Introduction
Hereditary multiple exostosis (HME) is an autosomal
dominant disorder with an incidence of 1 in 50,000 (1). Thus, it is one of the most common
inherited musculoskeletal disorders. Approximately 20% of reported
cases have no family history of multiple exostosis (2). There are 3 features of HME. First,
patients show multiple benign exostoses, typically located at the
juxta-epiphyseal region of bones. Second, it has an obvious
hereditary property. Third, and the most severe complication, is
the characteristic of malignant transformation (3).
Previous studies have reported that the EXT family
is responsible for HME. The EXT family was reported to encode
proteins linked to the biosynthesis of heparan sulfate (HS)
(4). HS is an essential molecule
and dysfunction could cause HME. The EXT family consists of
exostosin-1 (EXT1) (5), exostosin-2
(EXT2) (6), exostosin-3 (EXT3)
(7), exostosin-like 1 (EXTL1)
(8), exostosin-like 2 (EXTL2)
(9) and exostosin-like 3 (EXTL3)
(10). Among these, EXT1 has been
suggested to be responsible to a large extent for HME (9,11).
With the rapid development of DNA sequencing
technology, the power of exome sequencing for gene mapping of
diseases has been demonstrated (12–14).
Exome sequencing can help to identify novel causal genetic
variants, as it covers all exon regions.
In the present study, we performed exome sequencing
on 3 affected and 3 unaffected individuals from a HME family. After
filtering out the mutations from the dbSNP database (build 132),
the candidate mutations were validated on all of the patients from
the HME family by Sanger sequencing. Further validation was also
performed on three members from another unrelated HME family.
Additionally, immunohistochemisty was conducted to provide a deeper
insight into the importance of the identified causal gene mutation.
Furthermore, the mutation spectrum of the EXT1 gene was
analyzed.
Materials and methods
Subjects
For exome sequencing, we selected 3 affected cases
and 3 unaffected individuals (Table
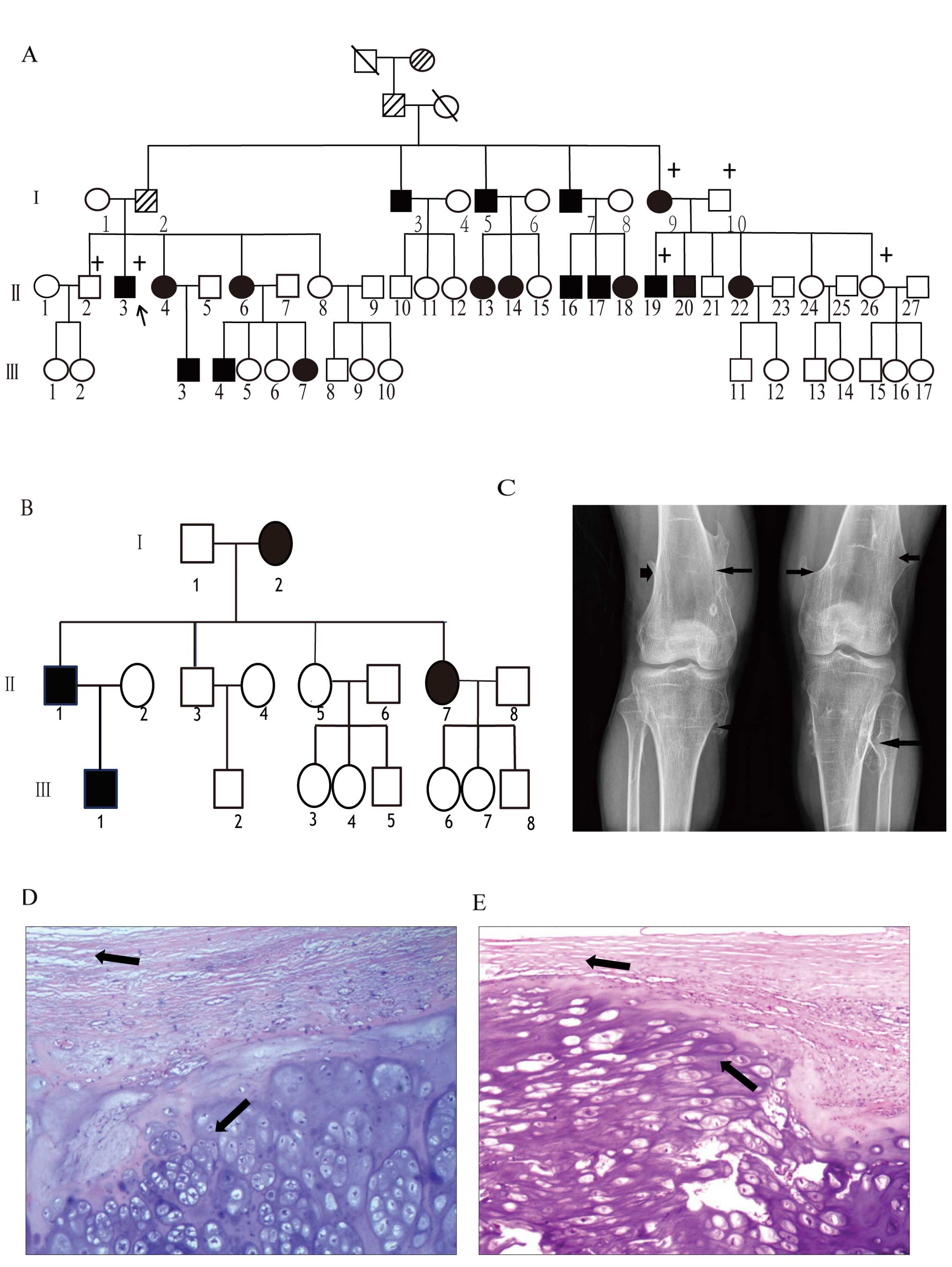
I) from a five-generation HME family (Fig. 1A) from the Henan Province of China.
Sixteen affected cases, 13 unaffected individuals in the family and
3 individuals (2 affected cases and 1 unaffected) from another
unrelated family (Fig. 1B) were
selected for further validation. All of the participants were
informed and consented with the specimen collection.
 | Table IClinical characteristics of the
subjects for exome sequencing. |
Table I
Clinical characteristics of the
subjects for exome sequencing.
| Sample no. | Position | Clinical
diagnosis | Blood | Age (years) | Gender | Analyzed by WES |
|---|
| 13JX00006WB3 | I10 | / | Yes | 65 | Male | Yes |
| 13JX00007WB2 | I9 | HME | Yes | 62 | Female | Yes |
| 13JX000011LC2 | II3 | HME | Yes | 27 | Male | Yes |
| 13JX00002LC2 | II2 | / | Yes | 29 | Male | Yes |
| 13JX000017WB2 | II19 | HME | Yes | 19 | Male | Yes |
| 13JX000019WB2 | II26 | / | Yes | 39 | Female | Yes |
After filling in the informed consent form, all of
the individuals were carefully examined by at least 2 experienced
doctors. All of the cases were diagnosed as HME, while all of the
unaffected individuals were healthy.
Venous blood samples were collected from all members
who participated. Genomic DNA was extracted from peripheral blood
by standard procedures for exome sequencing. In this study, all
steps were conducted according to the Declaration of Helsinki
Principles. The use of human subjects in this report was approved
by the Shenzhen Second People’s Hospital Institutional Review
Committee.
Exome capture and sequencing
Exome capture was performed with the Human All Exon
V3 (Agilent Technologies, Santa Clara, CA, USA) and the sequencing
was conducted with the HiSeq 2000 platform (Illumina Inc., San
Diego, CA, USA). Genomic DNA samples were randomly fragmented for
library conducted and base-pair peaks of 150–200 bp were selected
and then adapters were ligated to both ends of the fragments. The
adapter-ligated templates were purified by Agencourt AMPure SPRI
beads, and fragments with insert size ~200 bp were excised.
Extracted DNA was amplified by ligation-mediated polymerase chain
reaction (LM-PCR), purified and hybridized to the SureSelect
Biotinylated RNA Library (BAITS) for enrichment. Hybridized
fragments were bound to the strepavidin beads whereas
non-hybridized fragments were washed out after 24 h. The captured
LM-PCR product was subjected to the Agilent 2100 Bioanalyzer to
estimate the magnitude of enrichment. All of the steps were
performed according to the manufacturer’s recommendations. Each
captured library was then loaded onto the Hiseq 2000 platform and
sequencing was performed with read lengths of 90 bp, which provided
at least a 50× average depth for each sample.
Read mapping and variant detection
Reads with adapters, the N’s percent >10% and
reads whose low quality bases (quality value ≤5) were >50% were
filtered. The clean reads of each individual were aligned to the
human reference genome (NCBI Build 36.3, hg19) using
Burrows-Wheeler transform (BWA; Cambridge, UK); the parameter were
set as ‘-o 1 -e 50 -m 100,000 -t 4 -i 15 -q 10’. Reads that had
duplicated aligned sites were removed; the remaining reads mapped
on or near the target were collected for subsequent analyses and
variant calling.
For the SNP calling, we used Short Oligonucleotide
Analysis Package (SOAPsnp, China) (15). A consensus genotype with Phred-like
quality of at least 20 and at least 4× coverage depth was
considered to be a high-confidence genotype. The genotypes that
were different from the reference were extracted as candidate SNP
and the SNP list was filtered with the followed criterion:
Phred-like SNP quality ≥20, overall depth of 4 to 1,000×.
Genome Analysis Toolkit (GATK; Broad Institute,
Cambridge, MA, USA) (16) was used
to call InDel and the results were filtered as follows: After gap
aligning the sequence reads with default parameters to the human
reference (hg19), local realignment of the BWA-aligned reads using
the GATK IndelRealigner were performed and the final InDels were
called by GATK IndelGentotyperV2 according to the recommendations
of the software.
Functional annotation of genetic
variants
With ANNOVAR (USA) (17) the variants were annotated and
categorized into missense, nonsense, synonymous, splice-site and
insertion/deletion mutations. After filtering out synonymous and
nonframeshift, we obtained mutations which were likely to be
deleterious and then filtered against Chinese Han SNP data
available in the dbSNP database (build 132). We chose those
variants that were shared by all affected but not any unaffected
individual as candidate casual variants.
Sanger sequencing
Sanger sequencing was performed to confirm the
identified variants found by exome sequencing including 18 affected
and 14 unaffected individuals. Sanger sequencing was performed
according the standard protocol. The primer sequences were as
follows: 5′-GATGGACCCCATTAGAGTAG-3′ and 5′-AGAGTAGTGACTCTACCCTC-3′.
Sequence comparisons and analyses were performed using Chromas2
(Technelysium Pty Ltd., Tewantin, QLD, Australia).
Histochemical staining and
immunohistochemistry
The paraformaldehyde-fixed chondroma tissues from
the HME patient were fixed with 1% paraformaldehyde (PFA;
Sigma-Aldrich), rinsed and decalcified and embedded in paraffin.
Paraffin-embedded chondroma tissues were sectioned (5-μm thick) and
placed on glass slides. For histochemistry, the tissue slides of
HME were dewaxed in xylene, hydrated with graded ethanol and
stained by toluidine blue (TB) and hematoxylin and eosin (H&E),
respectively.
For immunohistochemistry, the dewaxed and hydrated
chondroma sections of HME were treated with 3% hydrogen peroxide
solution for 10 min, rinsed with PBS followed by 5% donkey serum
blocking and incubated with rabbit polyclonal anti-EXT1 antibody
(1:50 working dilution; Abcam Plc, UK) at 4°C overnight. The
chondroma sections were then incubated with the secondary antibody
(Golden Bridge Biotechnology, Zhongshan, China) at 37°C for 30 min
and stained using the DAB substrate kit (Vector Laboratories,
Burlingame, CA, USA) and Meyer’s hematoxylin.
EXT1 mutation spectrum
We downloaded the mutation data according to the
variant type of substitution, deletion, insertion, inversion,
insertion or deletion and translocation from the Multiple
Osteochondroma Mutation Database. Subsequently, we summarized the
mutations which were located in the exon regions and drew the
mutation spectrum with Circos plot.
Results
We observed that the tissue structure (Fig. 1C) of HME was represented with a
cartilage cap, covered by fibrous perichondrium and merged into an
underlying spongy bone. The stained chondroma sections with H&E
and TB are presented in Fig.
1D.
We used Human All Exon V3 Agilent (50 M) to capture
the exome of 3 affected (I9, II3 and II19) and 3 unaffected (I10,
II2 and II26) individuals in the HME family (Fig. 1A). After performing parallel
sequencing, ~120 million bases and 80 million reads per individual
in average were generated. The mapping rates against the human
being reference (hg19) of all individuals achieved 99%.
Approximately 92% of the target regions were sequenced at least 10
times (depth ≥10x) in each individual. For every individual,
~80,000 variants were identified within the captured regions.
We focused on those variants that were located in
exon, UTR and splicing sites. Taking the familial dominant model of
inheritance into account, the variants that appeared in all 3
affected individuals but not in any unaffected individual were
selected. We then filtered against the available public databases
(dbSNP). Furthermore, we only focused on nonsynonymous, stopgain,
stoplose and frameshift variants. After all the previous
filtrations, only 3 heterozygous variants, ATXN3, TRPM3 and
C11orf40, remained and only one heterozygous deletion (c.1325delC)
in EXT1 was probably damaging, as the EXT family has been reported
to be associated with HME. No reports were found at this position
in the Multiple Osteochondroma Mutation Database. Thus, we selected
it for further study.
To eliminate false-positive mutations, we examined
and validated the mutations by Sanger sequencing in the source
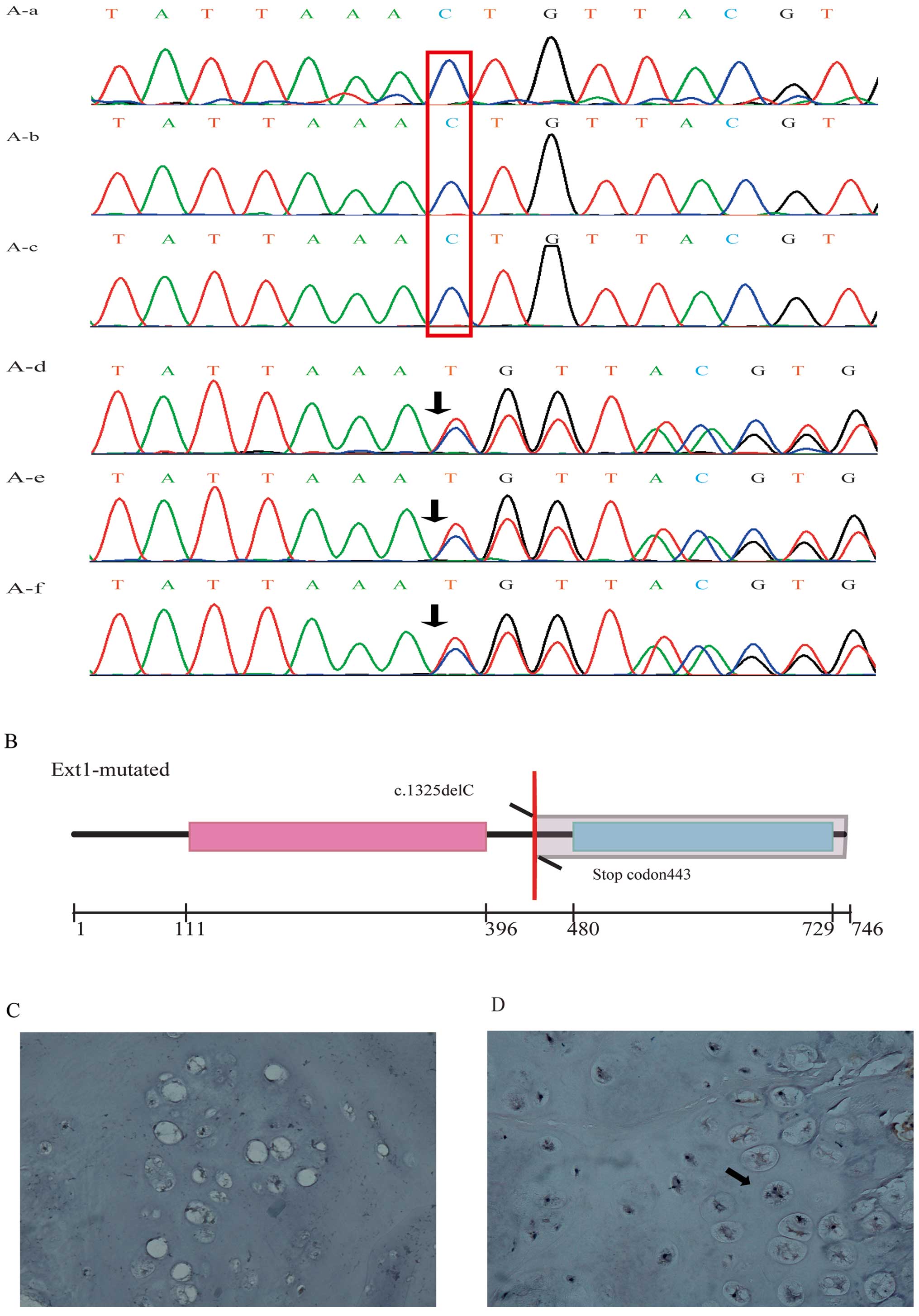
individuals. The heterozygous deletion g.chr8: 118834796 delC
(Fig. 2A) was confirmed to be
shared by all of the 3 studied patients but was absent in the
normal individuals. The mutation led to a frameshift at codon 442
resulting in a change of serine to isoleucine and generated a stop
codon at codon 443 causing the glycosyltransferase domain loss
(Fig. 2B).
To further verify that the deletion was the
disease-causing gene mutation in the HME family, we performed PCR
and Sanger sequencing on all the family members and another 3
individuals (I1, I2 and II7) from another unrelated family
(Fig. 1B). The mutation was
confirmed to be shared by all the affected individuals except
patient II22 and was absent in all of the healthy family
members.
Immunohistochemistry was used to detect the protein
expression level of EXT1 in these 2 unrelated families. We found
that the EXT1 protein level of a HME patient (Fig. 2C) with the novel mutation was less
than the level in an HME patient without the EXT1 mutation from
another unrelated family (Fig.
2D).
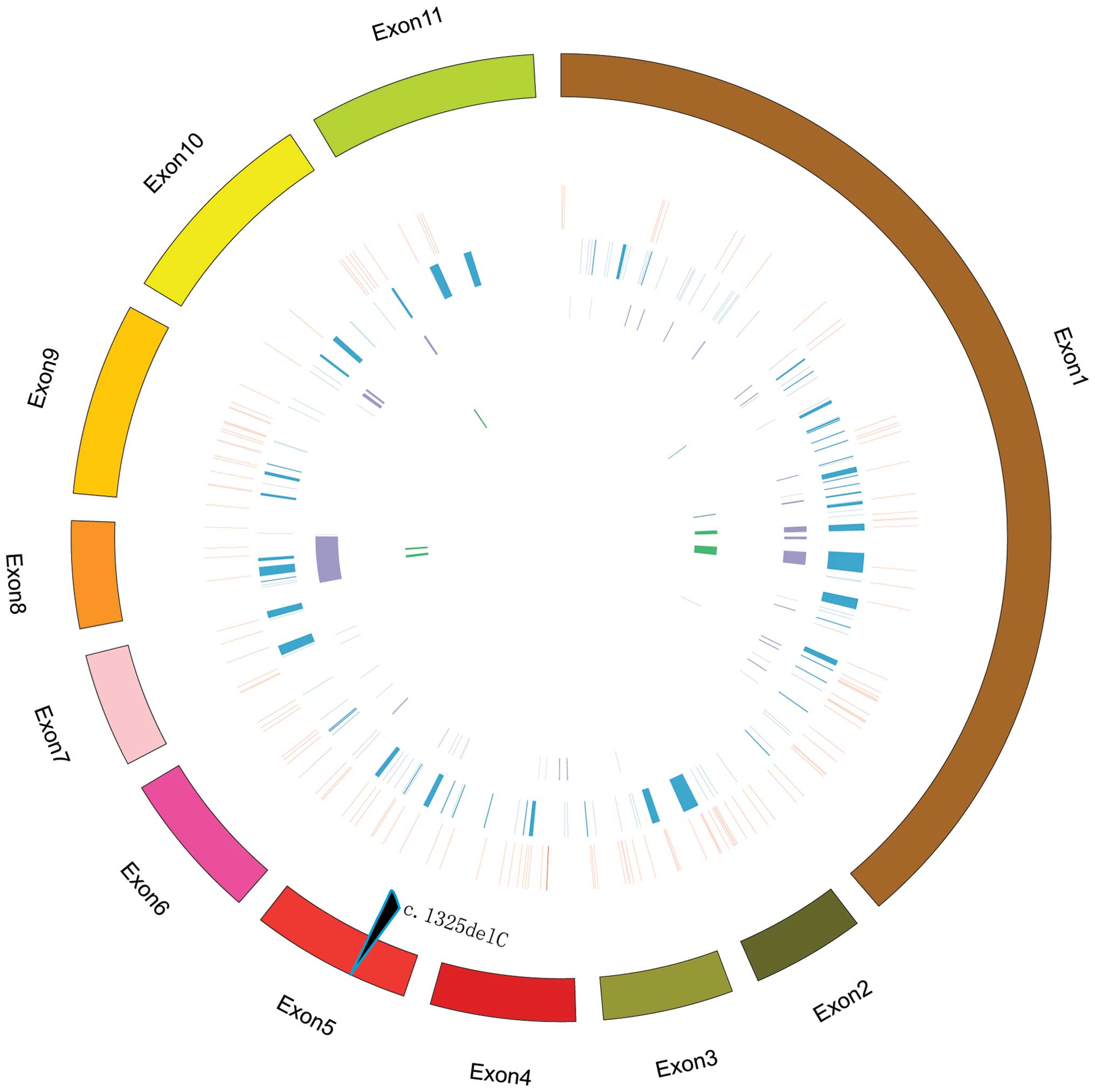
To further analyze the mutations in EXT1, we
gathered all of the mutations in its 11 exons from the Multiple
Osteochondroma Mutation Database. No inversion and translocation
were found on all of the 11 exons. Substitutions and deletions were
evenly distributed on the 11 exons, while small insertions were
rarely locate in exon 2, 9 and 11 (Fig.
3).
Discussion
The gene EXT1 locates on 8q24.1 and consists of 11
exons. EXT1 encodes a protein which possesses the activity of
glucuronic acid sugar-based transfer enzyme and N acetyl
glucosamine glycosyltransferase (4)
which catalyzes the polymerization of HS chains at the endoplasmic
reticulum and Golgi apparatus (18). HS is widely expressed on the cell
surface, and as the component of extracellular matrix glycoprotein
it mediates cell adhesion, signal transduction and the receptor
ligand binding process. Previous studies have indicated that HS is
an essential molecule and dysfunction could cause HME (19,20).
Herein, we found a novel c.1325delC mutation located
in exons 5. This frameshift mutation was validated to be shared by
all of the affected individuals except patient II22 and was absent
in the healthy family members. Additionally, the mutation was not
discovered in individuals from another HME family. As this damaging
deletion mutation was not present in patient II22, further study
will be carried out.
The c.1325delC mutation locates between the
exostosin and glycosyltransferase domains. It led to a frameshift
at codon 442 and resulted in a change of a serine to an isoleucine.
The promptly premature stop codon at codon 443 caused loss of the
glycosyltransferase domain, and the decrease in EXT1 protein
expression was confirmed by immunohistochemisty. Because of the
truncation, the EXT1 products may be unable to fold correctly and
would rapidly degrade (21). This
result is consistent with the reduced EXT1 protein observed in HME
patients in previous studies (22,23).
According to the statistics of all mutations on EXT1 in the
Multiple Osteochondroma Mutation Database (24), all variant types were nearly equally
distributed on the 11 exons.
Taken together, we report a novel frameshift
mutation c.1325delC in the gene EXT1 by whole-exome sequencing. The
mutation was validated by Sanger sequencing and its negative
influence on the expression of EXT1 was examined via
immunohistochemistry. Our results provide a new mutation in EXT1,
further emphasizing the dysfunction of the EXT gene family as a
cause of HME. This finding may be helpful for the early diagnosis
and prenatal genetic screening of HME.
Acknowledgements
We would like to thank all members for their
participation in the study. This study was mainly supported by
Grant support from: The Shenzhen Science and Technology Project
(project nos. CXZZ20130321152713220, GJHZ20130412153906739,
CXZZ20120614160234842, CXZZ20130321152218032,
JCYJ20130401114928183, JCYJ20130401114715714 and
JCYJ20130401112006994) and SOAPsnp (Short Oligonucleotide Analysis
Package); The Natural Science Foundation of China (nos. 81260161,
81000460 and 81301740), The Natural Science Foundation of Guangdong
Province, China (no. S2012010008129), The China Postdoctoral
Science Foundation Funded Project (no. 2013M530385) and The
Promotion Program for Shenzhen Key Laboratory, Shenzhen, China
(nos. CXB201104220049A and ZDSY20120614154551201).
References
|
1
|
Schmale GA, Conrad EU III and Raskind WH:
The natural history of hereditary multiple exostoses. J Bone Joint
Surg Am. 76:986–992. 1994.PubMed/NCBI
|
|
2
|
Chen WC, Chi CH, Chuang CC and Jou IM:
Three novel EXT1 and EXT2 gene mutations in Taiwanese patients with
multiple exostoses. J Formos Med Assoc. 105:434–437. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wicklund CL, Pauli RM, Johnston D and
Hecht JT: Natural history study of hereditary multiple exostoses.
Am J Med Genet. 55:43–46. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Busse M, Feta A, Presto J, et al:
Contribution of EXT1, EXT2 and EXTL3 to heparan sulfate chain
elongation. J Biol Chem. 282:32802–32810. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ahn J, Ludecke HJ, Lindow S, et al:
Cloning of the putative tumour suppressor gene for hereditary
multiple exostoses (EXT1). Nat Genet. 11:137–143. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stickens D, Clines G, Burbee D, et al: The
EXT2 multiple exostoses gene defines a family of putative tumour
suppressor genes. Nat Genet. 14:25–32. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Le Merrer M, Legeai-Mallet L, Jeannin PM,
et al: A gene for hereditary multiple exostoses maps to chromosome
19p. Hum Mol Genet. 3:717–722. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wise CA, Clines GA, Massa H, Trask BJ and
Lovett M: Identification and localization of the gene for EXTL, a
third member of the multiple exostoses gene family. Genome Res.
7:10–16. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wuyts W and Van Hul W: Molecular basis of
multiple exostoses: mutations in the EXT1 and EXT2 genes. Hum
Mutat. 15:220–227. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Van Hul W, Wuyts W, Hendrickx J, et al:
Identification of a third EXT-like gene (EXTL3) belonging to the
EXT gene family. Genomics. 47:230–237. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Signori E, Massi E, Matera MG, et al: A
combined analytical approach reveals novel EXT1/2 gene mutations in
a large cohort of Italian multiple osteochondromas patients. Genes
Chromosomes Cancer. 46:470–477. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu S, Sun X, Zhu W, et al: Evidence for
GAL3ST4 mutation as the potential cause of pectus excavatum. Cell
Res. 22:1712–1715. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang SQ, Jiang T, Li M, et al: Exome
sequencing identifies MVK mutations in disseminated superficial
actinic porokeratosis. Nat Genet. 44:1156–1160. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Daoud H, Dobrzeniecka S, Camu W, et al:
Mutation analysis of PFN1 in familial amyotrophic lateral sclerosis
patients. Neurobiol Aging. 34:e1311–e1312. 2013.
|
|
15
|
Li R, Yu C, Li Y, et al: SOAP2: an
improved ultrafast tool for short read alignment. Bioinformatics.
25:1966–1967. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
McKenna A, Hanna M, Banks E, et al: The
genome analysis toolkit: a MapReduce framework for analyzing
next-generation DNA sequencing data. Genome Res. 20:1297–1303.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang K, Li M and Hakonarson H: ANNOVAR:
functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lind T, Tufaro F, McCormick C, Lindahl U
and Lidholt K: The putative tumor suppressors EXT1 and EXT2 are
glycosyltransferases required for the biosynthesis of heparan
sulfate. J Biol Chem. 273:26265–26268. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Duncan G, McCormick C and Tufaro F: The
link between heparan sulfate and hereditary bone disease: finding a
function for the EXT family of putative tumor suppressor proteins.
J Clin Invest. 108:511–516. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Busse-Wicher M, Wicher KB and
Kusche-Gullberg M: The extostosin family: Proteins with many
functions. Matrix Biol. 35:25–33. 2014. View Article : Google Scholar
|
|
21
|
Wuyts W, Van Hul W, De Boulle K, et al:
Mutations in the EXT1 and EXT2 genes in hereditary multiple
exostoses. Am J Hum Genet. 62:346–354. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bernard MA, Hall CE, Hogue DA, et al:
Diminished levels of the putative tumor suppressor proteins EXT1
and EXT2 in exostosis chondrocytes. Cell Motil Cytoskeleton.
48:149–162. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang F, Liang J, Guo X, et al: Exome
sequencing and functional analysis identifies a novel mutation in
EXT1 gene that causes multiple osteochondromas. PLoS One.
8:e723162013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jennes I, Pedrini E, Zuntini M, et al:
Multiple osteochondromas: mutation update and description of the
multiple osteochondromas mutation database (MOdb). Hum Mutat.
30:1620–1627. 2009. View Article : Google Scholar : PubMed/NCBI
|