Introduction
Lung cancer has emerged as the third leading cause
of cancer-related mortality worldwide (1). In the USA, over 200,000 new lung
cancer cases are diagnosed annually, causing over 150,000 deaths in
one year (1). Approximately 85% of
lung cancer patients suffer from non-small cell lung cancer
(NSCLC). Due to the comparative therapeutic advantage,
cisplatin-based combination regimens are recommended as the optimal
choice currently for the majority of NSCLC patients indicative of
chemotherapy or adjuvant chemotherapy (2). However, the average survival time for
patients with advanced stage of NSCLC receiving cisplatin plus
gemcitabine treatment is ~16 months and may even be reduced to 12
months in those with cisplatin-resistance (3). The dilemma in managing late-stage
NSCLC requires the elucidation of the mechanisms in cisplatin
resistance to define novel, effective and applicable therapeutic
targets for lung cancer (2,3).
Several signal transduction pathways controlling
chemo-sensitivity are aberrantly activated in various types of
cancer, among which Wnt is of special significance (4). The role of Wnt1/Int-1 in the induction
of mouse mammary tumor as an integration site for mouse mammary
tumor virus (MMTV) was first recognized 30 years ago (4). Wnt/β-catenin signaling is initiated by
the binding of secreted Wnt proteins with the frizzled, a class of
seven-pass transmembrane receptors. Activation of the receptor
leads to the phosphorylation of the dishevelled protein which,
through its association with axin, prevents glycogen synthase
kinase 3β (GSK3β) from phosphorylating β-catenin and the negative
regulators axin and adenomatous polyposis coli (APC).
Unphosphorylated β-catenin escapes recognition, ubiquitinization
and degradation by β-TRCP, accumulates in the cytoplasm and
translocates to the nucleus, where it engages with transcription
factors such as TCF and LEF. The nuclear accumulation of β-catenin
switches on the TCF/LEF-controlled transcription of downstream
genes. Wnt effector genes include E-cadherin, MMP7, survivin, c-myc
and cyclin D1, all of which control the fate of cancer development,
progression and metastasis (4,5).
Abnormal activation of the Wnt/β-catenin signaling pathway has been
most extensively studied in colorectal neoplasias, and was realized
further in almost all types of solid organ and haematologic
malignancies in human beings, including lung adenocarcinoma
(6). Wnt inhibition, either by
monoclonal antibodies, small-interfering RNAs (siRNAs) targeting
Wnt components or the overexpression of Wnt antagonists, retards
lung cancer progression in various in vitro and in
vivo tumor models (5,6). Furthermore, inactivation of
Wnt/β-catenin signaling sensitizes chemotherapy by inducing
apoptosis and growth arrest in cancer cells, suggesting the
potential role of Wnt signaling inhibitors in the reversal of
cisplatin resistance (7–9).
As a soluble Wnt inhibitor, Dickkopf-3 (Dkk3)
is involved in molecular cancer therapy. Dkk3 binds with
LDL-receptor-related protein5/6 (LRP5/6) and destabilizes
cytoplasma β-catenin (10). Dkk3 is
downregulated in a variety of malignancies including hepatic
cancer, kidney carcinoma, urinary bladder cancer, pancreatic cancer
and lung cancer, earning its alias, the ‘reduced expression in
immortalized cells’ (REIC) (11).
Re-established Dkk3/REIC expression induces apoptosis in cancer
cell lines (12–14). Downregulation of Dkk3/REIC through
epigenetic hypermethylation is universal among lung cancer cell
lines and human lung cancer samples (15,16).
While DKK3 knockdown stimulates the proliferation of lung cancer
cells, DKK3 overexpression inhibits the growth of NSCLC cells by
inducing apoptosis and cell cycle disturbance via the
transactivation of c-myc and cyclin D1 through β-catenin/TCF-4
signaling (17). The
above-mentioned emphasize the tumor suppressive role of Dkk3 with
capacities of promoting apoptosis and inhibiting proliferation in
lung cancer cells (18). The
possible chemosensitizing effect of Dkk3 is suggested as its
homologue, Dkk1, has shown a proapoptotic activity that inhibits
the growth synergistically with cisplatin in cisplatin-resistant
head-neck and brain tumor cells (19,20).
The present study aimed to test the hypothesis that
Dkk3 may disturb the growth of lung cancer cells synergistically
with cisplatin. DKK3 expression levels in wild-type and
cisplatin-resistant NSCLC cell lines were subsequently transfected
with DKK3 siRNA or DKK3 gene. We then investigated whether the
downregulation or upregulation of DKK3 was able to disrupt the
growth of cisplatin-resistant NSCLC cells treated with cisplatin
in vitro and in vivo. To determine the possible
chemosensitization mechanisms of Dkk3 functioning, a
small-molecular Wnt activator was allocated prior to cisplatin
treatment (21). Then we compared
the biological behavior of cisplatin-resistant NSCLC cells with or
without DKK3 transfection, and the expression profile of genes and
proteins downstream of the Wnt signaling pathway, which possibly
regulate the cell cycle, cell proliferation and apoptosis. The
results may provide substantial evidence in support of the
therapeutic value of DKK3 for lung cancer refractory to
chemotherapy.
Materials and methods
Cell culture
Immortalized HEK293 and A549, Calu1 and H460 human
lung adenocarcinoma cell lines were purchased from the American
Type Culture Collection (ATCC; Manassas, VA, USA). These cell lines
were tested and authenticated prior to using short tandem repeat
(STR) loci by the Cell ID System (Progema, Madison, WI, USA).
Cisplatin-resistant cell sublines, A549cis, Calu1cis and H460cis,
were generated by continuous exposure to increasing concentrations
of cisplatin (from 10 to 50 μM) for 12 months as previously
described (21). The cells were
incubated at 37°C with 5% CO2 in atmosphere and grown in
RPMI-1640 supplemented with 10% fetal bovine serum, 1%
penicillin/streptomycin, and 5% MEM non-essential aminoacids
(HyClone Laboratories, Waltham, MA, USA). The medium was restored
three times a week, and subcultures were performed prior to cells
reaching 60–70% confluence. To identify the impact of GSK3β
inactivation on the concentration-effect curves with cisplatin,
A549cis and A549cis-DKK3 cells were treated with a GSK3β inhibitor
SB216763 [3-(2, 4-dichlorophenyl)-4-(1-methyl-1H-indol-3-y
l)-1H-pyrrole-2,5-dione] 2 h prior to cisplatin exposure (22).
DKK3 RNA interference
siRNA of DKK3 and a control scrambled RNA targeting
a sequence not sharing homology with the human genome (negative
control) was synthesized (Shanghai Invitrogen Ltd., Shanghai,
China). The oligonucleotide sequences targeting DKK-3 were:
5′-AAUGGUCUGGU ACUUAUUCCdGdC-3′ (forward), and 3′-dCdGUUACCAG
ACCAUGAAUAAGG-5′ (reverse). Calu1 and H460 lung carcinoma cells
were incubated with siRNAs and control scrambled RNA using the
Lipofectamine RNAiMAX reagent (Shanghai Invitrogen Ltd.). The
procedures were performed according to the manufacturer’s
instructions. After transfection for 6 h, the transfection mixture
was removed and the cells were incubated for 1–3 × 24 h prior to
detection.
Plasmid construction and DKK3
transfection
Genomic DNA was isolated from A549 lung cancer cell
lines using the All Prep DNA/RNA Mini Kit (Qiagen). The bisulfite
modification of genomic DNA was carried out with the EpiTect
Bisulfite kit (Qiagen) according to the manufacturer’s
instructions. Primers for bisulfite genomic sequencing PCR were
designed manually or by using the online MethPrimer program. The
DKK3 primers used were: forward, 5′-GGAGGAGGTTATTTTT AATGAGATGT-3′
and reverse, 5′-TCCAAACTTTTTAACA AAAAAACAAA-3′. The amplification
products were sequenced directly by an outside vendor (McLab, San
Francisco, CA, USA). The full-length cDNA was integrated into a
cosmid vector pAxCAwt and transferred into an adenovirus vector by
the COS-TPC method (Takara Bio, Shiga, Japan). An adenovirus vector
carrying the LacZ gene was used to monitor infection efficiency.
A549 and cisplatin-resistant A549cis lung carcinoma cells were
incubated with DMSO, Ad-DKK3 or Ad-GFP at various multiplicity of
infections (MOIs) (ranging from 0, 1, 5, 10, 20, 50 to 100). The
procedures were performed according to the manufacturer’s
instructions. After incubation for 6 h, the transfection mixtures
were removed. Clonal cells were nominated as A549-AdV, A549-DKK3,
A549cis-AdV and A549cis-DKK3. The cells were incubated for another
1–6 × 24 h prior to detection or in vivo injection.
Reverse transcriptase-polymerase chain
reaction (RT-PCR)
Total RNA was isolated using TRIzol reagent
(Invitrogen) according to the manufacturer′s instructions. The
primer sequences used were: DKK3, 5′-ATGCAGCGGCTTGGG
GCCACCCTGCTGTGC-3′ (forward), and 5′-GATGGTCCCA
TTGCTGCCCCTGGTGGCCAT-3′ (reverse); Wnt1, 5′-TGGT
TTGCAAAGACCACCTCCA-3′ (forward), and 5′-TGATTCC AGGAGGCAAACGCAT-3′
(reverse); Wnt3a, 5′-TGGTTTGC AAAGACCACCTCCA-3′ (forward), and
5′-TGATTCCAGG AGGCAAACGCAT-3′ (reverse); c-myc, 5′-CTGCGACGAG
GAGGAGGACT-3′ (forward), and 5′-GGCAGCAGCTCG AATTTCTT-3′ (reverse);
E-cadherin, 5′-TCCCATCAGC TGCCCAGAAA-3′ (forward), and
5′-TGACTCCTGTGT TCCTGTTA-3′ (reverse); and MMP-7, 5′-GGTCACCTACAG
GATCGTATCATAT-3′ (forward), and 5′-CATCACTGCAT TAGATCAGAGGAA-3′
(reverse). cDNA synthesis was normalized by PCR with GAPDH primers:
5′-CATCACTGC CACCCAGAAGA-3′ (forward), and 5′-TGAAGTCGCAGGA
GACAACC-3′ (reverse). PCR conditions were as follows: 45 cycles of
30 sec at 95°C, 30 sec at 58°C, and 60 sec at 72°C. Specificity of
amplification products was verified by agarose gel
electrophoresis.
Western blot analysis
Antibodies against β-catenin, Fas, caspase-3,
cleaved caspase-3, caspase-8, cleaved caspase-8, survivin, β-actin
and poly(ADP-ribose) polymerase (PARP) were produced by Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). Protein lysate was
separated electrophoretically on denaturing SDS-polyacrylamide gel,
transferred to nitrocellulose membranes, and probed with goat
polyclonal IgG antibodies (1:200 or 1:250 diluted). Blots were
exposed to secondary antibodies and detection was performed using
an enhanced chemiluminescence reagent (Amersham). For the loading
control, the membrane used in the initial western blotting was
placed in Restore Western blotting Stripping Buffer (Thermo
Scientific) for 15 min to remove the antibody (primary and
secondary antibodies). The membrane was then washed with water for
5 min, blocked with 5% milk for 1 h, and probed with β-actin.
Cell proliferation assay
Cell viability/proliferation assays were performed
using the Cell Counting Kit-8 assay (CCK-8; Dojindo Molecular
Technologies, Shanghai, China) which uses the bioreduction of WST-8
to orange-colored formazan to measure cell viability. Briefly, 100
μl of cells at 2×104/ml cells were plated on 96-well
culture plates. NSCLC cells transfected with DKK3siRNA or DKK3 were
cultured in cisplatin (Sigma-Aldrich, St. Louis, Mo, USA). At
various time-points over the next 72 h, the culture medium in
quadruplicate wells was replaced with fresh medium containing 5%
CCK-8 reagent, and after 1 h, absorbance at 450 nm was measured on
a Multiskan Plus (MTX Lab Systems®, Vienna, VA, USA)
plate reader. Absorbance values were corrected by subtracting those
of blank control wells that did not have cells. The linear
regression equations that correlated log (cisplatin concentration)
with cell viability were established by linear regression. The
IC50 values (50% inhibitory concentration) of cisplatin
for each condition were then calculated by applying the logarithmic
equations. The initial cisplatin concentration gradient was 0,
0.05, 0.1, 0.2, 0.4, 0.8, 1.6, 3.2, 6.4, 12.8, 25.6 and 51.2 μM,
and was subsequently adjusted based on the IC50
value.
Transwell invasion assay
Cell invasion assays were performed using 24-well
Transwell (Corning Life Sciences) coated with Matrigel (1 mg/ml, BD
Sciences). Cells (104/well) were seeded in the upper
chambers of the wells in 200 μl FBS-free medium, and the lower
chambers were filled with 500 μl 10% FBS medium to induce cell
migration. Following incubation for 24 h, the cells on the filter
surface were fixed with 4% formaldehyde, stained with 0.5% crystal
violet, and examined under a microscope. Cells in at least five
random microscopic fields (×200) were counted.
Immunofluorescence
The expression of Dkk3 or β-catenin in NSCLC cells
was investigated by immunofluorecent staining. Cells were plated
onto a cover glass (1×1 cm) placed in a 6-well culture plate at a
density of 1×105 cells/well. Cover glasses were washed
with 0.1 M phosphate-buffered saline (PBS), fixed in ice-cold
acetone for 5 min, and soaked in PBS containing 0.2% Triton X-100
for 5 min to increase their permeability to primary antibodies. The
glasses were rinsed in PBS for 15 min and blocked with 5% bovine
serum albumin at room temperature for 30 min, then conjugated with
primary antibodies (1:200) overnight at 4°C. After washing three
times with PBS, the cover glasses were incubated with fluorescein
isothiocyanate (FITC)-conjugated secondary antibodies (1:40) at
room temperature for 4–6 min. The glasses were then double-stained
with 0.5 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) for 5 min, and
sealed with glycerol. Immunofluorescent images were captured using
a Nikon C1si multifocal one-photon laser scanning microscope (Nikon
Ltd., Tokyo, Japan) within 30 min. Primary and secondary antibodies
were purchased from Santa Cruz Biotechnology, Inc. DAPI was from
Sigma-Aldrich Co.
Flow cytometric analysis
The cells were plated at a density of
1×105 cells/well in 6-well plates. The cell cycle
distribution of cells stained with propidium iodide (PI) was
analyzed. Apoptotic events were measured by Annexin V-FITC double
staining according to the manufacturer’s instructions (Nexins
Research, Kattendijk, The Netherlands). Analyses were performed on
a FACSCalibur instrument using the CellQuest or the ModFit 3.0
software packages (Becton-Dickinson, Mount View, CA, USA).
Apoptosis was also manifested by detecting PARP cleavage by
immunoblot as mentioned above.
In vivo tumorigenesis
Experimental procedures were approved by the Animal
Care and Use Committee of Zhengzhou University, and were in
accordance with the Guide for the Care and Use of Laboratory
Animals issued by the USA National Institutes of Health.
Four-week-old male nu/nu nude mice, weighing 19.3±2.2 g, obtained
from the Shanghai Institute of Drug of Chinese Academy of Sciences,
were housed in specific pathogen-free conditions. A549cis,
A549cis-AdV or A549cis-DKK3 cells were grown to near confluence,
digested and resuspended in PBS. PBS (0.1 ml) containing
1×106 cells was then injected subcutaneously into the
flanks of nude mice. Cisplatin was administered via intraperitoneal
injection at doses of 2.5 mg/kg/day for 5 days/week. The tumor size
was measured using a caliper. The tumor volume was calculated
according to the formula: V = 1/6 π ab2 (π, 3.14; a,
long axis; and b, short axis of the tumor). Growth curves were
plotted from the mean tumor volume ± SD from 10 animals in each
group. Six weeks after the injection, the animals were sacrificed
and tumors were harvested, measured, weighed and fixed in 10%
formalin. The wet tumor weight of each animal was calculated as
means ± SD from 10 animals in each group.
Statistical analysis
Data were analyzed by the SPSS 15.0 software (SPSS,
Inc., Chicago, IL, USA) and SigmaPlot 12.0 (Systat Software Ltd.,
Chicago, IL, USA). The Student’s t-test and one-way ANOVA were used
to examine the differences between or among groups. The
Kruskal-Wallis rank sum test was used alternatively when data were
inappropriate for ANOVA analysis (e.g., without homogeneity of
variance). Statistical tests were two-sided. P<0.05 was
considered to indicate a statistically significant result.
Results
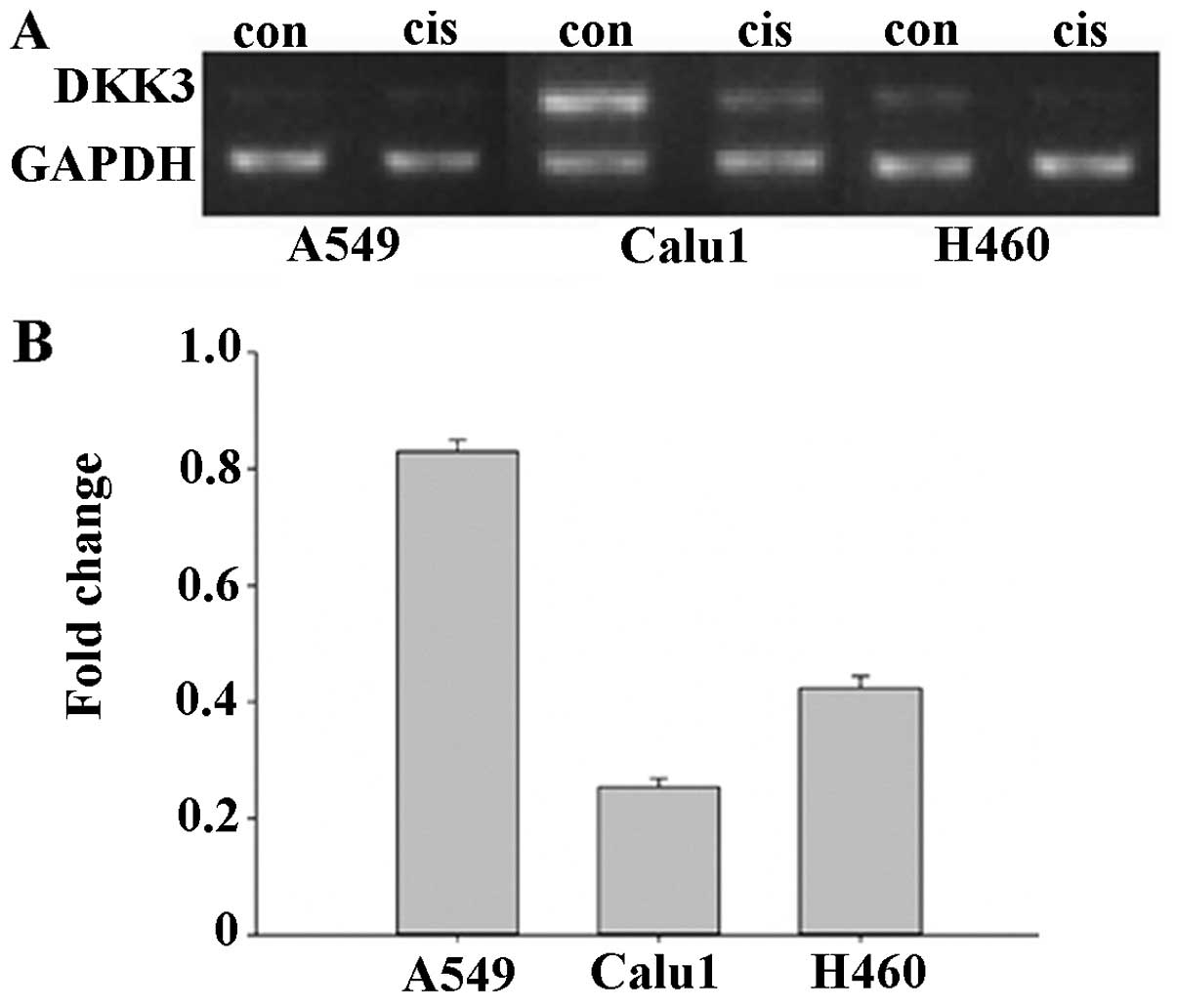
Decreased expression of DKK3 is
associated with cisplatin resistance in lung cancer cells
The cisplatin-resistant A549cis, Calu1cis and
H460cis NSCLC sublines, were characterized by the WST-8 viability
assay. Dkk3 expression was markedly reduced in Calu1cis (0.23-fold
compared with Calu1) and less in H460cis (0.41-fold compared with
Calu1) (Fig. 1). A549 expressed low
levels of Dkk3, with RT-PCR analysis shown a 1.2-fold decrease in
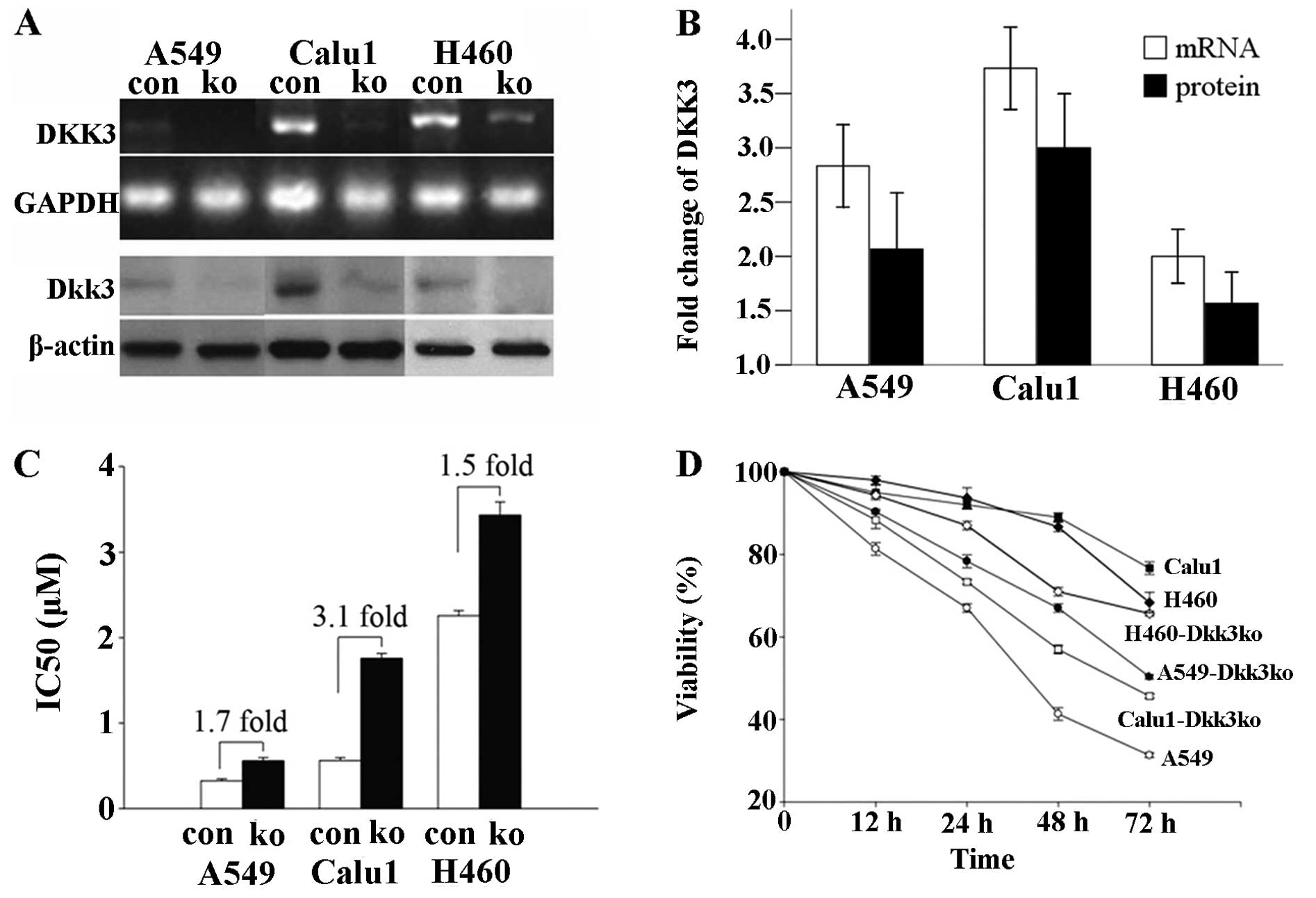
DKK3 mRNA with A549cis (data not shown). The impact of DKK3
knockdown (by siRNA trans-fection) on cisplatin-induced growth
arrest was determined. Compared to scrambled RNA transfection, DKK3
knockdown ameliorated cisplatin-induced growth arrest in the three
NSCLC cell lines, and rendered them resistant to cisplatin
(Fig. 2A and B). As shown in
Fig. 2C and D, the viability and
proliferation index of Calu1-DKK3ko and H460-DKK3ko were increased
as compared with Calu1 and H460 at the same cisplatin
concentrations (P<0.05 for both).
DKK3 transfection inhibits NSCLC cell
growth
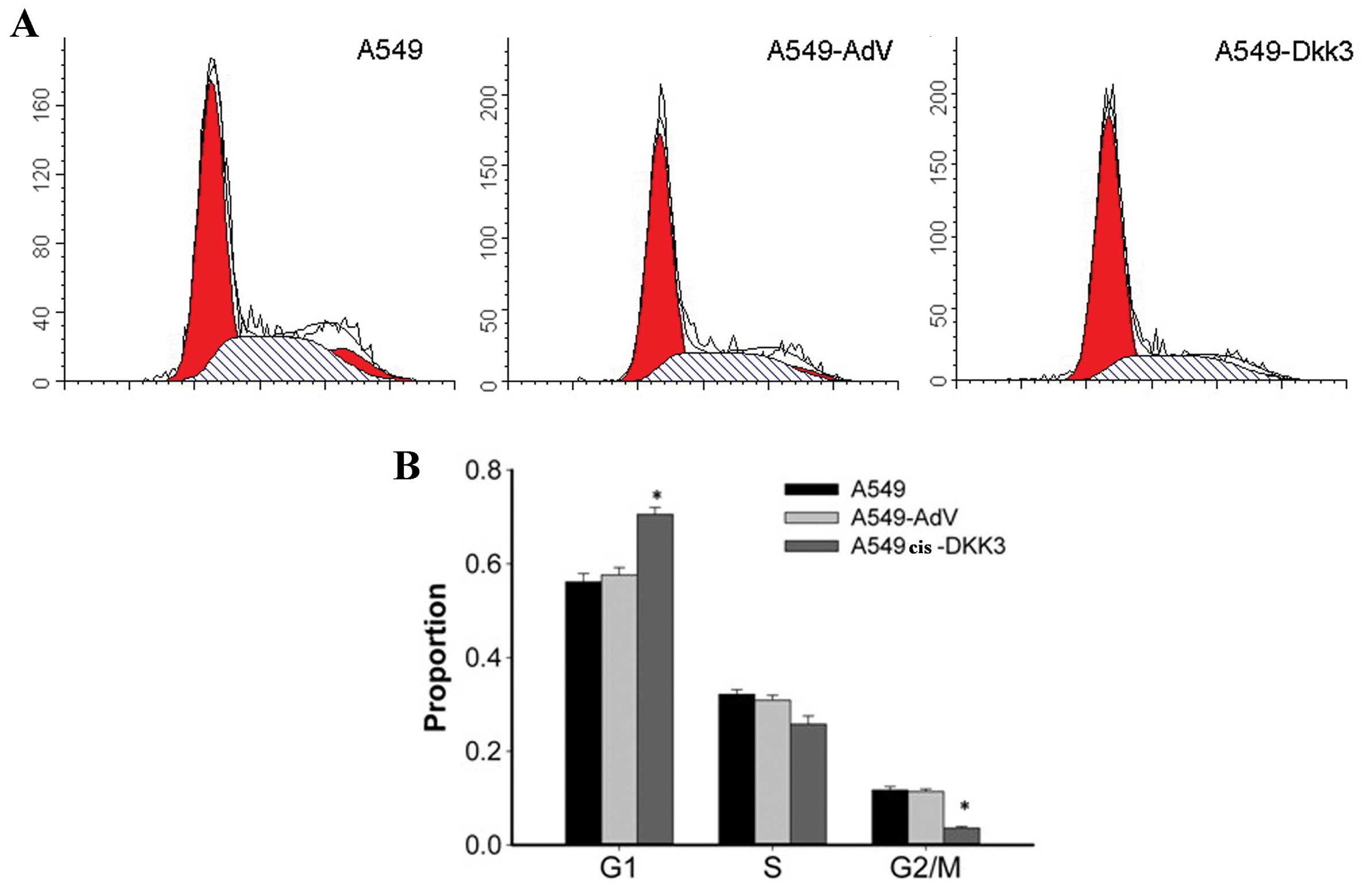
As shown in Fig. 3,
the DKK3 transgene altered the cell cycle in A549 cells, and
attenuated A549 cell mobility. An increase in the portion of cells
in the G0/G1 phase of the cell cycle by 17–20% in A549cis cells
transfected with DKK3 gene and with a corresponding decrease in
cells in the S and G2-M phase was observed. By contrast, for
A549cis transfected with the empty vector, no significant changes
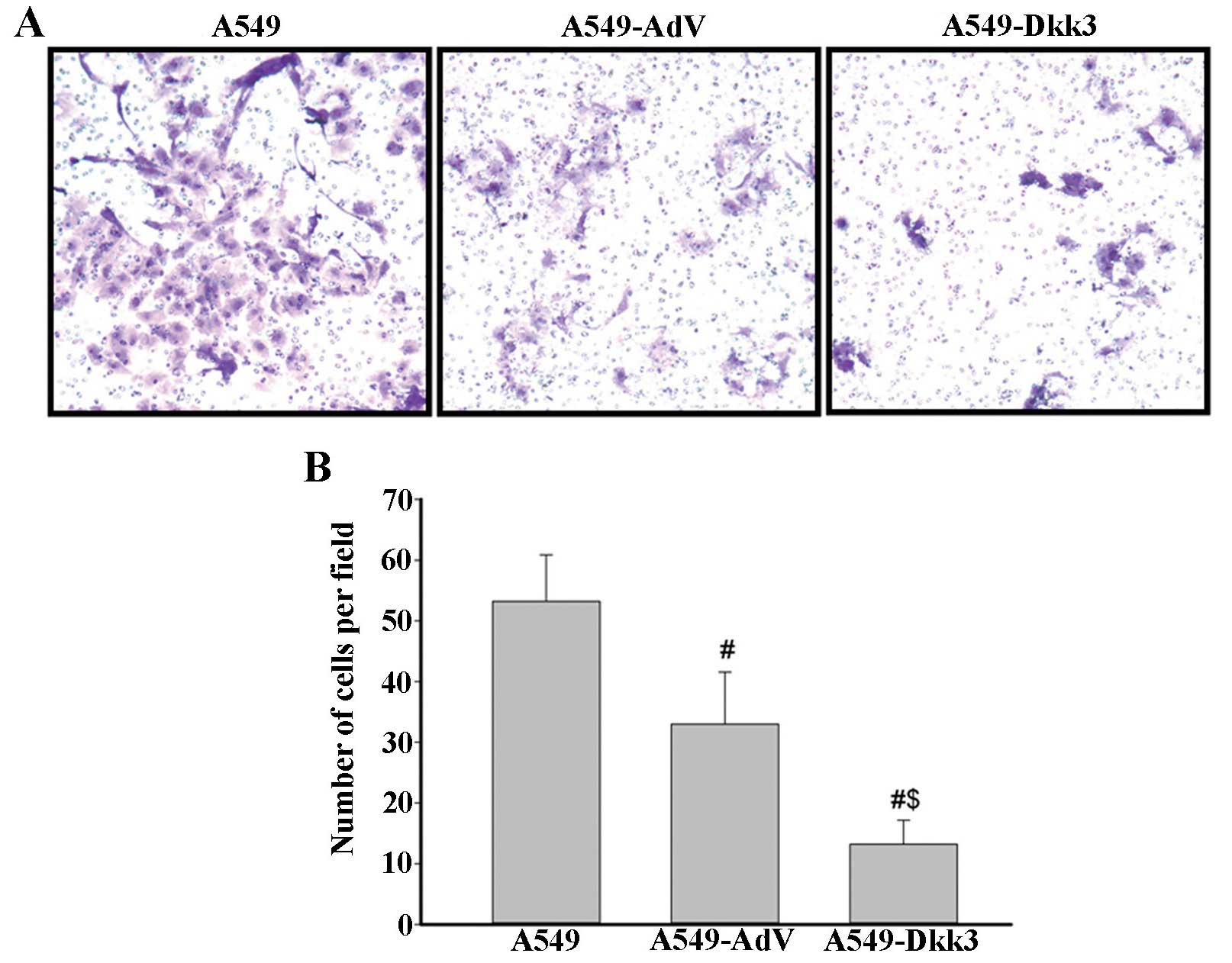
in cell cycle distribution were detected (Fig. 3). Cell invasion assay was performed
using a 24-well Transwell. Ectopic expression of DKK3 significantly
reduced the number of cells migrating through the Matrigel-coated
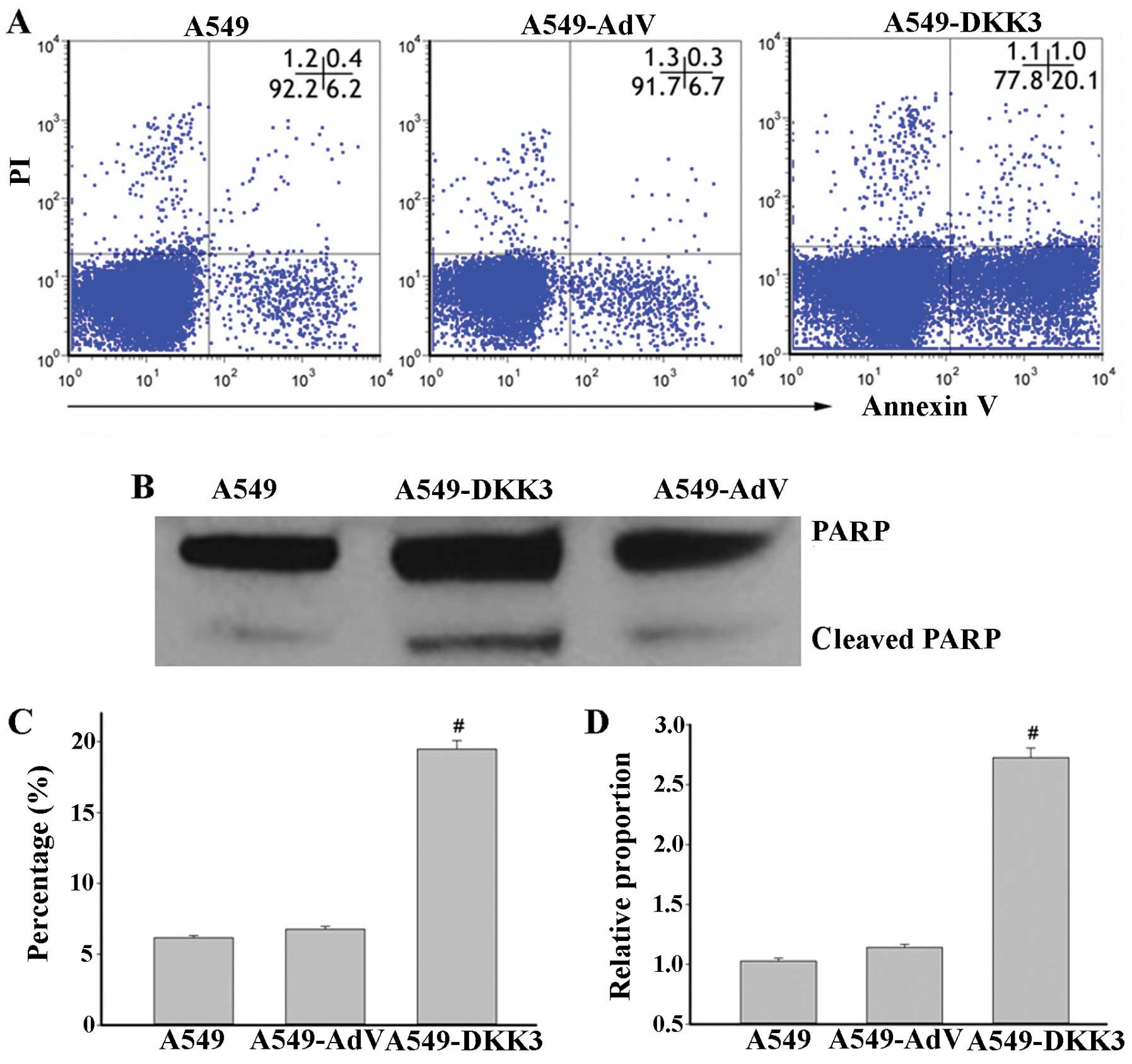
filters (Fig. 4). Cell apoptosis
was assayed by flow cytometry with Annexin V-FITC staining.
Overexpression of DKK3-induced apoptosis of lung cancer cells as
compared to scramble sequence-transfected cells in A549 cell lines
(Fig. 5A and C). The cleaved PARP
level was also significantly elevated with DKK3 transfection
(Fig. 5B and D). These data suggest
that DKK3 alone inhibits NSCLC cell growth.
DKK3 transfection plus cisplatin
synergistically suppresses the growth of resistant NSCLC cells in
vitro and in vivo
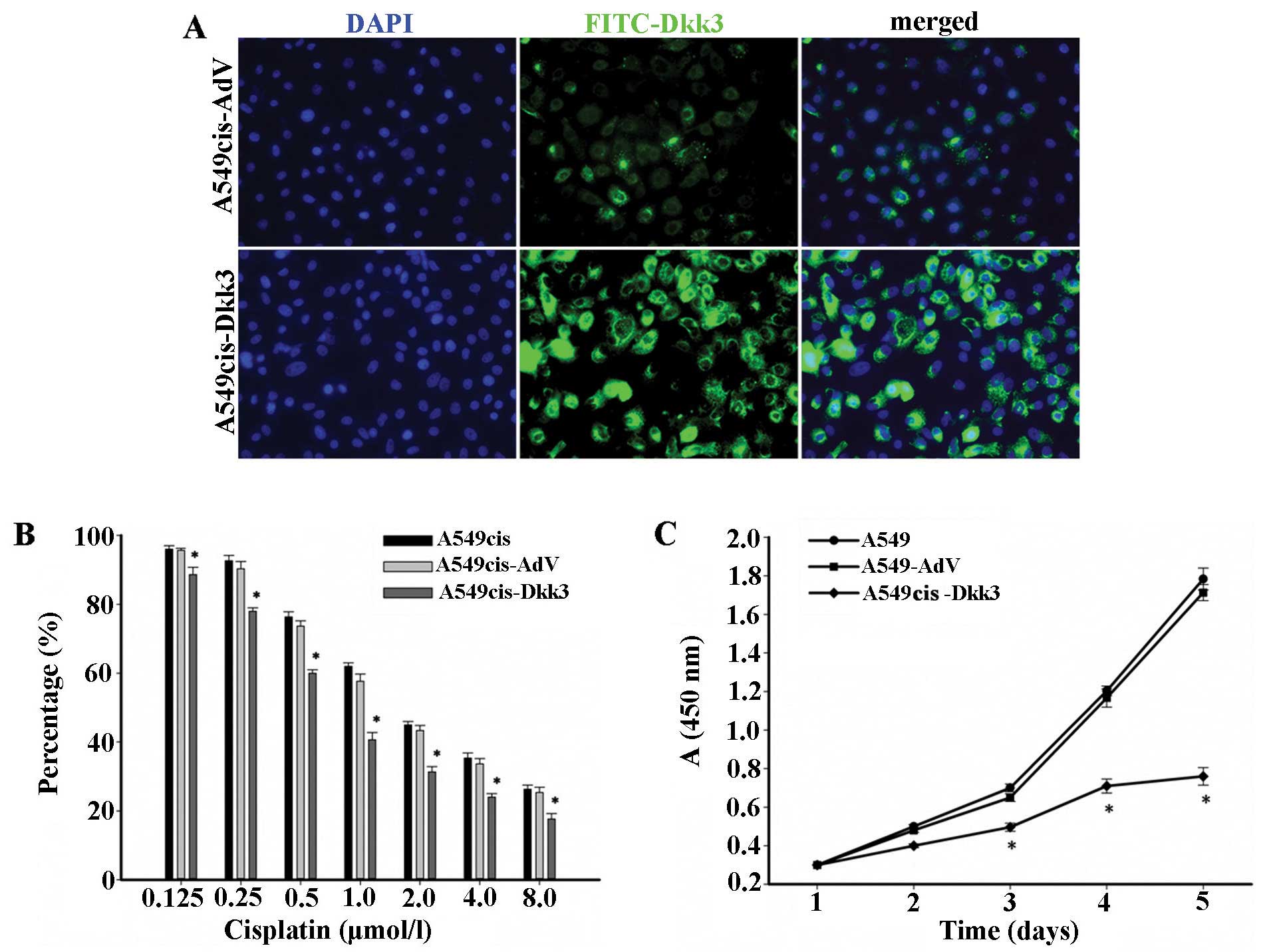
We transfected A549cis with DKK3, and assessed the
efficiency of DKK3 transfection by RT-PCR and immunofluorescence.
As a consequence of DKK3 transfection, DKK3 was localized mostly at
the cytoplasm, and elevated in A549cis lung cancer cells, as shown
at immunofluorescence (green) (Fig.
6A). DKK3 transgene increased the sensitivity to cisplatin in
A549cis cells (Fig. 6B and C). The
dose of cisplatin employed in the xeno-graft tumor model was
optimized by a dose-finding pre-study, in which xenografted
tumor-bearing mice were treated with cisplatin (0.5, 1.0, 2.0, 4.0
or 8.0 mg/kg/day) by i.p. injection of 1×106 A549cis
cells. The mice treated with 2.0 mg/kg/day of cisplatin showed a
moderate tumor growth delay and retained a survival rate of ~60% to
the 4-week treatment. The dose of cisplatin was then optimized to
2.0 mg/kg/day, once a day, 5 days a week for an additional
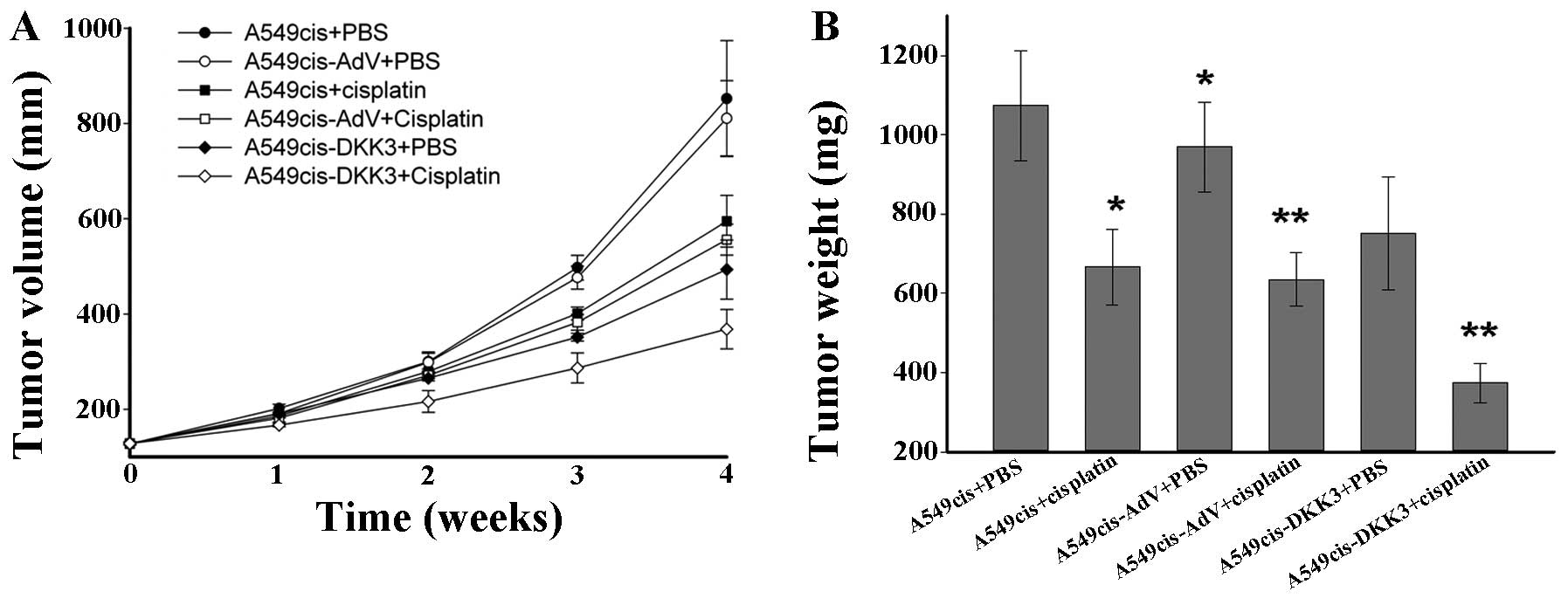
consecutive 4 weeks. Nude mice were randomized to six groups: the
A549cis+PBS, the A549cis+cisplatin, the A549-AdV+PBS, the
A549cis-AdV+cisplatin, the A549cis-DKK3+PBS and the
A549cis-DKK3+cisplatin groups. The tumor growth in vivo was
monitored daily and tumor volume and weight were measured. As shown
in Fig. 7, tumor growth was more
significantly retarded in the A549cis-DKK3+cisplatin group than the
A549cis-AdV+cisplatin and A549cis-DKK3+PBS groups, suggesting a
synergistic antitumor effect of DKK3 transfection with cisplatin in
cisplatin-resistant lung cancer cells in vivo.
DKK3 inhibits survivin expression via the
Wnt/β-catenin pathway in lung cancer cells
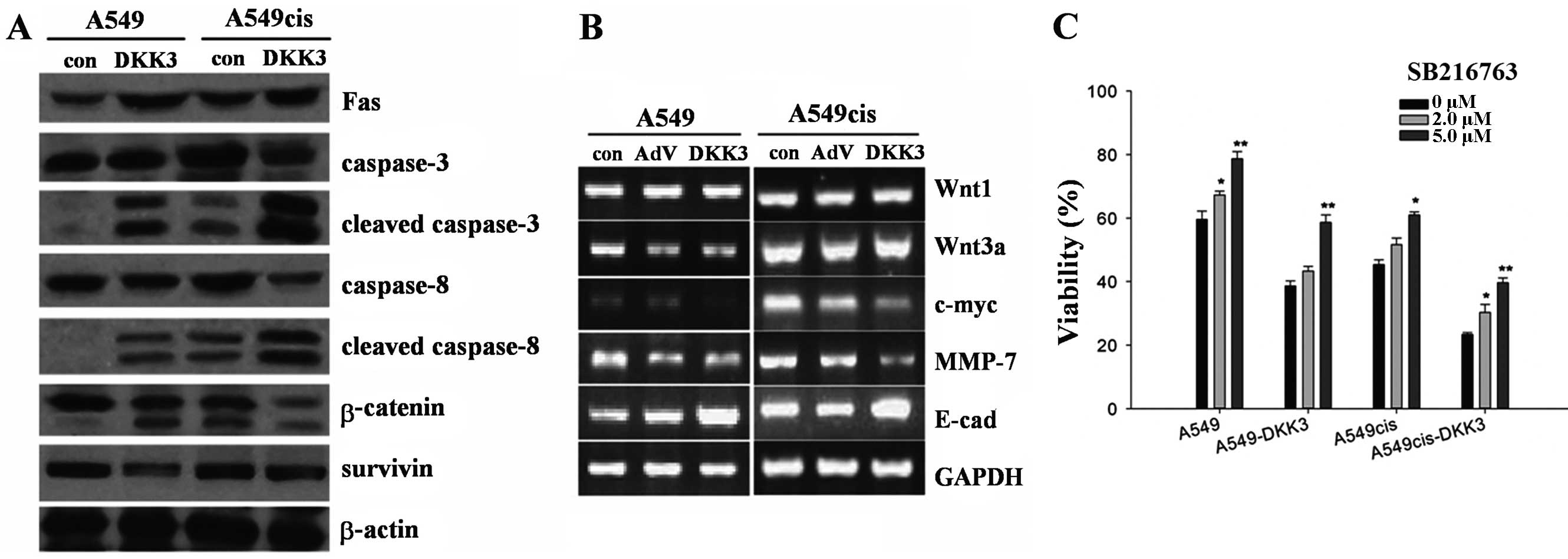
DKK3 transfection significantly enhanced the
expression of DKK3. DKK3 was localized at the cytoplasm. To analyze
the impact of DKK3 transfection on Wnt signaling, we compared the
genes and/or proteins controlled by Wnt signaling or associated
with cell cycle, growth and invasion in cultured A549 cells, as
shown in Fig. 8A and B. DKK3
suppressed Wnt signaling through β-catenin degradation, but not
directly through Wnt. As shown by the western blot analysis, DKK3
inhibited β-catenin expression and simultaneously enhanced
phosphorylation of β-catenin (Fig.
8A). DKK3 transfection also downregulated the expression of
survivin (Fig. 8A). Results of the
RT-PCR assay, showed that DKK3 overexpression was responsible for a
decrease of c-myc and MMP7, a moderate increase of E-cadherin and a
mild increase of β-catenin, although fewer changes for the WNT1 and
WNT3a expression were evident (Fig.
8B). To analyze the effect of GSK3β inactivation on the
concentration-effect curves with cisplatin, A549cis and
A549cis-DKK3 cells were treated with a GSK3β inhibitor SB216763 2 h
prior to cisplatin exposure. Viability was assayed 72 h later.
SB216763 treatment is dose-dependently associated with an increased
viability in the two cell types (Fig.
8C).
Discussion
Cisplatin chemotherapy is sensitized by antagonizing
aberrantly activated Wnt signaling in lung cancer. Incubation with
anti-DKK1 antibody induces apoptosis in A549 cells through the
caspase-dependent pathway in vitro and suppresses the growth
of A549 and H2170 lung cancer cells in nude mice (23). The tumor suppressor role of Dkk3 in
NSCLC is twofold: DKK3 knockdown accelerates NSCLC cell
proliferation, whereas Dkk3 overexpression hinders the growth of
NSCLC cells through the induction of apoptosis and cell cycle
disturbance (15–18). It is thus conceivable that Dkk3, a
Wnt inhibitor harboring tumor-suppressive activity, may exert
similar functions such as its homologue Dkk1, which hinders NSCLC
cell growth synergistically with cisplatin (18). To the best of our knowledge however,
the present study is the first one to show an association of Dkk3
and cisplatin resistance in lung cancer. This association is
demonstrated by: i) cisplatin-resistant NSCLC cells expressing low
levels of DKK3 (Fig. 1); ii)
siRNA-mediated knockdown of DKK3 decreasing the sensitivity of
NSCLC cells to cisplatin (Fig. 2);
iii) DKK3 transfection enhancing cisplatin treatment by inhibiting
the growth of and inducing apoptosis in cisplatin-resistant NSCLC
cells (Figs. 6 and 7).
Dkk3 is also known as the ‘reduced expression in
immortalized cells’ (REIC). The dysregulated functioning status of
Dkk3 contributes to carcinogenesis in the lung, as DKK3 is
downregulated in immortalized NSCLC cell lines and human lung
cancer tissues (11). DKK3
hypermethylation is universal among lung cancer cell lines and
human lung tissue samples and is associated with a poor prognosis
(15,16,24).
Our data show that Dkk3 expression is further suppressed in NSCLC
cell lines with chemoresistance obtained by chronic cisplatin
exposure, however the manner in which Dkk3 is reduced is unclear.
One possible explanation is the feedback modulating the
Wnt/β-catenin pathway, i.e., the Wnt/β-catenin pathway is activated
in cisplatin resistance and, activation of Wnt/β-catenin signaling
in turn reduces DKK1 expression (25). It is unknown whether DKK3 expression
is also negatively regulated by Wnt/β-catenin signaling, however
the determinant mechanism should be investigated (10).
The role of Wnt/Dkk3 signaling has been well
documented in chemoresistance. To the best of our knowledge, for
the first time it has been shown that Dkk3 enhances apoptosis and
retards growth alone or synergistically with cisplatin in resistant
NSCLC cells. This is explained by the direct induction of caspases
and indirect inhibition of apoptosis antagonists, with both
mechanisms being Wnt-dependent. This study has shown that Wnt
inhibition by Dkk3 increased caspase-3, -8 and -9 and decreased
c-myc, cyclin D1 and survivin in NSCLC cells. Wnt1 inhibited
cytochrome c release and subsequent caspase-9 activation in
colorectal cancer cells treated with chemotherapeutic agents,
rendering them resistant to chemotherapy-mediated apoptosis,
whereas chemosensitivity was regained through transcriptional
blockage using a dominant-negative mutant of Tcf-4 (26). c-Myc is a nuclear phosphoprotein
that responds directly to mitogenic signals and plays a critical
role in the cell-cycle progression, particularly during the
transition from the G1 to the S phase (27). Wnt signaling promotes oncogenic
transformation by inhibiting c-myc-regulated apoptosis (28,29).
Cyclin D1 is a rate-limiting signal in the G1-S phase transition
which controls the length of the cellular proliferation cycle. Wnt
activation triggers the excessive expression of cyclin D1, which
has been observed in lung cancer cells separated from patients as
well as in immortalized cell lines, and is responsible for rapid
growth and proliferation in lung cancer cells (30,31).
Survivin, also known as the baculoviral inhibitor of apoptosis
repeat-containing 5 (BIRC5), is a member of the inhibitor of
apoptosis (IAP) family identified in the majority of human tumors.
Survivin binds with caspase-3 and -7 and attenuates Bax-, Fas- and
etoposide-induced apoptosis in cancer cell lines (32,33).
As it is abnormally transactivated in lung cancer cells, survivin
has been used in the clinic as a prognostic factor and a
therapeutic target using antisense oligonucleotides, siRNA,
ribozymes, immunotherapy and small molecules (34). Antisense oligonucleotides targeting
survivin induces apop-tosis and sensitize lung cancer cells to
chemotherapy (35). Wnt/β-catenin
signaling promotes survivin expression, as a Wnt2 antibody induces
apoptosis in A549 cells via the inactivation of survivin (36). Cisplatin is a cytotoxic agent
targeting DNA, which suppresses RNA transcription and induces
cell-cycle derangement and apoptosis. Cisplatin resistance in
vivo is largely attributed to increased DNA repair, imbalance
of pro-apoptotic-anti-apoptotic signals, which are mediated by Wnt
signaling as aforementioned (37,38).
The current study shows that Dkk3 downregulated survivin in lung
cancer cells via Wnt inhibition, which was accompanied with an
elevated expression of pro-apoptotic caspase-3, -8 and -9 and may
account for the apoptosis-inducing capabilities of Dkk3. This, Wnt
blockage by Dkk3 results in a shift favoring a pro-apoptotic
protein expression and discouraging anti-apoptotic proteins,
particularly survivin, which disrupts the
pro-apoptotic-anti-apoptotic balance and accounts for the
chemosensitizing activity of Dkk3.
It is noteworthy that Wnt signaling defines the fate
of cancer stem cells (39).
Wnt/β-catenin signaling controls proliferation, clone formation,
migration and drug resistance abilities as well as the expression
of cyclin D1 and OCT-4 in A549-derived lung cancer stem cells
(40). In addition, survivin
expression is downregulated by β-catenin/TCF-4 signaling inhibition
with APC, another Wnt inhibitor, which increases apoptosis of
cancer stem cells and other proliferative cells in the lower crypt,
thereby preventing the expansion of cancer stem cells and
initiation of colon carcinogenesis (41). Cancer stem cells are considered to
be refractory to chemotherapy and radiotherapy, which largely
results in the treatment failure of cancer (42). In this regard, the inhibition of
Wnt/β-catenin signaling by Dkk3 may change the style of survivin
expression as well as proliferation, survival and chemosensitivity
of lung cancer stem cells. This is an important issue requiring
further investigations, as our experiment does not provide direct
rationale thereof.
The Dkk family members include Dkk1, 2, 3 and 4,
which may be divided into two groups. While Dkk2 activates the
Wnt/β-catenin signaling pathway (the canonical Wnt signaling
pathway), Dkk1, 3 and 4 inactivate Wnt signaling. Dkk3 primarily
binds with LRP5/6 and acts as an antagonist of the Wnt/β-catenin
signaling pathway (the canonical Wnt signaling pathway) (4,10,18).
It is also reported that Dkk3 inhibits other Wnt transduction
pathways such as the Wnt/JNK signaling pathway in tumor cells
(43). In the present study, DKK3
overexpression attenuated β-catenin nuclear translocation, as well
as the expression of c-myc, cyclin D1 and survivin, which are
downstream genes controlled by Wnt/β-catenin/TCF (LEF) signaling.
In addition, the chemosensitizing effect of DKK3 was blocked by Wnt
activation with a GSK3 inhibitor SB216763 which stabilizes
β-catenin prior to cisplatin exposure (Fig. 8C). The overall data demonstrate that
Dkk3 sensitized cisplatin fundamentally through inhibition of the
Wnt/β-catenin signaling pathway. However, this does not
sufficiently exclude other mechanisms, since a number of apoptotic
signaling pathways are involved in the progression of lung cancer
(37,38). In another respect, Dkk3 regulates
cell proliferation and apoptosis in a complex manner, as Dkk3 may
also directly or indirectly modulate apoptosis via the
mitochondrial pathway and c-Jun N-terminal kinase (JNK) signaling
pathway (44–46). The genome-wide assay has also shown
that Wnt inhibition by β-catenin knockout alters over 130 gene
expressions and regulates the PI3K/Akt, NF-κB and p53 pathways
which play a crucial role in cell apoptosis (47). Additional studies may identify the
exact mechanism of Dkk3 involved in the reversal of cisplatin
resistance.
Results in this study are controversial. Although
considered a tumor suppressor, the inactivation of Dkk3 alone is
not sufficient for tumor initiation as the knockout of Dkk3 in mice
did not increase tumor incidence (48). Moreover, instead of favoring tumor
growth as assumed, Dkk3 knockdown favors apoptosis as in the case
of Dkk3 overexpression in NSCLC cells (10,15–18,49).
According to Jung et al, siRNA-mediated DKK3 knockdown
enables H460 cells to detach from the bottom of the culture plate,
as well as to initiate the apop-totic process as marked by an
increase of cyclin-dependent kinases (CDK) D1 and E. DKK3 knockdown
also increases the intracellular levels of reactive oxygen species
(ROS) and the expression of P53, p21 and Bax (49). These results suggest that DKK3 with
undetectable levels may act as a proapoptotic signal by regulating
the ROS-mediated expression of P53 and other pro- or anti-apoptotic
proteins. The inconsistency between different studies necessitates
future investigations to delineate the mechanisms by which Dkk3
regulates Wnt/β-catenin signaling as well as chemosensitivity.
Efforts should be made to narrow the gap between building a
therapeutic target in the bench (animal and cell) study, and the
eventual usage in the bedside towards NSCLC.
Taken together, we report on the effect of Dkk3
synergistically with cisplatin on inhibiting NSCLC cell growth,
motility in soft agar and inducing apoptosis via inhibition of the
Wnt/β-catenin signaling pathway. This effect is possibly attributed
to the reactivation of apoptotic pathways by the attenuation of
survivin expression. Based on its chemosensitizing potential, Dkk3
is a promising target for the development of therapeutic and
preventive strategies against cisplatin-resistant NSCLC. Studies
should also be conducted to delineate the exact action mechanisms
and to realize the beneficial use of Dkk3 clinically in patients
with cisplatin-resistant NSCLC.
Acknowledgements
The present study was sponsored by the Chinese
Ministry of Education Grant WKJ-2009-2-013, and a research fund
from the Ministry of Health of Henan Province. The authors would
like to thank Zhao-Gang Dong and Li Zhang for their technical
support and kind review, and of the assistance on the
manuscript.
Abbreviations:
|
DKK/Dkk
|
Dickkopf
|
|
NSCLC
|
non-small cell lung cancer
|
|
PARP
|
poly(ADP-ribose) polymerase
|
|
REIC
|
reduced expression in immortalized
cells
|
References
|
1
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Spira A and Ettinger DS: Multidisciplinary
management of lung cancer. N Engl J Med. 350:379–392. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
de las Peñas R, Sanchez-Ronco M, Alberola
V, et al: Polymorphisms in DNA repair genes modulate survival in
cisplatin/gemcitabine-treated non-small-cell lung cancer patients.
Ann Oncol. 17:668–675. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Logan CY and Nusse R: The Wnt signaling
pathway in development and disease. Annu Rev Cell Dev Biol.
20:781–810. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mazieres J, He B, You L, Xu Z and Jablons
DM: Wnt signaling in lung cancer. Cancer Lett. 222:1–10. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Klaus A and Birchmeier W: Wnt signalling
and its impact on development and cancer. Nat Rev Cancer.
8:387–398. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang H, Fan L, Xia X, et al: Silencing
Wnt2B by siRNA interference inhibits metastasis and enhances
chemotherapy sensitivity in ovarian cancer. Int J Gynecol Cancer.
22:755–761. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dowejko A, Bauer R, Bauer K,
Müller-Richter UD and Reichert TE: The human HECA interacts with
cyclins and CDKs to antagonize Wnt-mediated proliferation and
chemo-resistance of head and neck cancer cells. Exp Cell Res.
318:489–499. 2012. View Article : Google Scholar
|
|
9
|
Su HY, Lai HC, Lin YW, et al: Epigenetic
silencing of SFRP5 is related to malignant phenotype and
chemoresistance of ovarian cancer through Wnt signaling pathway.
Int J Cancer. 127:555–567. 2010. View Article : Google Scholar
|
|
10
|
Li Y and Bu G: LRP5/6 in Wnt signaling and
tumorigenesis. Future Oncol. 1:673–681. 2005. View Article : Google Scholar
|
|
11
|
Tsuji T, Miyazaki M, Sakaguchi M, Inoue Y
and Namba M: A REIC gene shows down-regulation in human
immortalized cells and human tumor-derived cell lines. Biochem
Biophys Res Commun. 268:20–24. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ueno K, Hirata H, Majid S, et al: Wnt
antagonist DICKKOPF-3 (Dkk-3) induces apoptosis in human renal cell
carcinoma. Mol Carcinog. 50:449–457. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mizobuchi Y, Matsuzaki K, Kuwayama K, et
al: REIC/Dkk-3 induces cell death in human malignant glioma. Neuro
Oncol. 10:244–253. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hoang BH, Kubo T, Healey JH, et al:
Dickkopf 3 inhibits invasion and motility of Saos-2 osteosarcoma
cells by modulating the Wnt-β-catenin pathway. Cancer Res.
64:2734–2739. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hsieh SY, Hsieh PS, Chiu CT and Chen WY:
Dickkopf-3/REIC functions as a suppressor gene of tumor growth.
Oncogene. 23:9183–9189. 2004.PubMed/NCBI
|
|
16
|
Yue W, Sun Q, Dacic S, et al:
Downregulation of Dkk3 activates β-catenin/TCF-4 signaling in lung
cancer. Carcinogenesis. 29:84–92. 2008. View Article : Google Scholar
|
|
17
|
Tsuji T, Nozaki I, Miyazaki M, et al:
Antiproliferative activity of REIC/Dkk-3 and its significant
down-regulation in non-small-cell lung carcinomas. Biochem Biophys
Res Commun. 289:257–263. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Veeck J and Dahl E: Targeting the Wnt
pathway in cancer: the emerging role of Dickkopf-3. Biochim Biophys
Acta. 1825:18–28. 2012.
|
|
19
|
Gosepath EM, Eckstein N, Hamacher A, et
al: Acquired cisplatin resistance in the head-neck cancer cell line
Cal27 is associated with decreased DKK1 expression and can
partially be reversed by overexpression of DKK1. Int J Cancer.
123:2013–2019. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shou J, Ali-Osman F, Multani AS, Pathak S,
Fedi P and Srivenugopal KS: Human Dkk-1, a gene encoding a Wnt
antagonist, responds to DNA damage and its overexpression
sensitizes brain tumor cells to apoptosis following alkylation
damage of DNA. Oncogene. 21:878–889. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hong WS, Saijo N, Sasaki Y, et al:
Establishment and characterization of cisplatin-resistant sublines
of human lung cancer cell lines. Int J Cancer. 41:462–467. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Coghlan MP, Culbert AA, Cross DA, et al:
Selective small molecule inhibitors of glycogen synthase kinase-3
modulate glycogen metabolism and gene transcription. Chem Biol.
7:793–803. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sato N, Yamabuki T, Takano A, et al: Wnt
inhibitor Dickkopf-1 as a target for passive cancer immunotherapy.
Cancer Res. 70:5326–5336. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Suzuki M, Shigematsu H, Nakajima T, et al:
Synchronous alterations of Wnt and epidermal growth factor receptor
signaling pathways through aberrant methylation and mutation in
non-small cell lung cancer. Clin Cancer Res. 13:6087–6092. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Niida A, Hiroko T, Kasai M, et al: DKK1, a
negative regulator of Wnt signaling, is a target of the
β-catenin/TCF pathway. Oncogene. 23:8520–8526. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen S, Guttridge DC, You Z, et al: Wnt-1
signaling inhibits apoptosis by activating β-catenin/T cell
factor-mediated transcription. J Cell Biol. 152:87–96. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Spencer CA and Groudine M: Control of
c-myc regulation in normal and neoplastic cells. Adv Cancer Res.
56:1–48. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
You Z, Saims D, Chen S, et al: Wnt
signaling promotes oncogenic transformation by inhibiting
c-Myc-induced apoptosis. J Cell Biol. 157:429–440. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
He TC, Sparks AB, Rago C, et al:
Identification of c-MYC as a target of the APC pathway. Science.
281:1509–1512. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tetsu O and McCormick F: β-Catenin
regulates expression of cyclin D1 in colon carcinoma cells. Nature.
398:422–426. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hirata H, Hinoda Y, Nakajima K, et al: Wnt
antagonist DKK1 acts as a tumor suppressor gene that induces
apoptosis and inhibits proliferation in human renal cell carcinoma.
Int J Cancer. 128:1793–1803. 2011. View Article : Google Scholar
|
|
32
|
Perkins C, Kim CN, Fang G and Bhalla KN:
Arsenic induces apoptosis of multidrug-resistant human myeloid
leukemia cells that express Bcr-Abl or overexpress MDR, MRP, Bcl-2,
or Bcl-xL. Blood. 95:1014–1022. 2000.PubMed/NCBI
|
|
33
|
Ambrosini G, Adida C, Sirugo G and Altieri
DC: Induction of apoptosis and inhibition of cell proliferation by
survivin gene targeting. J Biol Chem. 273:11177–11182. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ryan BM, O’Donovan N and Duffy MJ:
Survivin, a new target for anti-cancer therapy. Cancer Treat Rev.
35:553–562. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Olie RA, Simões-Wüst AP, Baumann B, et al:
A novel antisense oligonucleotide targeting survivin expression
induces apoptosis and sensitizes lung cancer cells to chemotherapy.
Cancer Res. 60:2805–2809. 2000.PubMed/NCBI
|
|
36
|
You L, He B, Xu Z, et al: Inhibition of
Wnt-2-mediated signaling induces programmed cell death in
non-small-cell lung cancer cells. Oncogene. 23:6170–6174. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Siddik ZH: Cisplatin: mode of cytotoxic
action and molecular basis of resistance. Oncogene. 22:7265–7279.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Singhal S, Miller D, Ramalingam S and Sun
SY: Gene expression profiling of non-small cell lung cancer. Lung
Cancer. 60:313–324. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
He B and Jablons DM: Wnt signaling in stem
cells and lung cancer. Ernst Schering Found Symp Proc. 2006:27–58.
2006.
|
|
40
|
Teng Y, Wang X, Wang Y and Ma D:
Wnt/β-catenin signaling regulates cancer stem cells in lung cancer
A549 cells. Biochem Biophys Res Comm. 392:373–379. 2010. View Article : Google Scholar
|
|
41
|
Zhang T, Otevrel T, Gao Z, et al: Evidence
that APC regulates survivin expression: a possible mechanism
contributing to the stem cell origin of colon cancer. Cancer Res.
61:8664–8667. 2001.PubMed/NCBI
|
|
42
|
Kim CF, Jackson EL, Woolfenden AE, et al:
Identification of bronchioalveolar stem cells in normal lung and
lung cancer. Cell. 121:823–835. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Niehrs C: Function and biological roles of
the Dickkopf family of Wnt modulators. Oncogene. 25:7496–7481.
2006. View Article : Google Scholar
|
|
44
|
Yang ZR, Dong WG, Lei XF, Liu M and Liu
QS: Overexpression of Dickkopf-3 induces apoptosis through
mitochondrial pathway in human colon cancer. World J Gastroenterol.
18:1590–1601. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Abarzua F, Sakaguchi M, Takaishi M, et al:
Adenovirus-mediated overexpression of REIC/Dkk-3 selectively
induces apoptosis in human prostate cancer cells th rough
activation of c-Jun-NH2-kinase. Cancer Res.
65:9617–9622. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kashiwakura Y, Ochiai K, Watanabe M, et
al: Down-regulation of inhibition of differentiation-1 via
activation of activating transcription factor 3 and Smad regulates
REIC/Dickkopf-3-induced apoptosis. Cancer Res. 68:8333–8341. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Huang M, Wang Y, Sun D, et al:
Identification of genes regulated by Wnt/β-catenin pathway and
involved in apoptosis via micro-array analysis. BMC Cancer. 6:221.
2006. View Article : Google Scholar
|
|
48
|
del Barrantes IB, Montero-Pedrazuela A,
Guadaño-Ferraz A, et al: Generation and characterization of
dickkopf3 mutant mice. Mol Cell Biol. 26:2317–2326. 2006.
View Article : Google Scholar
|
|
49
|
Jung IL, Kang HJ, Kim KC and Kim IG:
Knockdown of the Dickkopf 3 gene induces apoptosis in a lung
adenocarcinoma. Int J Mol Med. 26:33–38. 2010.PubMed/NCBI
|