Introduction
Colorectal cancer is the third most prevalent cancer
worldwide, and its incidence rate in Asia increases each year.
Surgery is the primary means to effectively treat colorectal
cancer, however, the relapse rate can be as high as 30% for
partially terminal colorectal cancer patients even after radical
surgery and multidisciplinary treatment. Of these patients, only
7–20% of recurrences can be treated with surgery again (1–3).
Radiation therapy is another important type of cancer treatment.
Results of previous studies showed that concurrent radiotherapy and
chemotherapy prior to surgery can downstage advanced colorectal
cancer, improve the surgical resection rate, reduce the local
recurrence rate and improve progression-free survival benefits
(4,5). In current clinical practice,
radiosensitisers for colorectal cancer mainly include
fluoropyrimidine-based medicines. However, their treatment toxicity
is a major factor that limits the application of concurrent
radiotherapy and chemotherapy (6).
The recent emergence of targeting drugs provides a
more safe and effective way to achieve radiosensitisation.
Epidermal growth factor receptor (EGFR), which belongs to the ErbB
receptor tyrosine kinase family, is an important growth factor.
EGFR binding to its ligand phosphorylates the intracellular
tyrosine kinase receptor through conformational changes, activates
a number of signalling pathways and thus plays an important role in
tumour proliferation, differentiation, metastasis, and angiogenesis
(7,8). Approximately 60–80% of colorectal
cancer tumour tissue expresses EGFR, and its expression is closely
associated with cancer progression, poor prognosis, treatment
resistance and especially radiation resistance (9,10).
Thus, the EGFR signalling pathway is an important target for cancer
treatment, and it has been shown that EGFR inhibitors combined with
radiotherapy can enhance the efficacy of radiotherapy, rendering it
an effective radiosensitising drug (11,12).
Two types of drugs have been designed for selective
blockade of EGFR signalling, including monoclonal antibodies (mAbs)
specific for EGFR, such as cetuximab and nimotuzumab, and small
molecule inhibitors of the tyrosine kinase activity of EGFR
(EGFR-TKI), such as gefitinib and erlotinib. Although the former
type of drug exerted a radiosensitising effect in some colon cancer
cell lines, the effect is subject to the expression of certain
genes, such as K-RAS and BRAF, which increases
treatment uncertainty (13,14). In 2002, Williams et al found
that the small molecule TKI gefitinib can promote the reaction of
colon cancer cell lines (LoVo) to radiation in vitro
(15). Subsequent studies of a
mouse in vivo tumour model found that gefitinib in
combination with radiation therapy significantly inhibited tumour
proliferation compared with radiotherapy alone, which confirms the
radiosensitisation of gefitinib in colorectal cancer (15). However, in studies of other TKI
inhibitors, such as the radiosensitisation of colorectal cancer by
erlotinib and (ZEGFR19072), the sensitising effects of
erlotinib and (ZEGFR19072) were not equivalent to that
of gefitinib (16). Icotinib
hydrochloride (IH) is a new oral epidermal growth factor receptor
tyrosine kinase inhibitor, a quinazoline-type drug, with targets
and mechanisms similar to those of gefitinib (17,18).
It is a reversible EGFR intracellular TKI with an efficacy equal to
that of gefitinib both in vitro and preclinical studies
(18). This drug has already shown
a significant inhibitory effect on colon cancer at the cell level
(17). However, its superiority or
equivalence to the effect of gefitinib when combined with radiation
therapy remains unclear.
Owing to the lethal damage induced by radiation
therapy-DNA double-strand breaks (DSBs), interfering with DSBs
repair has become an important radiosensitisation strategy
(19,20). EGFR-TKI has been found to affect the
key components of intracellular DNA repair to sensitise cells to
radiotherapy (21). Therefore, this
present study was performed to evaluate the radiosensitisation
efficacy of a combination of IH and radiotherapy in human
colorectal cancer cell lines via in vitro and in vivo
models and to investigate whether the effects of the
radiosensitisation are correlated with changes in tumour cell
apoptosis, cell cycle and DNA repair, by examining the expression
of the phosphorylated histone H2AX (γ-H2AX) and 53BP1 (P53 binding
protein 1) proteins during DSB repair.
Materials and methods
Cell lines and cultures
HT29 and HCT116 cells were obtained from the
laboratory of the General Surgical Department, Union Hospital,
Tongji Medical College, Huazhong University of Science and
Technology (Hubei, China) and maintained in RPMI-1640 medium
supplemented with 10% foetal bovine serum. The cells were cultured
in a humidified incubator with 5% CO2 at 37°C. IH was
provided by Zhejiang Beta Pharma Ltd. (Hangzhou, China), and
dissolved in 100% dimethyl sulfoxide (DMSO) (Sigma, St. Louis, MO,
USA) to a final concentration of 30 mg/ml and stored at −20°C.
MTT assay
A total of 0.5-1×105 exponentially
growing cells were seeded in 96-well micro-titre plates (Corning
Inc., New York, NY, USA) and were treated with different
concentrations of IH (0.01, 0.03, 0.1, 0.3 and 0.9 mg/ml) following
incubation in growth medium overnight at 37°C. After 24 h of IH
addition, a microculture tetrazolium (MTT) assay was performed by
adding 20 μl of 3-(4,5-diethyl-2-thiazolyl)-2,5-diphenyltetrazolium
bromide (5 mg/ml) (Sigma) to each well for 4 h at 37°C to allow
metabolically active cells to generate formazan crystals from MTT.
The medium was aspirated and 150 μl of DMSO (Sigma) was added to
dissolve the formazan. After 10 min, the above mixture was measured
in a microplate reader (Bio-Tek, Winooski, VT, USA) at a wavelength
of 490 nm. The percentage inhibition rate was calculated as: (1 -
OD value of experimental group/control group OD) ×100%. The
IC20 was selected as the subsequent experiment
concentration.
Clonogenic assays
HCT116 and HT29 cells were collected from
exponential phase cultures by trypsinisation, counted, and then
seeded in 6-well plates (Corning Inc.) with densities varying from
1×102 to 5×103 cells/well depending on the
intended radiation dose. According to the MTT assay, the
IC20 was selected for subsequent experiments. IH (0.03
and 0.06 mg/ml for HCT116 and HT29 cells, respectively) was added
24 h prior to radiation exposure with single doses ranging from 0
to 10 Gy. Irradiation treatments in this study were performed on a
clinically calibrated Siemens Oncor accelerator using 6 MV photons
at a nominal dose rate of 3 Gy/min (Siemens Medical Solutions,
Concord, CA, USA). After 24 h, the medium containing IH or DMSO was
substituted with medium that only contained 10% foetal bovine
serum. After 10–14 days, the cells were fixed with methanol (Guge,
Wuhan, China) and stained with 10% crystal violet (Guge). The
colonies were counted, and a grouping of >50 cells was
considered a colony. The plating efficiency (PE) was the percentage
of cells seeded that grew into colonies under special culture
conditions. The survival fraction, expressed as a function of the
radiation dose, was calculated as: Survival fraction = colonies
counted/(cells seeded × PE/100). Experiments were repeated three
times.
Assessment of cell apoptosis
After 24 h of seeding, the cells were exposed to IH
(0.06 and 0.03 mg/ml for HT29 and HCT116 cells) for 24 h,
radiotherapy (10 Gy) for 24 h, or the combination treatment. The
trypsinised cells were re-suspended in 1X binding buffer at a
concentration of 2×105 cells/ml, and Annexin V-FITC and
propidium iodide (PI) (Sigma) were added. The cells were incubated
for an additional 15 min at room temperature in the dark and then
subjected to analysis with flow cytometry (BD Biosciences, San
Jose, CA, USA). A minimum of 10,000 cells in each sample were
analysed, and the data were analysed using the Cell Quest software
(BD Biosciences).
Cell cycle analysis
Cells were collected after 24 h of exposure to IH,
10 Gy radiation, or the combination treatment. The cells were
washed with PBS (Boster, Wuhan, China) and harvested by
trypsinisation. After centrifugation at ? for ?, the cell pellets
were fixed in 70% cold ethanol. Following removal of the ethanol by
centrifugation, the cells were stained with a DNA staining solution
(20 μg/ml of propidium iodide and 10 μg/ml of RNase A) for 30 min.
The stained cells were then suspended and immediately subjected to
analysis using a flow cytometer (BD Biosciences). The resulting DNA
distribution was analysed by ModFit for the proportion of cells in
the sub-G0, G1, S, and G2-M phases of the cell cycle. A minimum of
10,000 cells was counted in each sample, and the cell cycle
distribution was calculated using the Cell Quest software (BD
Biosciences).
Immunofluorescent staining for γ-H2AX and
53BP1
Cells (2×105) were plated in chamber
slides. IH was added following cell adhesion, resulting in a final
concentration of 0.06 and 0.03 mg/ml for HT29 and HCT116 cells,
respectively. After 24 h, the cells were treated with radiation.
After another 24 h, the medium was aspirated and the cells were
fixed with 4% paraformaldehyde. The cells were rinsed with PBS
three times and permeabilised with 0.2% Triton-X-100 (Boster) for
20 min at 4°C. The cells were rinsed with PBS three times again and
blocked with 5% BSA for 1 h at room temperature. The cells were
again washed with PBS three times and anti-γ-H2AX antibody (1:200,
Abcam, Cambridge, MA, USA) and anti-53BP1 antibody (1:200, Bethyl,
Inc., Montgomery, TX, USA) were added at a dilution of 1:800 and
1:200 in 1% BSA, respectively. Subsequently, the slides were
incubated overnight at 4°C. The cells were rinsed with PBS prior to
incubation with cyanine 3 (CY3)-labelled secondary antibody
(Protientech Group, Chicago, IL, USA) at a dilution of 1:200 in 1%
BSA for 1 h in the dark. The secondary antibody was aspirated, and
the cells were rinsed with PBS three times and incubated with
4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Vector
Laboratories Inc., Burlingame, CA, USA) in the dark for 10 min. The
slides were examined using a fluorescent microscope. The images
were captured with a confocal laser scanning microscope (CLSM)
(Olympus, Tokyo, Japan) equipped with a camera. For each sample,
the γ-H2AX and 53BP1 foci were determined in ≥100 cells.
Western blot analyses
The cells were seeded and allowed to adhere in
complete medium overnight. They were then treated with or without
IH (0.06 and 0.03 mg/ml for HT29 and HCT116 cells) 24 h prior to
irradiation with a 10 Gy dose, and the cells were lysed with a
lysis buffer (Beyotime, Wuhan, China). The protein lysates were
harvested and centrifuged, and the supernatants were collected. The
proteins were separated via 10 or 12% sodium dodecyl sulphate
(SDS)/polyacrylamide gel electrophoresis and then transferred to
polyvinyl difluoride membranes (Millipore, Billerica, MA, USA). The
membranes were then blocked with 5% bovine serum albumin or milk
and finally incubated with γ-H2AX (Abcam), 53BP1 antibody (Bethyl,
Inc.) or β-actin antibody (Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA). The membranes were incubated with the primary
antibodies overnight at 4°C and rinsed with TBST three times,
followed by incubation with secondary antibodies labelled with
horseradish peroxidase (HRP) (Protientech Group). The bands were
then visualised using enhanced chemiluminescence (ECL) (Pierce,
Rockford, IL, USA).
Assay for tumour growth in athymic nude
mouse model
Exponential phase HT29 and HCT116 cells were
prepared at a concentration of 2×107 cells/ml in
serum-free RPMI medium. Female athymic nude mice (nu/nu, body
weight, 20–25 g, 8–12 weeks of age) were obtained from Beijing HFK
Bioscience Co., Ltd. The mice were provided with sterilised food
and water and housed in a barrier facility with 12-h light/dark
cycles in laminar flow hoods at a constant temperature and humidity
for the entire course of the experiments and supplied with a
standard laboratory diet and water. The tumour xenografts were
established via the subcutaneous injection of a 0.2 ml volume of
the prepared cell stock into the right hind leg. After 7 days, when
the diameter of each tumour increased to 10 mm, the mice were
pooled and randomly assigned to 4 groups (control, IH alone,
radiation alone, and radiation in combination with IH) of 6 animals
each. IH (35 mg/kg) was administered via oral gavage once a day for
5 days, and locoregional irradiation was administered in a single 2
Gy fraction once a day (6-MV linear accelerator, MDX, Siemens) for
5 days. The tumour size was measured using callipers every two
days. Animal protocols and studies were conducted in accordance
with the guidelines of the Institutional Animal Care and Use
Committee of the Korea Institute of Radiological and Medical
Sciences, Korea.
The tumour volumes (V) were determined according to
the two axes of the tumour (L, longest axis; W, shortest axis). The
volume was calculated according to the formula: Tumour volume
(mm3) = (L × W2)/2 mm3, where L
and W are the shortest and the longest diameter.
Statistical analysis
SPSS 13.0 software was used for the data analysis.
The data are reported as the mean ± SEM and analysed using one-way
ANOVA to compare means in multiple groups. A q-test was used for
the pairwise comparison among groups. A difference was regarded as
significant if P<0.05.
Results
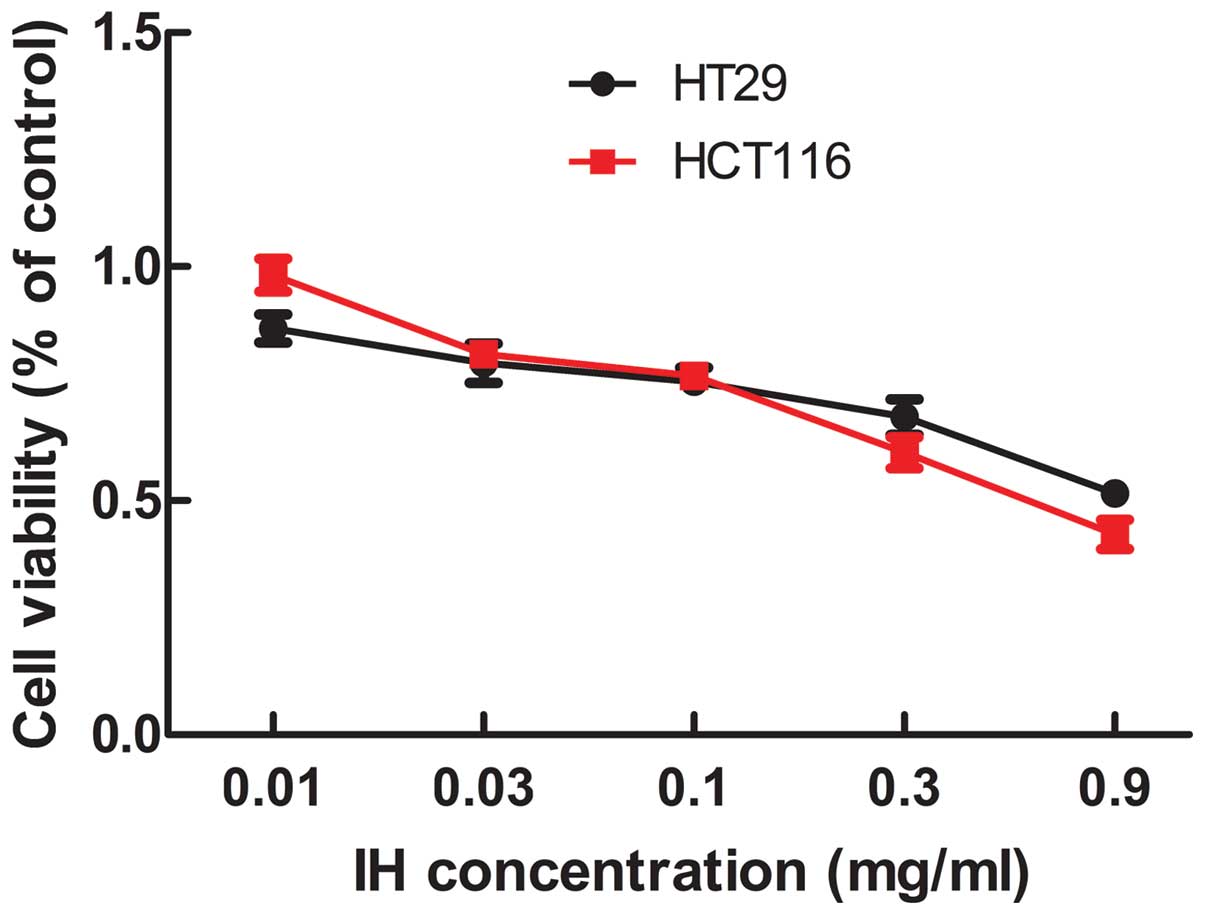
Effect of icotinib on cell viability
The effect of icotinib on cell viability was
measured with the MTT assay. The inhibitory effect of icotinib
positively correlates with the drug concentration (Fig. 1). The IC50 (median
inhibition concentration) and IC20 (inhibiting
concentration 20) values were obtained. The IC20 value
was selected as the drug concentration for subsequent experiments,
with values of 0.06 and 0.03 mg/ml for HT29 and HCT116.
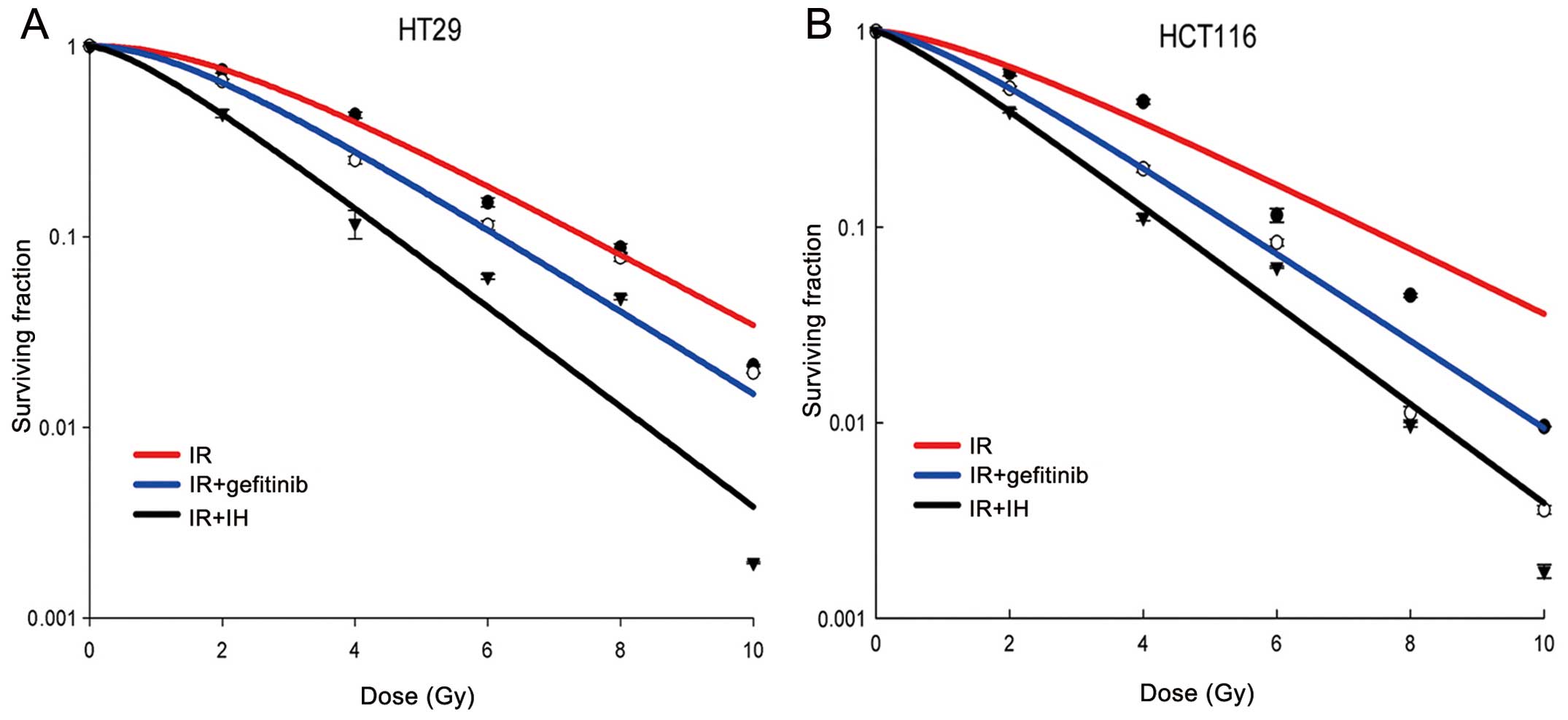
Influence of icotinib on the radiation
sensitivity of human colorectal cancer cell lines
To determine the radiosensitising effect of icotinib
on the HT29 and HCT116 colorectal cell lines, a clonogenic
formation assay was performed. The result was compared to gefitinib
combined with radiation, similar to icotinib. IC20
values of gefitinib on HT29 and HCT116 cells were explored and
applied for further study of radiosensitisation. The L-Q model was
used to investigate the difference in radiosensitivity from
gefitinib or IH combined with irradiation or irradiation alone. The
results showed that the Dq, Do and N values were reduced in
response to treatment with gefitinib or icotinib combined with
irradiation when compared with irradiaton alone for HT29 and
HCT116. Furthermore, icotinib exhibited a stronger ability to
reduce the Dq, Do and N values compared to gefitinib. Radiotherapy
alone decreased the SF2 values of HT29 and HCT116 to 76 and 66%,
whereas when gefitinib was added, the SF2 values decreased to 64
and 51%, respectively. Addition of icotinib resulted in the
reduction of SF2 values to 44 and 39%, respectively, suggesting
that icotinib more effectively sensitises colorectal cancer cells
to radiation as compared to gefitinib (Table I, Fig.
2).
 | Table IThe main parameters of cell survival
curves of HT29 and HCT116 following irradiation. |
Table I
The main parameters of cell survival
curves of HT29 and HCT116 following irradiation.
| HT29 | HCT116 |
|---|
|
|
|
|---|
| Parameter | IR | Gefitinib+IR | IH+IR | IR | Gefitinib+IR | IH+IR |
|---|
| D0 | 2.31 | 2.00 | 1.65 | 2.58 | 1.94 | 1.71 |
| Dq | 1.80 | 1.51 | 0.99 | 1.51 | 1.15 | 0.81 |
| N | 2.61 | 2.26 | 1.63 | 1.75 | 1.63 | 1.32 |
| SF2 | 0.76 | 0.64 | 0.44 | 0.66 | 0.51 | 0.39 |
| SER | | 1.19 | 1.73 | | 1.29 | 1.69 |
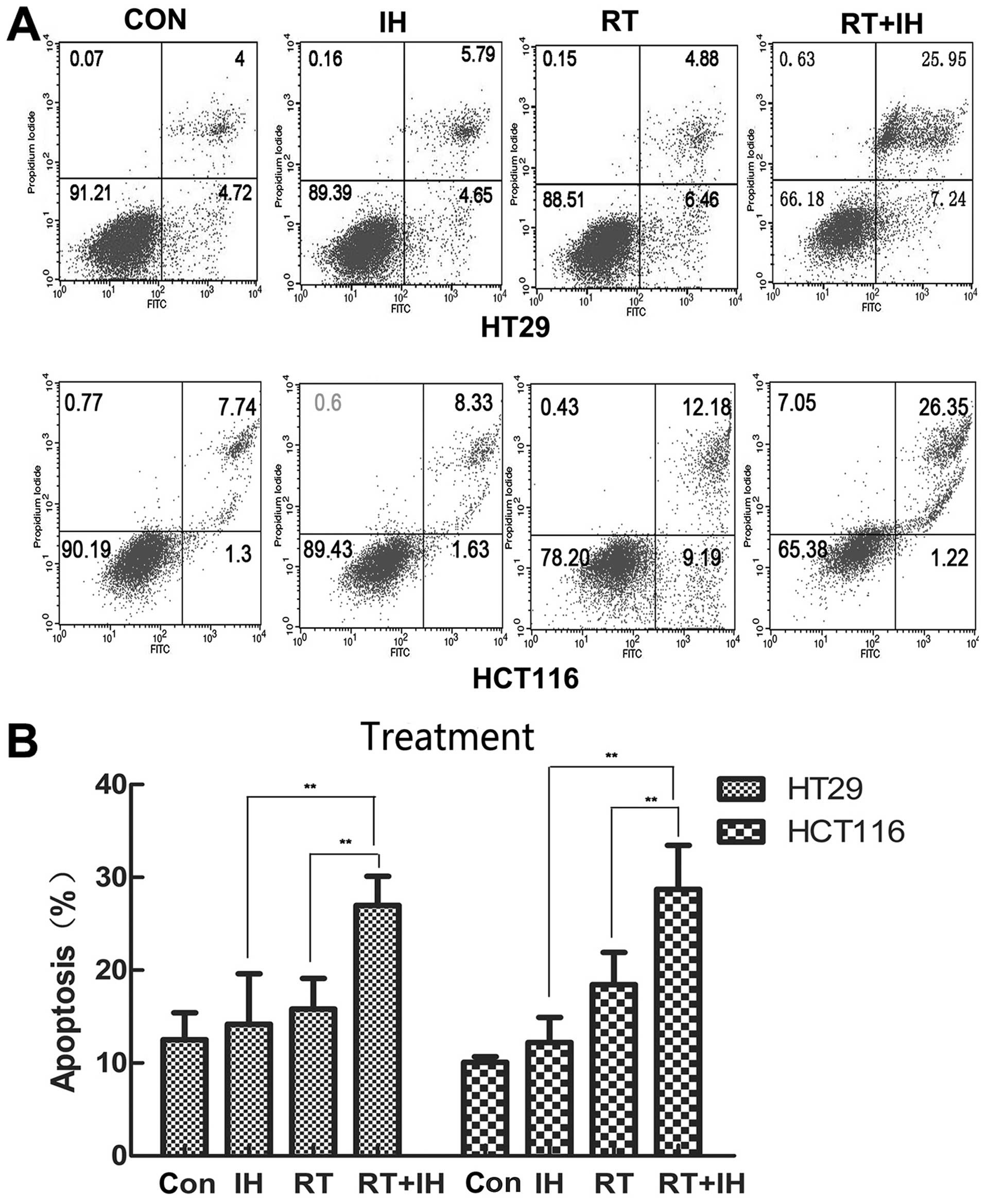
Effect of icotinib in combination with
radiotherapy on cell apoptosis
To determine whether icotinib combined with
radiotherapy increased apoptosis, HT29 or HCT116 cells were treated
with vehicle or IH with or without radiotherapy [control; IH alone;
radiotherapy (RT) alone; IH+RT], and the cell apoptotic rate was
then detected via flow cytometry 24 h after the treatment (Fig. 3). In HT29 cells, the apoptotic rate
of the combined group was 26.97±7.15%, while it was 14.17±5.44% in
the monotherapy group, 15.81±3.31% in the RT group and 12.5±2.93%
in the control group. Significant differences were observed between
the combined group and the remaining three groups (P<0.01). For
HCT116 cells in the control group, the apoptotic rate was
10.08±0.62%, while it was 12.21±2.71% in the IH alone group,
18.45±3.45% in the RT group and 28.72±4.73% in the combined group.
Compared with the remaining three groups, the apoptotic rate of the
combined group was significantly increased (P<0.05).
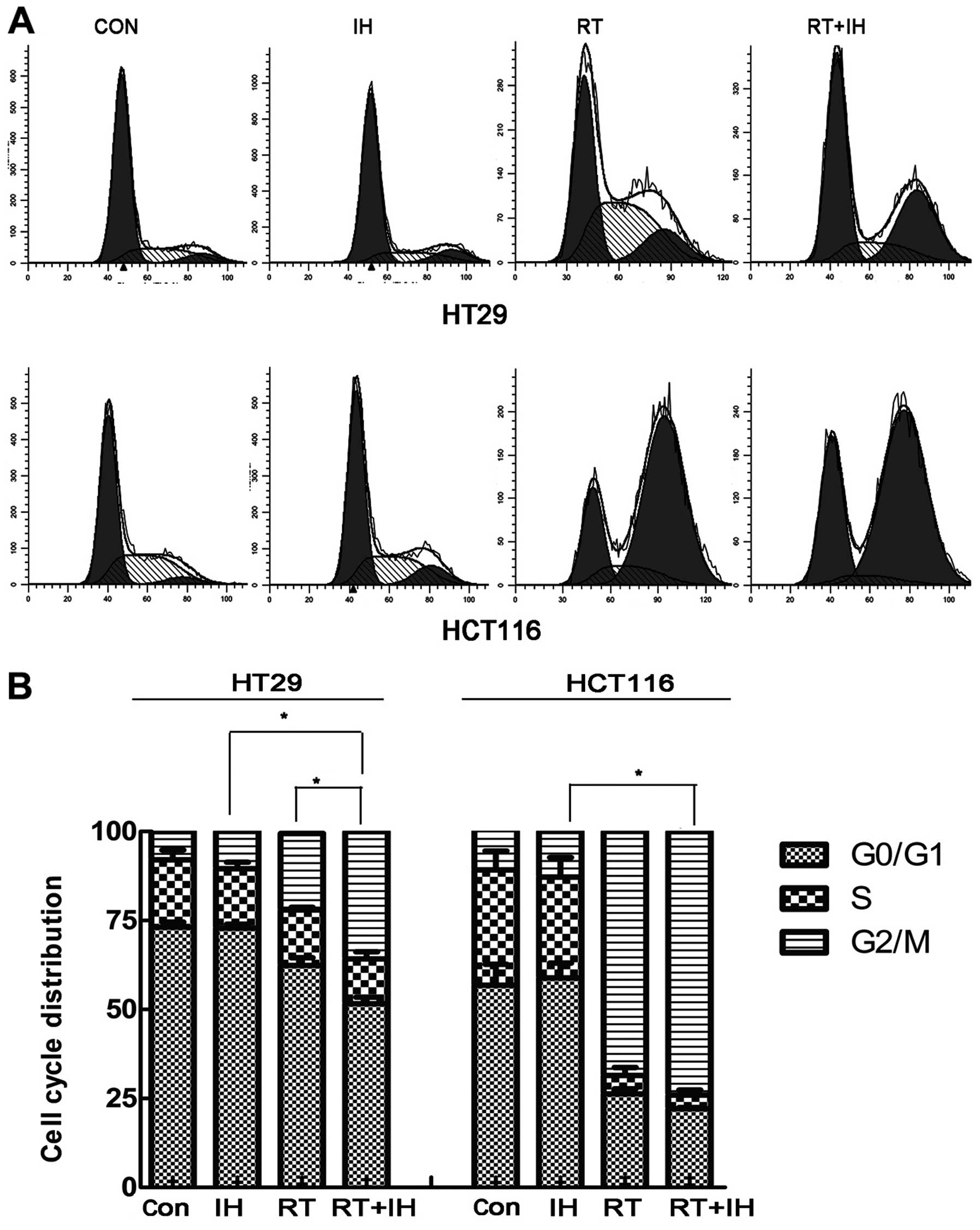
Effect of icotinib on cell cycle
following radiation
The influence of icotinib in combination with or
without radiotherapy on the HT29 and HCT116 cell cycle was detected
via flow cytometry (Fig. 4). In
HT29 cells, the G2/M phase of the single drug group was
10.45±0.89%, while it was 21.50±1.99% in the RT alone group and
35.79±1.10% in the combined group. The percentage of cells in the
G2/M phase in the combined group was higher than that of the single
drug or RT alone group (P<0.05). For HCT116 cells, the G2/M
phase of the single drug group was 12.77±2.03%, while that of the
RT alone group was 68.53±2.49% and that of the combined group was
74.00±1.33%. The combined group showed significantly longer arrest
in the G2/M phase compared with the single drug group and a
tendency to stagnate in the G2/M phase compared with the RT group,
although without significant differences (P>0.05). Icotinib
combined with RT treatment re-adjusted the cell cycle distribution,
significantly increased sensitive cells and improved the effect of
radiotherapy.
Effect of icotinib on γ-H2AX and 53BP1
foci formation following radiotherapy
The present results showed that, H2AX was rapidly
phosphorylated in the presence of DSB and aggregated at the DSB,
making it an important symbol of DNA DSB, as well as an important
method to detect DNA DSB repairability. Twenty-four hours after the
various treatments, the γ-H2AX foci per cell were detected
(Fig. 5). The results showed that
the combination groups showed a significantly higher number of
unrepaired double-stranded DNA in the HT29 and HCT116 cell lines,
while the RT alone or combined treatment with icotinib group showed
significant differences as compared to the combined group
(P<0.01). In the drug-RT combination group, the number of γ-H2AX
foci was significantly higher than that in the RT alone group.
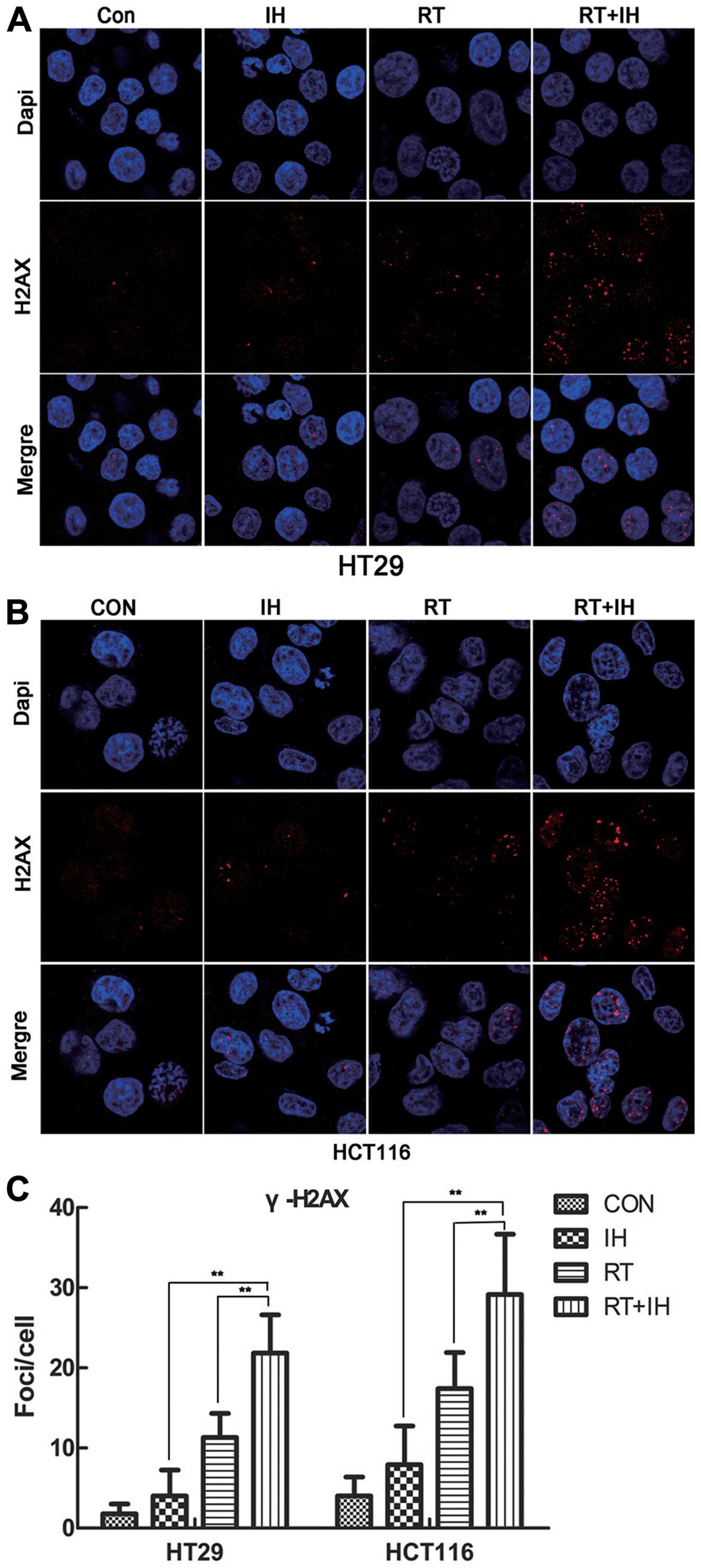 | Figure 5γ-H2AX foci expression was detected in
HT29 and HCT116 cells by immunofluorescent staining. (A)
Immunofluorescent staining of γ-H2AX in HT29 cells (x1,000). (B)
Immunofluorescent staining of γ-H2AX in HCT116 cells (x1,000). (C)
Quantitative detection of γ-H2AX foci number, Columns, means; Error
bars, SEM, from three independent experiments.
**P<0.01; Con, control group; IH, icotinib alone; RT,
radiotherapy alone; RT+IH, combined group. |
53BP1 has been proven to participate in
double-stranded DNA repair, and is an important early regulator of
DNA damage. To further determine whether 53BP1 is involved in
double-stranded DNA repair during tumour suppression, the present
study detected 53BP1 expression in HCT116 and HT29 cells treated in
different groups (Fig. 6). Similar
to γ-H2AX expression in different treatment groups, 53BP1 exhibited
the same trends in the two differently treated cell lines. Tumour
cells in the icotinib-RT combined group showed high levels of 53BP1
foci/cell that significantly differed from the RT, single drug and
control groups. 53BP1 expression was significantly increased in the
icotinib-RT combined group compared to the RT alone group and drug
treatment group (P<0.01). The results are in concordance with
the γ-H2AX data presented for each group, suggesting that 53BP1 and
γ-H2AX are involved in DNA double-strand repair and are involved in
the tumour response to treatment and radiosensitisation.
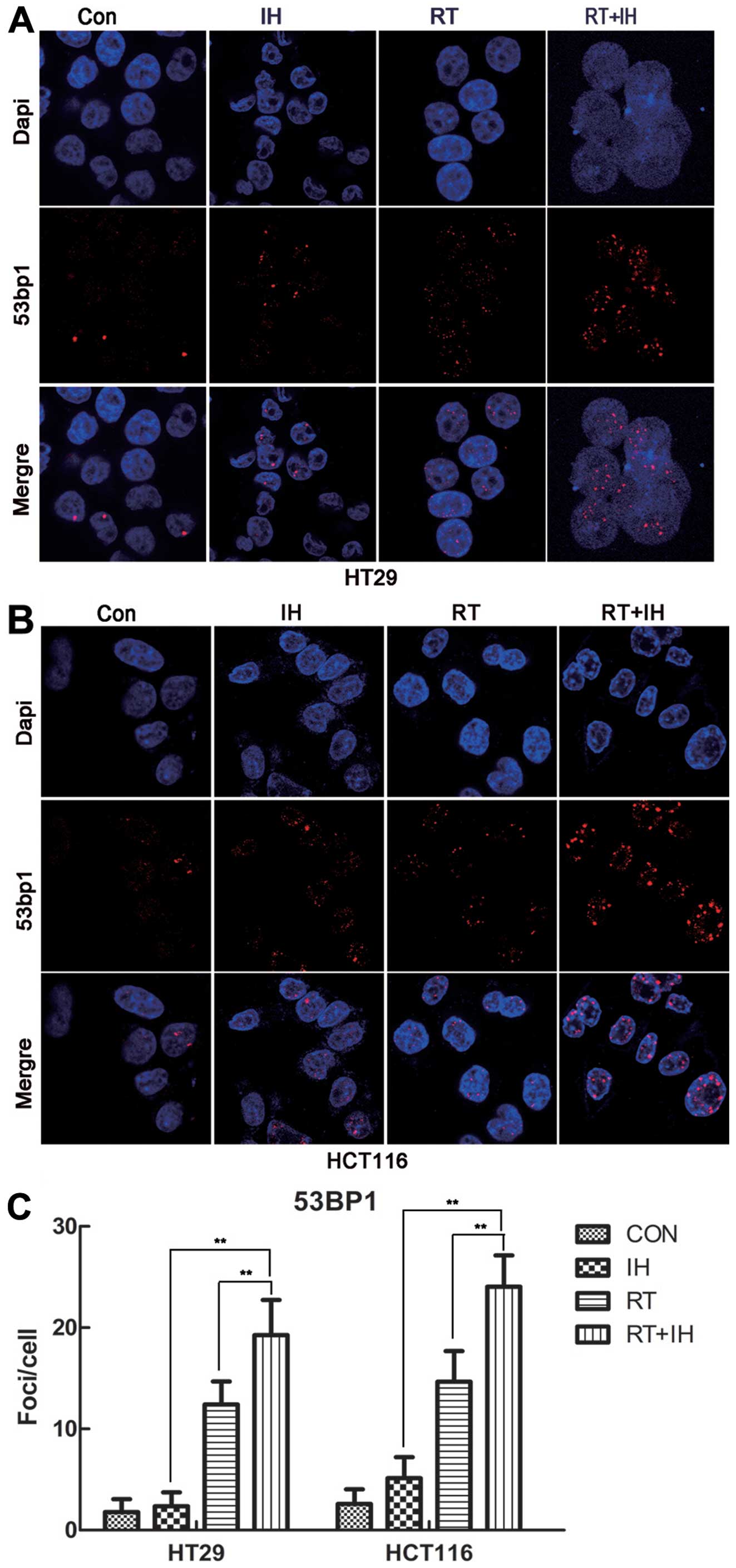 | Figure 653BP1 foci expression was detected in
HT29 and HCT116 cells by immunofluorescence staining. (A)
Immunofluorescent staining of 53BP1 in HT29 cells (x1,000). (B)
Immunofluorescent staining of 53BP1 in HCT116 cells (x1,000). (C)
Quantitative detection of 53BP1 foci number, Columns, means; Error
bars, SEM, from three independent experiments.
**P<0.01; Con, control group; IH, icotinib alone; RT,
radiotherapy alone; RT+IH, combined group. |
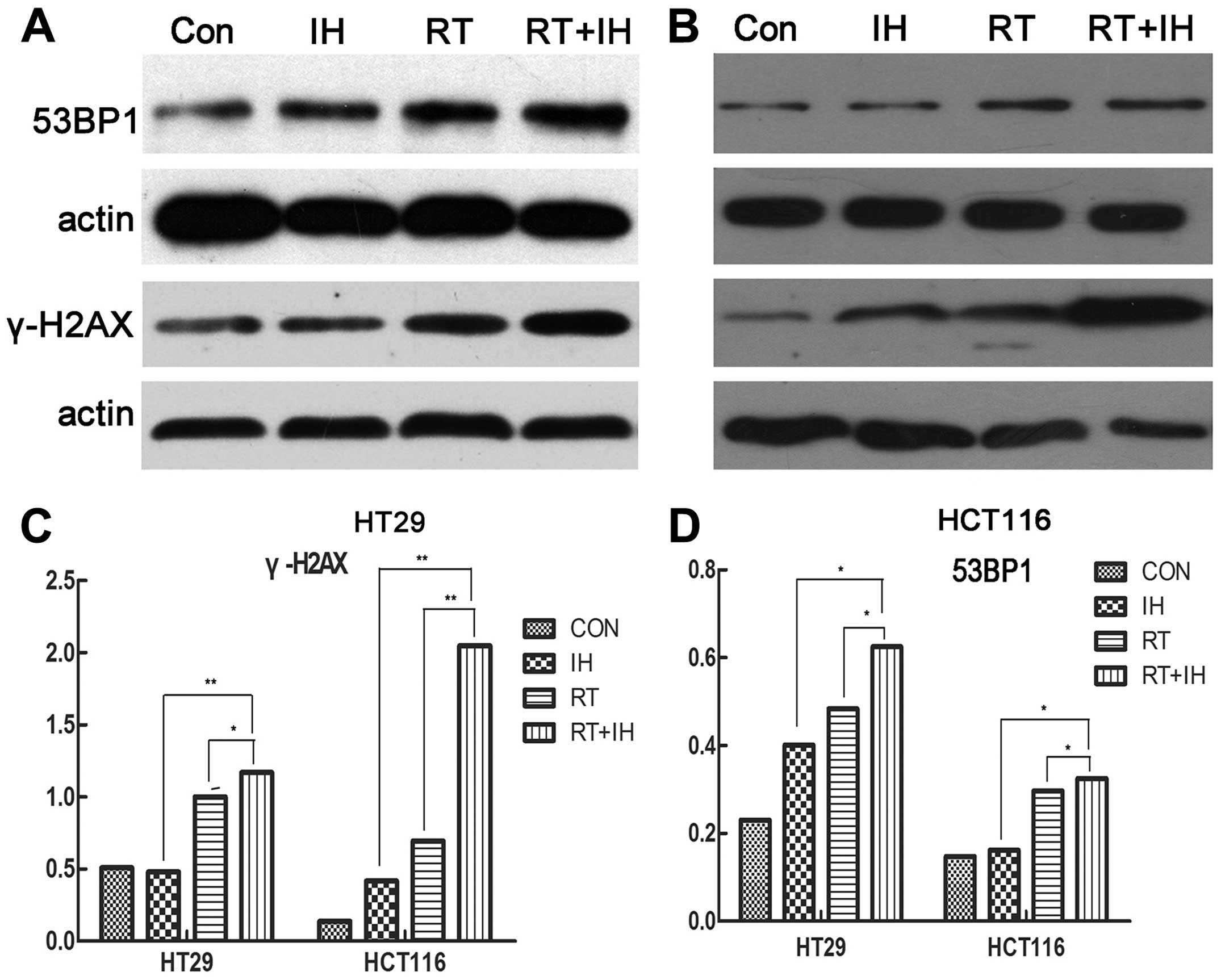
Icotinib hydrochloride increases the
expression of 53BP1 and γ-H2AX when combined with radiotherapy
To determine the factors that contribute to impaired
DSB repair, the responses of the 53BP1 and γ-H2AX proteins, which
play key roles in DSB repair, were determined. Western blots were
used to detect the expression of γ-H2AX and 53BP1 protein in human
colorectal cancer cells following interference in the different
groups. As shown in Fig. 7,
icotinib-RT combined treatment significantly increased the
expression levels of γ-H2AX and 53BP1 proteins, suggesting that the
combined treatment increased the DNA double-strand breaks,
attenuated DNA repair and improved the effect of radiotherapy.
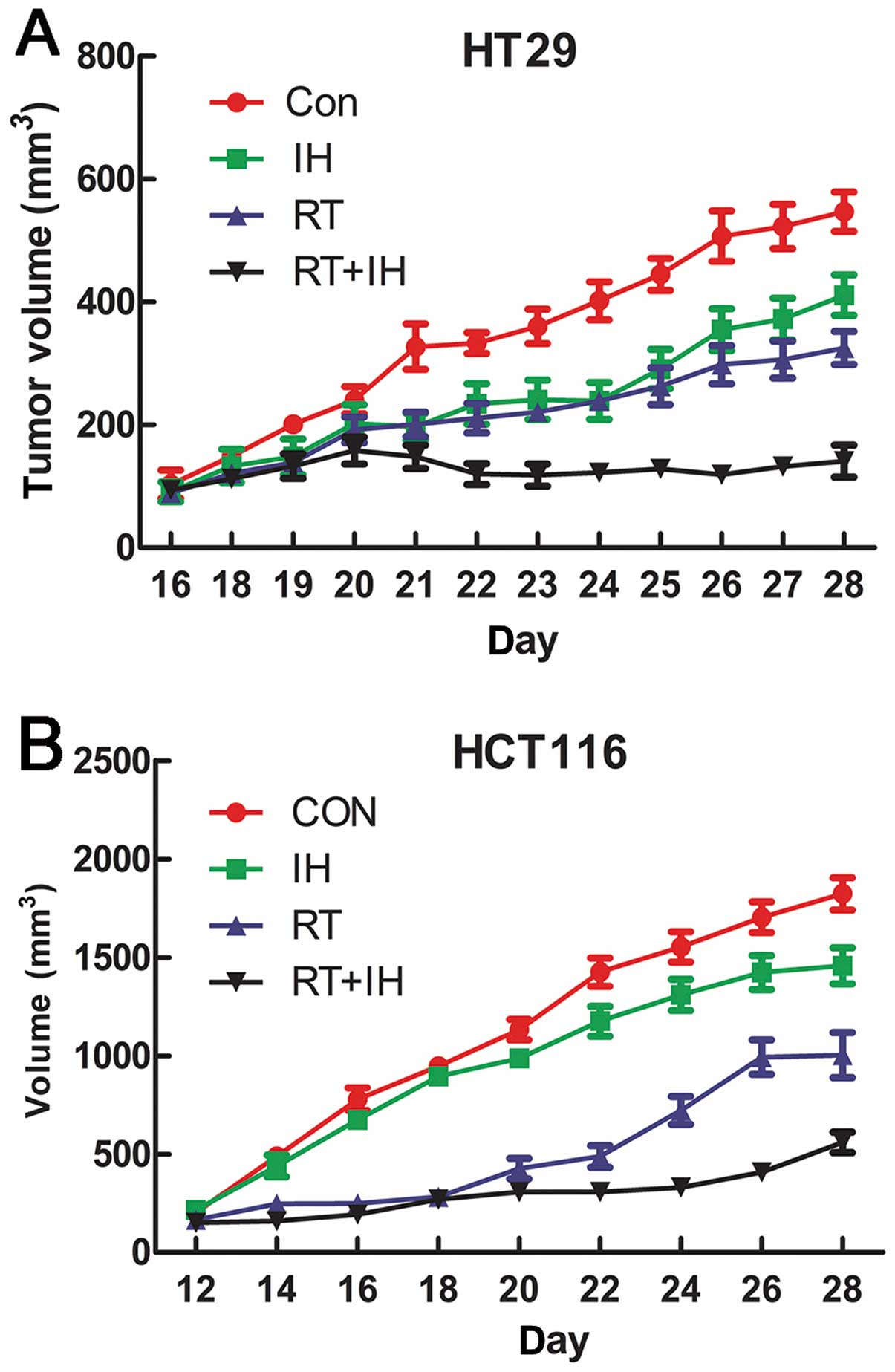
Antitumour activity of icotinib combined
with radiotherapy on human colorectal cancer subcutaneous tumour
xenograft
IH was shown to significantly inhibit HT29 and
HCT116 cell xenograft growth (Fig.
8). The final tumour volume of the combination group was
statistically significantly smaller than that of the other groups
(P<0.05) in both the HCT116 and HT29 models. This finding
indicates that icotinib in combination with radiotherapy
significantly inhibited the growth of colorectal cancer
tumours.
Discussion
EGFR is a tyrosine kinase receptor of the ErbB
family, and the signalling pathways in which it is involved
regulate many important cell functions, including cell
proliferation and apoptosis (22).
Inhibition of the EGFR signalling pathway can reportedly improve
the radiation effects (23,24), but inhibition of the EGFR signalling
pathway for the enhancement of the efficacy and mechanisms of
colorectal cancer radiotherapy requires additional study. IH is a
new oral epidermal growth factor receptor tyrosine kinase
inhibitor, a quinazoline-type drug, with targets and mechanism
similar to those of gefitinib. It is a reversible EGFR
intracellular tyrosine tyrosine kinase inhibitor and has shown
efficacy equal to that of gefitinib (17,18).
In vitro studies have already demonstrated a significant
inhibitory effect on colon cancer cells. However, its superiority
or equivalence to the effect of gefitinib when combined with
radiation therapy remains unclear.
Since the abnormal expression of certain genes (such
as K-RAS) in the EGFR signalling pathway affects the
efficacy of EGFR inhibitors (13,14),
the present study was conducted on the K-RAS gene expression
status. K-RAS wild-type HT-29 cell line and the K-RAS mutant
HCT-116 cell line were selected to establish wild- and mutant-type
colorectal cancer in vitro and in vivo models. At the
cell level, the impact of icotinib on the radiosensitivity of these
two colorectal cancer lines was studied via a clone formation
experiment in the present study. Moreover, icotinib with gefitinib
was compared to understand sensitising differences between the two
TKI inhibitors in colorectal cancer. Furthermore, tumour-bearing
mouse models were established to understand the effect of icotinib
combined radiotherapy treatment on tumour proliferation. The
results showed that icotinib and gefitinib promoted the
radiosensitivity of the two colon cancer cells in vitro
compared to radiotherapy alone, and their sensitising effect was
not influenced by K-RAS gene expression. Compared with
gefitinib, icotinib significantly increased the radiosensitivity of
tumour cells, confirming the superiority of icotinib to gefitinib.
In vivo, icotinib was shown to produce significant
inhibition of HT29 and HCT116 cell xenograft growth. In addition,
the inhibitory effect in the combination treatment with icotinib
and radiotherapy group was better than that of icotinib alone group
and better than the radiotherapy alone group. Thus, icotinib
combined with radiotherapy induced a highly significant inhibition
of tumour growth and realized the radiosensitisation effectively to
colorectal cancer in the present study.
Generally, radiosensitivity is governed by the
capacity of the cell for efficient repair of radiation-induced
lesions in the DNA, mainly the repair of DSBs. DNA double-strand
breaks are a lethal cell injury, and radiation can induce DSB
formation in tumour cells, interfering with DSB repair. Thus,
increasing the extent of DSB damage is a radiosensitisation
strategy (21,25). In the presence of DSB, histone H2AX
rapidly phosphorylates tryptophan 139 and aggregates at the DSB,
forming visible fluorescence foci (foci) (26). Other components of DNA damage,
including phosphorylated ATM, phosphorylated DNA-PKcs, 53BP1,
BRCA1, MDC1, RAD51 and MRE11/RAD50/NBS1 (MRN complex), all
subsequently participate in the repair (25). Previous studies have shown that the
initial number of foci is associated with the theoretical number of
radiation-induced foci, and γ-H2AX will subsequently be
dephosphorylated and abolished with the elimination of DNA damage
(such as being repaired) (27).
Therefore, fluorescently labelled γ-H2AX was considered to be an
effective tool to distinguish intracellular DSB, where the
intracellular amount of residual γ-H2AX foci, which represents the
cell damage repair capacity, depended on the cell type and is
closely associated with cell radio-sensitivity (28).
In our study, tumour cell apoptosis in different
treatment groups was detected by flow cytometry. The results showed
that icotinib increased the apoptotic rate of HCT29 and HT116 cells
following radiotherapy treatment compared to the drug and
radiotherapy alone groups. Furthermore, the residual γ-H2AX foci in
different treatment groups were examined in HCT29 and HT116 cells
using immunofluorescent staining. The results showed that icotinib
combined with radiotherapy significantly increased the
intracellular γ-H2AX foci compared with the drug group and
radiotherapy alone group. Western blot analysis indicated that
icotinib significantly increased the intracellular expression of
γ-H2AX protein after radiotherapy treatment, which suggests that
icotinib in combination with radiotherapy increases the
intracellular DSB, thereby inducing tumour cell apoptosis.
Studies have shown that EGFR-TKI can affect the key
components of the DNA repair pathway to increase DNA damage in
order to achieve a radiosensitisation effect (21). 53BP1 has been proven to play an
important intermediary role in DSB repair, while it is important in
the cell response to treatment (29,30).
The 53BP1 protein consists of two Tudor structural domains and a
C-end BRCT domain, with the former domain allowing it to aggregate
at DSB, while the latter domain is involved in the interactive
response with other DNA damage repair proteins (31,32).
DNA DSBs activate ataxia mutations (ataxia telangiectasia-mutated,
ATM) of capillaries, induce a strong phosphorylation of 53BP1, and
aggregate at DSB via the Tudor end and phosphorylated histone H2AX
(γ-H2AX) and other proteins associated with repair damage. The
C-terminal end activates cell cycle checkpoint kinase 1 (checkpoint
kinase-1, CHK1) and cell cycle checkpoint kinase 2 (checkpoint
kinase-2, CHK2) to regulate the cell cycle G1/S, S and G2/M
checkpoints (32,33). ATM-CHK2 activation further
phosphorylated P53 and induced apoptosis, which depends on P53.
Studies have reported that different levels of 53BP1 expression can
interfere with cell cycle distribution and influence the treatment
response. For example, Li et al found that a high expression
of 53BP1 can induce the breast cancer cell cycle to stagnate in the
G2/M phase, thus enhancing the sensitivity of tumour cells to
subsequent drug treatment. However, cells with a low expression of
53BP1 exhibited resistance to treatment (34). EGFR inhibitors can reportedly cause
tumour cell cycle redistribution and increase the percentage of
G2/M- or G1-phase cells while decreasing the proportion of S-phase
cells (35). Therefore, EGFR
inhibitors may influence the expression of 53BP1 and radiosensitise
cells by interfering with the cell cycle distribution.
Using flow cytometry to detect cell cycle
distribution, we found that icotinib in combination with
radiotherapy adjusts the cell cycle distribution of wild-type HT29
colon cancer cells and reduces the proportion of cells in the G0/G1
phase in order to maximise the number of cells in G2/M arrest and
facilitate the apoptosis or necrosis of radiosensitive cells. For
HCT116 colon cancer cells, radiotherapy treatment or icotinib
combined with radiotherapy treatment decreased the proportions of
cells in the G0/G1 and S phases and significantly increased
G2/M-phase arrest. Although the two treatments did not produce
statistically significant differences, an apparent increasing trend
for G2/M-phase arrest was observed in the combined treatment
group.
After further testing the 53BP1 foci of HCT29 and
HT116 cells following different interventions in the treatment
using immunofluorescent staining, the results showed that icotinib
in combination with radiotherapy significantly increased
intracellularly by the 53BP1 foci compared with the drug group and
radiotherapy alone group. Western blot analysis revealed that
icotinib significantly increased the intracellular 53BP1 expression
after radiotherapy, suggesting that icotinib may alter the cell
cycle by increasing the intracellular 53BP1 expression, and
radiosensitise cells by inducing apoptosis.
In conclusion, we have shown that IH, a potent
inhibitor of EGFR tyrosine kinase activity, at pharmacologically
achievable levels, may sensitise tumour cells to radiation by
influencing key DSB repair proteins and delaying DNA damage repair.
This finding suggests an interaction between EGFR signaling
pathways and the regulation of DSBs repair in the nucleus.
Examination of these interactions may reveal additional strategies
for radiosensitising human tumour cells or biomarkers for
identifying patients who may benefit from the combination of
molecularly targeted agents and radiotherapy.
References
|
1
|
Secco GB, Fardelli R, Rovida S, et al: Is
intensive follow-up really able to improve prognosis of patients
with local recurrence after curative surgery for rectal cancer? Ann
Surg Oncol. 7:32–37. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Salo JC, Paty PB, Guillem J, Minsky BD,
Harrison LB and Cohen AM: Surgical salvage of recurrent rectal
carcinoma after curative resection: a 10-year experience. Ann Surg
Oncol. 6:171–177. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pilipshen SJ, Heilweil M, Quan SH,
Sternberg SS and Enker WE: Patterns of pelvic recurrence following
definitive resections of rectal cancer. Cancer. 53:1354–1362. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shin SJ, Yoon HI, Kim NK, et al: Upfront
systemic chemotherapy and preoperative short-course radiotherapy
with delayed surgery for locally advanced rectal cancer with
distant metastases. Radiat Oncol. 6:992011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bosset JF, Collette L, Calais G, et al:
Chemotherapy with preoperative radiotherapy in rectal cancer. N
Engl J Med. 355:1114–1123. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rodel C, Liersch T, Becker H, et al:
Preoperative chemoradiotherapy and postoperative chemotherapy with
fluorouracil and oxaliplatin versus fluorouracil alone in locally
advanced rectal cancer: initial results of the German
CAO/ARO/AIO-04 randomised phase 3 trial. Lancet Oncol. 13:679–687.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Baumann M and Krause M: Targeting the
epidermal growth factor receptor in radiotherapy: radiobiological
mechanisms, preclinical and clinical results. Radiother Oncol.
72:257–266. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ullrich A and Schlessinger J: Signal
transduction by receptors with tyrosine kinase activity. Cell.
61:203–212. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Galizia G, Lieto E, Ferraraccio F, et al:
Prognostic significance of epidermal growth factor receptor
expression in colon cancer patients undergoing curative surgery.
Ann Surg Oncol. 13:823–835. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Porębska I, Harłozińska A and Bojarowski
T: Expression of the tyrosine kinase activity growth factor
receptors (EGFR, ERB B2, ERB B3) in colorectal adenocarcinomas and
adenomas. Tumour Biol. 21:105–115. 2000. View Article : Google Scholar
|
|
11
|
Milas L, Mason K, Hunter N, et al: In vivo
enhancement of tumor radioresponse by C225 antiepidermal growth
factor receptor antibody. Clin Cancer Res. 6:701–708.
2000.PubMed/NCBI
|
|
12
|
Harari PM and Huang SM: Head and neck
cancer as a clinical model for molecular targeting of therapy:
combining EGFR blockade with radiation. Int J Radiat Oncol Biol
Phys. 49:427–433. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dewdney A, Cunningham D, Tabernero J, et
al: Multicenter randomized phase II clinical trial comparing
neoadjuvant oxaliplatin, capecitabine, and preoperative
radiotherapy with or without cetuximab followed by total mesorectal
excision in patients with high-risk rectal cancer (EXPERT-C). J
Clin Oncol. 30:1620–1627. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shin HK, Kim M-S, Lee JK, et al:
Combination effect of cetuximab with radiation in colorectal cancer
cells. Tumori. 96:7132010.
|
|
15
|
Williams KJ, Telfer BA, Stratford IJ and
Wedge SR: ZD1839 (‘Iressa’), a specific oral epidermal growth
factor receptor-tyrosine kinase inhibitor, potentiates radiotherapy
in a human colorectal cancer xenograft model. Br J Cancer.
86:1157–1161. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Häggblad Sahlberg S1, Spiegelberg D,
Lennartsson J, Nygren P, Glimelius B and Stenerlöw B: The effect of
a dimeric Affibody molecule (ZEGFR:1907)2 targeting EGFR
in combination with radiation in colon cancer cell lines. Int J
Oncol. 40:176–184. 2012.
|
|
17
|
Tan F, Shen X, Wang D, et al: Icotinib
(BPI-2009H), a novel EGFR tyrosine kinase inhibitor, displays
potent efficacy in preclinical studies. Lung Cancer. 76:177–182.
2012. View Article : Google Scholar
|
|
18
|
Shi Y, Zhang L, Liu X, et al: Icotinib
versus gefitinib in previously treated advanced non-small-cell lung
cancer (ICOGEN): a randomised, double-blind phase 3 non-inferiority
trial. The Lancet Oncol. 14:953–961. 2013. View Article : Google Scholar
|
|
19
|
Santivasi WL and Xia F: Ionizing
radiation-induced DNA damage, response, and repair. Antioxid Redox
Signal. 21:251–259. 2014. View Article : Google Scholar
|
|
20
|
Qu YY, Hu SL, Xu XY, et al: Nimotuzumab
enhances the radiosensitivity of cancer cells in vitro by
inhibiting radiation-induced DNA damage repair. PLoS One.
8:e707272013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tanaka T, Munshi A, Brooks C, Liu J, Hobbs
ML and Meyn RE: Gefitinib radiosensitizes non-small cell lung
cancer cells by suppressing cellular DNA repair capacity. Clin
Cancer Res. 14:1266–1273. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Herbst RS and Bunn PA Jr: Targeting the
epidermal growth factor receptor in non-small cell lung cancer.
Clin Cancer Res. 9:5813–5824. 2003.PubMed/NCBI
|
|
23
|
Dittmann K, Mayer C, Fehrenbacher B, et
al: Radiation-induced epidermal growth factor receptor nuclear
import is linked to activation of DNA-dependent protein kinase. J
Biol Chem. 280:31182–31189. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sano D, Kawakami M, Fujita K, et al:
Antitumor effects of ZD6474 on head and neck squamous cell
carcinoma. Oncol Rep. 17:289–295. 2007.PubMed/NCBI
|
|
25
|
Rothkamm K and Löbrich M: Evidence for a
lack of DNA double-strand break repair in human cells exposed to
very low x-ray doses. Proc Natl Acad Sci USA. 100:5057–5062. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Burma S, Chen BP, Murphy M, Kurimasa A and
Chen DJ: ATM phosphorylates histone H2AX in response to DNA
double-strand breaks. J Biol Chem. 276:42462–42467. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mah LJ, Orlowski C, Ververis K, Vasireddy
RS, El-Osta A and Karagiannis TC: Evaluation of the efficacy of
radiation-modifying compounds using γH2AX as a molecular marker of
DNA double-strand breaks. Genome Integr. 2:32011. View Article : Google Scholar
|
|
28
|
Lobrich M, Shibata A, Beucher A, et al:
gammaH2AX foci analysis for monitoring DNA double-strand break
repair: strengths, limitations and optimization. Cell Cycle.
9:662–669. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ward IM, Difilippantonio S, Minn K, et al:
53BP1 cooperates with p53 and functions as a haploinsufficient
tumor suppressor in mice. Mol Cell Biol. 25:10079–10086. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bouwman P, Aly A, Escandell JM, et al:
53BP1 loss rescues BRCA1 deficiency and is associated with
triple-negative and BRCA-mutated breast cancers. Nat Struct Mol
Biol. 17:688–695. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nuciforo PG, Luise C, Capra M, Pelosi G
and d’Adda di Fagagna F: Complex engagement of DNA damage response
pathways in human cancer and in lung tumor progression.
Carcinogenesis. 28:2082–2088. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Clarke A, Jones N, Pryde F, Adachi Y and
Sansom O: 53BP1 deficiency in intestinal enterocytes does not alter
the immediate response to ionizing radiation, but leads to
increased nuclear area consistent with polyploidy. Oncogene.
26:6349–6355. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Morales JC, Franco S, Murphy MM, et al:
53BP1 and p53 synergize to suppress genomic instability and
lymphomagenesis. Proc Natl Acad Sci USA. 103:3310–3315. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li X, Xu B, Moran MS, et al: 53BP1
functions as a tumor suppressor in breast cancer via the inhibition
of NF-κB through miR-146a. Carcinogenesis. 33:2593–2600. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Park JS, Jun HJ, Cho MJ, et al:
Radiosensitivity enhancement by combined treatment of celecoxib and
gefitinib on human lung cancer cells. Clin Cancer Res.
12:4989–4999. 2006. View Article : Google Scholar : PubMed/NCBI
|