
through a variety of genetic alterations affecting different
signaling molecules, such as the FLT3 and KIT receptor tyro-sine
kinases (RTKs) and members of the RAS family of guanine
nucleotide-binding proteins. These mutant signaling proteins are
attractive therapeutic targets; however, the development of
targeted therapies for each genotypic variant and determining the
relationships between different genotypes and critical functional
dependencies of leukemic cells remain major challenges. As the
large number of mutant signaling proteins that have been identified
in AML are likely to reflect activation of a more limited number of
downstream effector pathways, such as the RAF/MEK/ERK and PI3K/AKT
cascades, targeting these unifying pathways may represent a more
broadly applicable therapeutic strategy. Furthermore, integrative
genomic studies combining DNA sequencing, DNA copy number analysis,
transcriptional profiling and functional genetic approaches hold
great promise for identifying additional signaling abnormalities in
AML that are relevant to leukemogenesis and can be exploited
therapeutically. Eventually, it may become possible to use
pathogenesis-oriented combinations of signal transduction
inhibitors to improve the cure rate in AML patients. Biological
therapies, including gene therapy, have shown great promise for the
treatment of AML. However, present strategies are not effective
enough to eliminate cancer completely, particularly in human
patients. Therefore, investigation of potent vectors and
therapeutic genes for this purpose are needed.
Research has uncovered a specific gene, melanoma
differentiation-associated gene-7/interleukin-24 (MDA-7/IL-24),
displaying cancer-specific apoptosis-inducing properties. Isolation
using this scheme has now come into the limelight as a new gene
therapy for divergent types of cancers. Although the mechanism of
cancer cell selectivity of MDA-7/IL-24 remains to be verified,
numerous attributes enable this gene to be an effective therapy for
cancer, including the ability to discriminate between normal and
cancer cells, to induce apoptosis in diverse tumor cells, to
promote ‘bystander’ antitumor effects, to inhibit tumor growth and
angiogenesis in animal models, to synergize with radiation and to
modulate immune responses. These unique features combined with
successful transition into the clinic provides hope that IL-24, as
a single or more likely as part of a combinatorial approach, may
provide profound therapeutic benefit for cancer patients. Our
previous study confirmed that IL-24 exerted strong antitumor
activity in vitro and in vivo. A clinical trial with
a replication-defective adenovirus expressing IL-24 has shown a
good safety profile and some partial responses in patients with
cancers. In the present study, we aimed to ascertan whether the
‘magical bullet’ IL-24 could be used as a therapeutic gene for the
treatment of AML.
To achieve the maximal transduction efficiency in
cancer cells, application of the replication-competent viral
vectors is required. It has been shown that oncolytic adenoviruses
transduce tumor cells effectively and exhibit an adequate safety
profile. In our previous studies, we constructed the
double-regulated oncolytic adenoviral vectors (AdCN205-EGFP and
AdCN205-IL-24) that could target human telomerase reverse
transcriptase and retinoblastoma pathways (18,32,33).
At the same time, we constructed oncolytic adenoviral vectors in
which fibers on the human adenovirus type 5 (Ad5) were replaced
with the human adenovirus type 11 (Ad11) that can only target
specific selective replication of recombinant adenoviruses in
leukemia cells. This vector only selectively replicates in AML
cells (34,35). Therefore, we used this vector to
deliver the IL-24 gene (AdCN205-11-EGFP and AdCN205-11-IL-24) for
the therapy of AML.
Our data revealed that AdCN205-11-IL-24 exhibited a
strong cytotoxic effect on AML cells and this cytotoxic effect was
much higher than that induced by Ad-wt, AdCN205-EGFP or
AdCN205-IL-24. Most importantly, AdCN205-11-IL-24 could not
replicate in normal cells and it did not induce any cytotoxicity in
normal cells. We also demonstrated that transfer of IL-24 by
AdCN205-11-IL-24 resulted in marked expression in AML cells, and
AdCN205-IL-24 was barely detected. These results indicate that the
strong cytotoxic effect of AdCN205-11-IL-24 in AML cells was caused
by alteration of the cell cycle and induction of cell apoptosis.
Our data further demonstrated that AdCN205-11-IL-24 induced cell
apoptosis by induction of the cleavage of caspase 3 and PARP, the
main and important mechanisms for induction of cell apoptosis. The
present study revealed that AdCN205-11-IL-24 induced AML cell
apoptosis through the activation of the extrinsic pathway.
Taken together, AdCN205-11-IL-24, a chimeric
oncolytic adenovirus harboring IL-24, was successfully generated.
This vector selectively replicates in AML cells and exhibits marked
antitumor activity in vitro. Restoration of IL-24 by
chimeric oncolytic adenovirus could induce AML cell apoptosis
through the activation of the extrinsic pathway in vitro.
This study provides promise for the development of a novel gene
therapeutic strategy for AML.
Acknowledgements
We thank Dr Cheng Qian for his kind assistance.
References
|
1
|
Estey E and Dohner H: Acute myeloid
leukaemia. Lancet. 368:1894–1907. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Frohling S, Scholl C, Gilliland DG and
Levine RL: Genetics of myeloid malignancies: pathogenetic and
clinical implications. J Clin Oncol. 23:6285–6295. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Appelbaum FR, Gundacker H, Head DR, et al:
Age and acute myeloid leukemia. Blood. 107:3481–3485. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Downing JR: The core-binding factor
leukemias: lessons learned from murine models. Curr Opin Genet Dev.
13:48–54. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Scholl C, Gilliland DG and Frohling S:
Deregulation of signaling pathways in acute myeloid leukemia. Semin
Oncol. 35:336–345. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kornblau SM, Womble M, Qiu YH, et al:
Simultaneous activation of multiple signal transduction pathways
confers poor prognosis in acute myelogenous leukemia. Blood.
108:2358–2365. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Steelman LS, Pohnert SC, Shelton JG,
Franklin RA, Bertrand FE and McCubrey JA: JAK/STAT, Raf/MEK/ERK,
PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis.
Leukemia. 18:189–218. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sangro B and Prieto J: Gene therapy for
liver cancer: clinical experience and future prospects. Curr Opin
Mol Ther. 12:561–569. 2010.PubMed/NCBI
|
|
9
|
Denefle PP: Introduction to gene therapy:
a clinical aftermath. Methods Mol Biol. 737:27–44. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wadhwa PD, Zielske SP, Roth JC, Ballas CB,
Bowman JE and Gerson SL: Cancer gene therapy: scientific basis.
Annu Rev Med. 53:437–452. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pfeifer A and Verma IM: Gene therapy:
promises and problems. Annu Rev Genomics Hum Genet. 2:177–211.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Qian C, Sangro B and Prieto J: New
strategies to enhance gene therapy efficiency. Gastroenterology.
123:639–642. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wong HH, Lemoine NR and Wang Y: Oncolytic
viruses for cancer therapy: overcoming the obstacles. Viruses.
2:78–106. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Qian C, Liu XY and Prieto J: Therapy of
cancer by cytokines mediated by gene therapy approach. Cell Res.
16:182–188. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kirn D, Martuza RL and Zwiebel J:
Replication-selective virotherapy for cancer: biological
principles, risk management and future directions. Nat Med.
7:781–787. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Singer O and Verma IM: Applications of
lentiviral vectors for shRNA delivery and transgenesis. Curr Gene
Ther. 8:483–488. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Berk AJ: Recent lessons in gene
expression, cell cycle control, and cell biology from adenovirus.
Oncogene. 24:7673–7685. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang W, Cai R, Luo J, et al: The
oncolytic adenovirus targeting to TERT and RB pathway induced
specific and potent anti-tumor efficacy in vitro and in vivo for
hepatocellular carcinoma. Cancer Biol Ther. 6:1726–1732. 2007.
View Article : Google Scholar
|
|
19
|
Sherr CJ and McCormick F: The RB and p53
pathways in cancer. Cancer Cell. 2:103–112. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cui Q, Jiang W, Wang Y, et al: Transfer of
suppressor of cytokine signaling 3 by an oncolytic adenovirus
induces potential antitumor activities in hepatocellular carcinoma.
Hepatology. 47:105–112. 2008. View Article : Google Scholar
|
|
21
|
Papadakis ED, Nicklin SA, Baker AH and
White SJ: Promoters and control elements: designing expression
cassettes for gene therapy. Curr Gene Ther. 4:89–113. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Luo J, Xia Q, Zhang R, et al: Treatment of
cancer with a novel dual-targeted conditionally replicative
adenovirus armed with mda-7/IL-24 gene. Clin Cancer Res.
14:2450–2457. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Segerman A, Mei YF and Wadell G:
Adenovirus types 11p and 35p show high binding efficiencies for
committed hematopoietic cell lines and are infective to these cell
lines. J Virol. 74:1457–1467. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lyons M, Onion D, Green NK, et al:
Adenovirus type 5 interactions with human blood cells may
compromise systemic delivery. Mol Ther. 14:118–128. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yu L, Shimozato O, Li Q, et al: Adenovirus
type 5 substituted with type 11 or 35 fiber structure increases its
infectivity to human cells enabling dual gene transfer in
CD46-dependent and -independent manners. Anticancer Res.
27:2311–2316. 2007.PubMed/NCBI
|
|
26
|
Pestka S, Krause CD, Sarkar D, Walter MR,
Shi Y and Fisher PB: Interleukin-10 and related cytokines and
receptors. Annu Rev Immunol. 22:929–979. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lebedeva IV, Emdad L, Su ZZ, et al:
mda-7/IL-24, novel anticancer cytokine: Focus on bystander
antitumor, radiosensitization and antiangiogenic properties and
overview of the phase I clinical experience (Review). Int J Oncol.
31:985–1007. 2007.PubMed/NCBI
|
|
28
|
Fisher PB: Is mda-7/IL-24 a ‘magic bullet’
for cancer? Cancer Res. 65:10128–10138. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sauane M, Gopalkrishnan RV, Sarkar D, et
al: MDA-7/IL-24: novel cancer growth suppressing and apoptosis
inducing cytokine. Cytokine Growth Factor Rev. 14:35–51. 2003.
View Article : Google Scholar
|
|
30
|
Qian W, Liu J, Tong Y, et al: Enhanced
antitumor activity by a selective conditionally replicating
adenovirus combining with MDA-7/interleukin-24 for B-lymphoblastic
leukemia via induction of apoptosis. Leukemia. 22:361–369. 2008.
View Article : Google Scholar
|
|
31
|
Zhao L, Dong A, Gu J, et al: The antitumor
activity of TRAIL and IL-24 with replicating oncolytic adenovirus
in colorectal cancer. Cancer Gene Ther. 13:1011–1022. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Aghi M and Martuza RL: Oncolytic viral
therapies - the clinical experience. Oncogene. 24:7802–7816. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mathis JM, Stoff-Khalili MA and Curiel DT:
Oncolytic adeno-viruses - selective retargeting to tumor cells.
Oncogene. 24:7775–7791. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wong HH, Jiang G, Gangeswaran R, et al:
Modification of the early gene enhancer-promoter improves the
oncolytic potency of adenovirus 11. Mol Ther. 20:306–316. 2012.
View Article : Google Scholar :
|
|
35
|
Segerman A, Atkinson JP, Marttila M,
Dennerquist V, Wadell G and Arnberg N: Adenovirus type 11 uses CD46
as a cellular receptor. J Virol. 77:9183–9191. 2003. View Article : Google Scholar : PubMed/NCBI
|