Introduction
Hormonal influence on breast tissue and in breast
cancer development has long been recognized. Examinations of ~70%
of breast cancer patients have been found to be positive for
estrogen receptor (ER)/progesterone (PR) receptor (1,2).
Additionally, the receptor status has been used as a biomarker for
therapy and prognosis. Currently, ER/PR+ breast cancer
patients are treated with anti-estrogen drugs, such as tamoxifen
(Tam) or aromatase inhibitors (AIs) (3,4). Tam
binds to ER, thereby preventing the binding of estrogen and
subsequent activation of ER. Aromatase inhibitors (AI) suppress the
activity of aromatase enzyme and thus, the production of estrogen.
Although these inhibitors were shown to be effective, some patients
develop resistance and do not respond to Tam therapy or other
targeted endocrine therapies. The de novo resistance appears
to be due to the up-regulation/overexpression of the HER2 receptor
and/or Akt protein. Clinical and in vitro studies have also
suggested that overexpression of the HER2 receptor is associated
with resistance to hormone therapy (5–7). In
addition, the majority of breast cancer patients with ER expression
initially respond to endocrine therapy but, develop ‘acquired’
resistance over a period of time (8). MCF-7 and T47D breast cancer cell lines
have been used as experimental models to investigate the
mechanism(s) involved in acquired resistance to Tam (5,9,10). The
results from such studies reported by our and other research groups
have shown that drug resistance in these cells can be acquired
through various mechanisms, including: i) loss of ERα expression,
ii) altered activity of co-regulators, iii) cross-talk between the
ERα and growth factor signaling pathways and, iv) overexpression of
PKCα or PKCδ (3,5,11).
These anti-estrogen-resistant models are also useful for the
identification of other ‘potential’ therapeutic agents that can
inhibit or eliminate anti-estrogen-resistant breast tumors.
Tumor necrosis factor-related apoptosis-inducing
ligand (TRAIL) induced apoptosis through the death receptors, DR4
and DR5, which are present at the surface of target cells (12,13).
Recombinant human TRAIL was shown to induce apoptosis
preferentially in cancer over normal cells, and appear to have
little or no overt toxicity when systemically administered to
animals (12,14). Consequently, numerous clinical
trials targeting TRAIL receptors, including TRAIL, as well as
agonistic antibodies to DR4 and DR5 have been conducted. However, a
significant portion of tumor cells, including breast cancer cells,
were reported to be resistant to these agents (13,15–17).
In this context, the use of platinum complexes for breast cancer
therapy has emerged as a new treatment modality. Cisplatin, either
independently or in combination with other chemotherapeutic drugs,
was shown to be effective in the treatment of breast cancer
(18,19). Cisplatin induces intra- and
inter-strand cross-links in DNA leading to cell death. The aim of
the present study was to investigate whether cisplatin plus TRAIL
treatment induced cell death and apoptosis in Tam-resistant cells
and whether such a combinatorial treatment provides a new strategy
for future therapeutic intervention of patients with
anti-estrogen-resistant breast cancer.
Materials and methods
Cell lines and reagents
The MCF-7 human breast carcinoma cell line was
obtained from the American Type Culture Collection (ATCC; Manassas,
VA, USA) and maintained in Dulbecco’s modified Eagle’s medium
(DMEM), supplemented with 5% FCS and insulin, and penicillin and
streptomycin (Life Technologies, Gaithersburg, MD, USA). Stably
transfected MCF-7 cells with full-length HER2 cDNA (MCF-7/HER2–18)
or control vector (MCF-7/neo) have been previously described
(9). Tam/MCF-7 cells were grown in
the presence of 4-OH Tam (10−6 M) (20). The cells obtained from ATCC were
immediately expanded and frozen, and restarted every 3–4 months
from a frozen vial of the same batch of cells and no additional
authentication was done on these cells. 4-OH Tam was purchased from
Sigma Chemical Co. (St. Louis, MO, USA). The
anti-glyceraldehyde-3-phosphate dehydrogenase (G3PDH) rabbit
polyclonal was purchased from Trevigen (Gaithersburg, MD, USA).
Monoclonal antibodies of PARP and Bid were purchased from BD
Biosciences (San Diego, CA, USA). Caspase-9 antibodies were
purchased from Cell Signaling Technology, Inc. (Boston, MA, USA).
Caspase-3 inhibitor Z-DEVD-FMK, Caspase-8 inhibitor Z-IETD-FMK,
Caspase-9 inhibitor Z-LEHD-FMK, and pan-caspase inhibitor Z-VAD-FMK
were purchased from R&D System (Minneapolis, MN, USA). Unless
otherwise specified, any other chemicals were obtained from Sigma
Chemical Co. at highest suitable purities.
MTT assay
Briefly, 5×104 cells were added in
96-well tissue culture plates. After 24 h, the cells were treated
with TRAIL (10 ng/ml), cisplatin (10 μg/ml), or a combination of
TRAIL plus cisplatin for another 24 or 48 h. Then, 100 μl of
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
(1 mg/ml) was added into each sample and incubated for 3 h under 5%
CO2 and 37°C. The cell viability was measured by MTT,
which was converted by succinate dehydrogenase in the mitochondria
of viable cells to yield a purple formazan dye. The formazan dye
was dissolved in dimethyl sulfoxide (DMSO) and measured by
absorption at a wavelength of 550 nm using Benchmark®
microplate reader from Bio-Rad (Hercules, CA, USA).
Apoptosis assay
Apoptosis was assessed using the Cell Death
Detection ELISAplus kit (Roche Applied Science, Indianapolis, IN,
USA) according to the manufacturer’s instructions. This kit
quantitatively detects cytosolic histone-associated DNA fragments.
Briefly, the cells were treated with cisplatin (10 μg/ml) and TRAIL
(10 ng/ml) or a combination of cisplatin (10 μg/ml) plus TRAIL (10
ng/ml) for 16 h of treatment. Apoptosis was quantified by ELISA and
normalized to values measured in untreated cells. Data were
presented as the mean ± SEM of triplicate determination.
Western immunoblot analysis
The cells were grown in 6-well plates, to near
confluence in the presence or absence of various treatments. The
cells were lysed and western blotting was performed as previously
described (5) using a standard
protocol. Briefly, the cell extracts were obtained by lysing the
cells in RIPA buffer (20 mM Hepes, 100 mM NaCl, 0.1% SDS, 1%
Nonidet P-40, 1% deoxycholate, 1 mM Na3VO4, 1
mM EGTA, 50 mM NaF, 10% glycerol, 1 mM EDTA, 1 mM
phenylmethylsulfonyl fluoride, and 1X protease inhibitor mixture).
The samples containing 100 μg of total protein were electrophoresed
on 8% or 15% SDS-polyacrylamide gels and transferred on to PVDF
membranes by electroblotting. The membranes were probed with
antibodies as indicated, followed by HRP-conjugated mouse or rabbit
secondary antibodies (Amersham Amersham Biosciences, Piscataway,
NJ, USA). Anti-G3PDH was used for loading controls.
Statistical analysis
Data are presented as mean ± SEM, and statistically
analyzed using the unpaired Student’s t-test. Differences were
considered statistically significant when P<0.05.
Results
Cisplatin plus TRAIL enhances cell death
in anti-estrogen-resistant and -sensitive cells without
significantly effecting normal breast cells
To develop new strategies to eliminate
anti-estrogen-resistant and -sensitive breast cells, we examined
the effect of cisplatin on Tam/MCF-7 and Tam/T47D cells. Cell
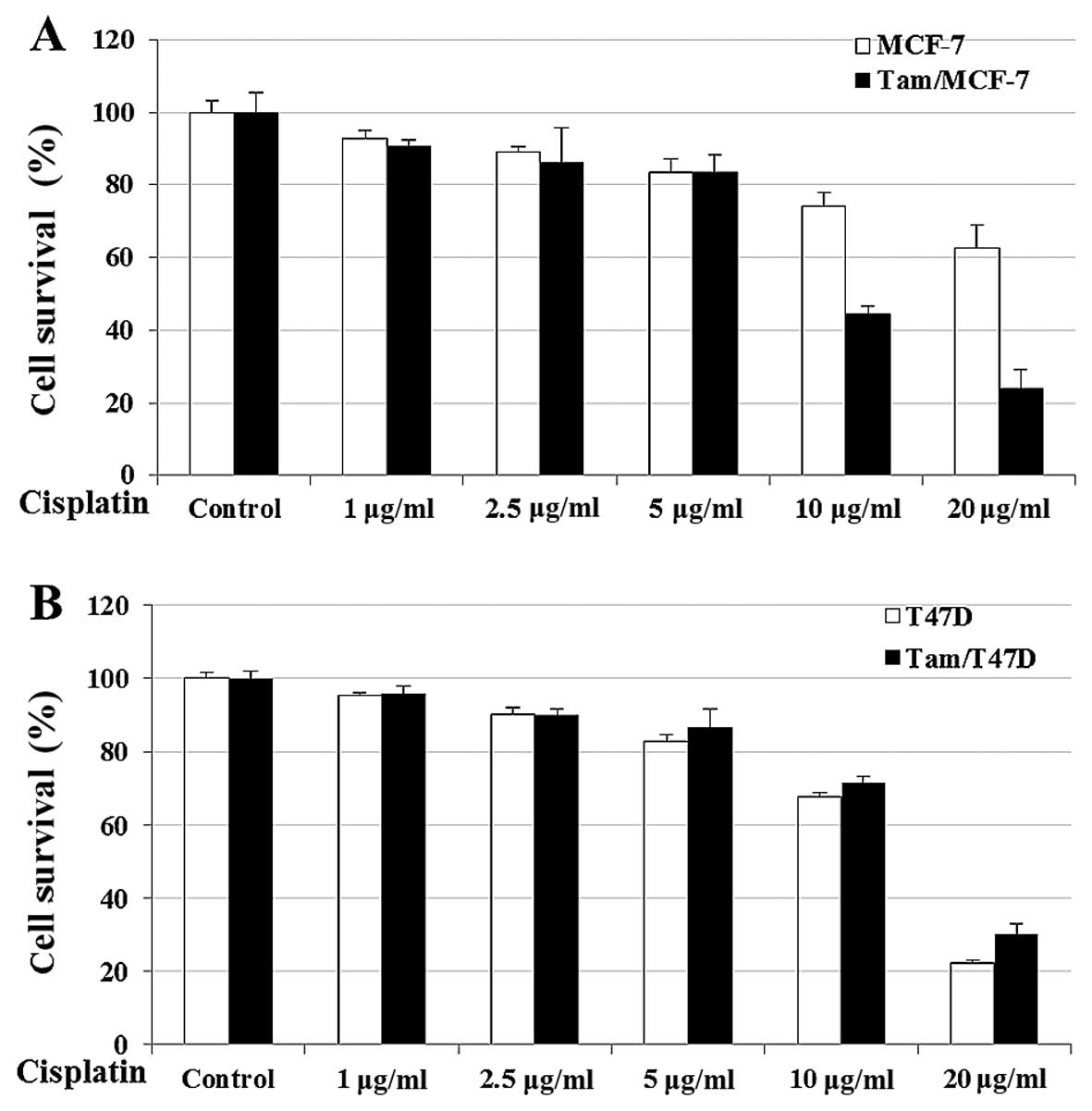
survival is shown in Fig. 1.
Treatment of the cells with Cisplatin induced a significant
decrease in cell survival in the Tam/MCF-7 and Tam/T47D cells. The
efficacy of cisplatin was dose-dependent; however, a significant
risk of nephrotoxicity frequently hindered the use of higher doses
to maximize the antitumor effects of cisplatin (21). Our data showed that 10 μg/ml of
cisplatin exhibited optimum inhibition in MCF-7- and T47D-derived
anti-estrogen-resistant cells (Fig.
1). In addition, the results showed that cisplatin was more
effective in killing anti-estrogen-resistant cells when compared to
sensitive cells in the MCF-7 cell line. However, in the
T47D-derived cell lines Tam-resistant and -sensitive cells were
killed (Fig. 1). The reasons for
this discrepancy in ER-positive breast cell lines remains to be
determined.
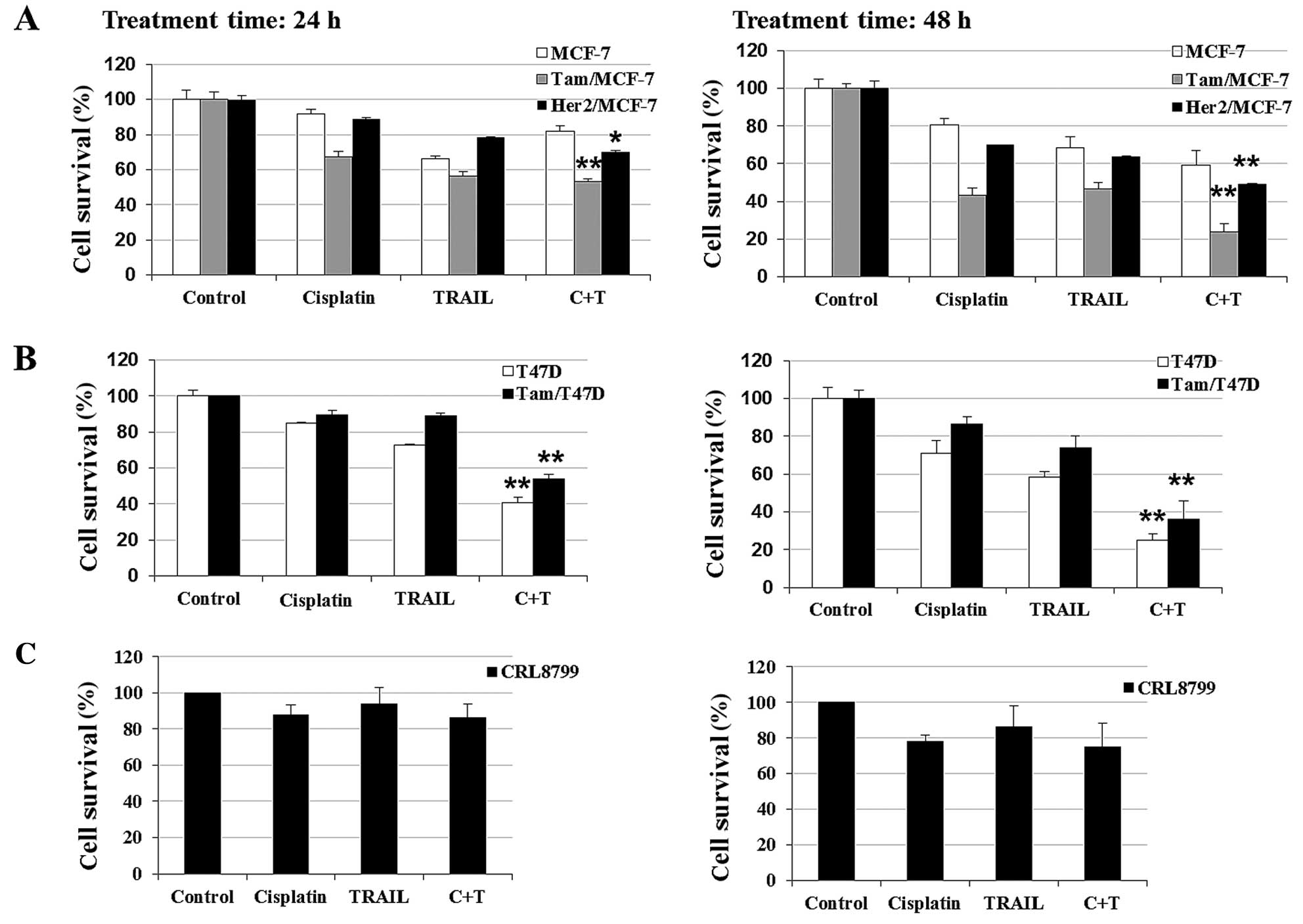
We evaluated whether a combination of cisplatin plus
TRAIL enhanced killing in anti-estrogen-resistant cells. Fig. 2 shows that cisplatin or TRAIL
treatment decreased cell survival in Tam/MCF-7 (acquired resistance
to Tam), HER2/MCF-7 (de novo resistance) and Tam/T47D
(acquired resistance to Tam) cells to varying degrees. However,
cisplatin plus TRAIL had a maximum effect on
anti-estrogen-resistant cells by 48 h. A similar treatment of
CRL8799 normal breast cells resulted in a moderate increase in cell
death.
Cisplatin plus TRAIL enhances apoptosis
in Tam-resistant cells
Most chemotherapy agents and irradiation trigger
apoptosis through the cell-intrinsic pathway, as an indirect
consequence of cell damage. The intrinsic pathway usually requires
functional p53. However, inactivation of p53, either directly
through mutation(s) or indirectly through p53 modulation through
the MDM2 protein, is common in many human tumors. The extrinsic
pathway induces apoptosis in response to the engagement of death
receptors by their ligands such as TRAIL. This pathway stimulates
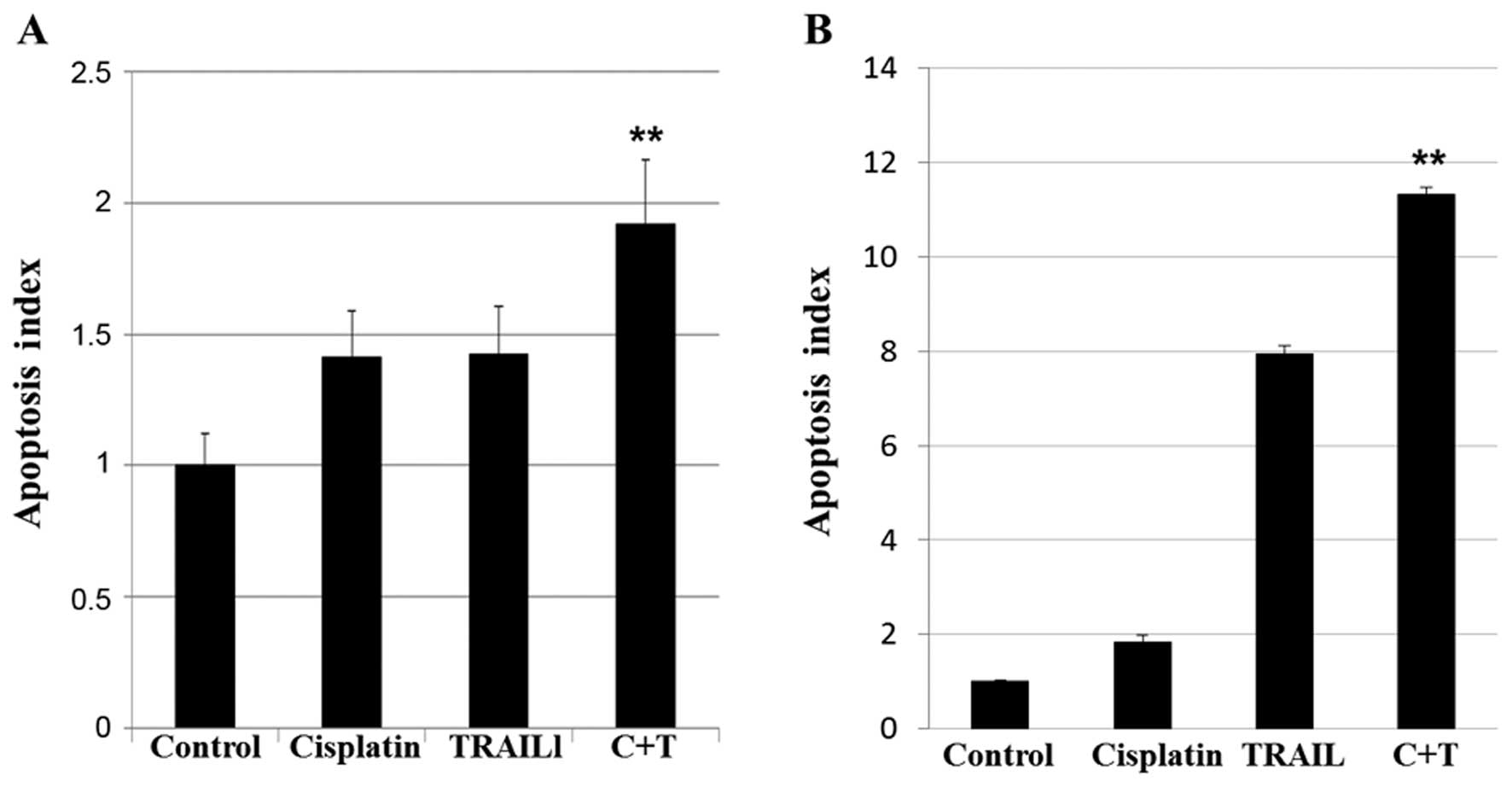
the apoptotic caspase mechanism independent of p53 (22). The effect of cisplatin, TRAIL, and
cisplatin plus TRAIL treatment on apoptosis in Tam/MCF-7 and
Tam/T47D cells are shown in Fig. 3.
Although treatment with cisplatin or TRAIL increased apoptosis to
varying degrees, a combination of cisplatin plus TRAIL had a
maximum effect on apoptosis in Tam/MCF-7 (Fig. 3A), and Tam/T47D (Fig. 3B) cells (p<0.05). These data
suggested that cisplatin plus TRAIL increased apoptosis in
anti-estrogen-resistant cells compared with the untreated
controls.
Caspase inhibition reduces cisplatin plus
TRAIL-induced cell death
Caspases have been previously shown to play an
important role in TRAIL-induced apoptosis (23). It was previously shown that 75% of
human breast tumors lack caspase-3 transcripts, whereas other
caspases, such as caspase-8 and -9 were shown to be normal
(24). MCF-7 cells lack caspase-3
expression as a result of a functional deletion mutation in the
caspase-3 gene, while the expression of other caspase-8 and -9
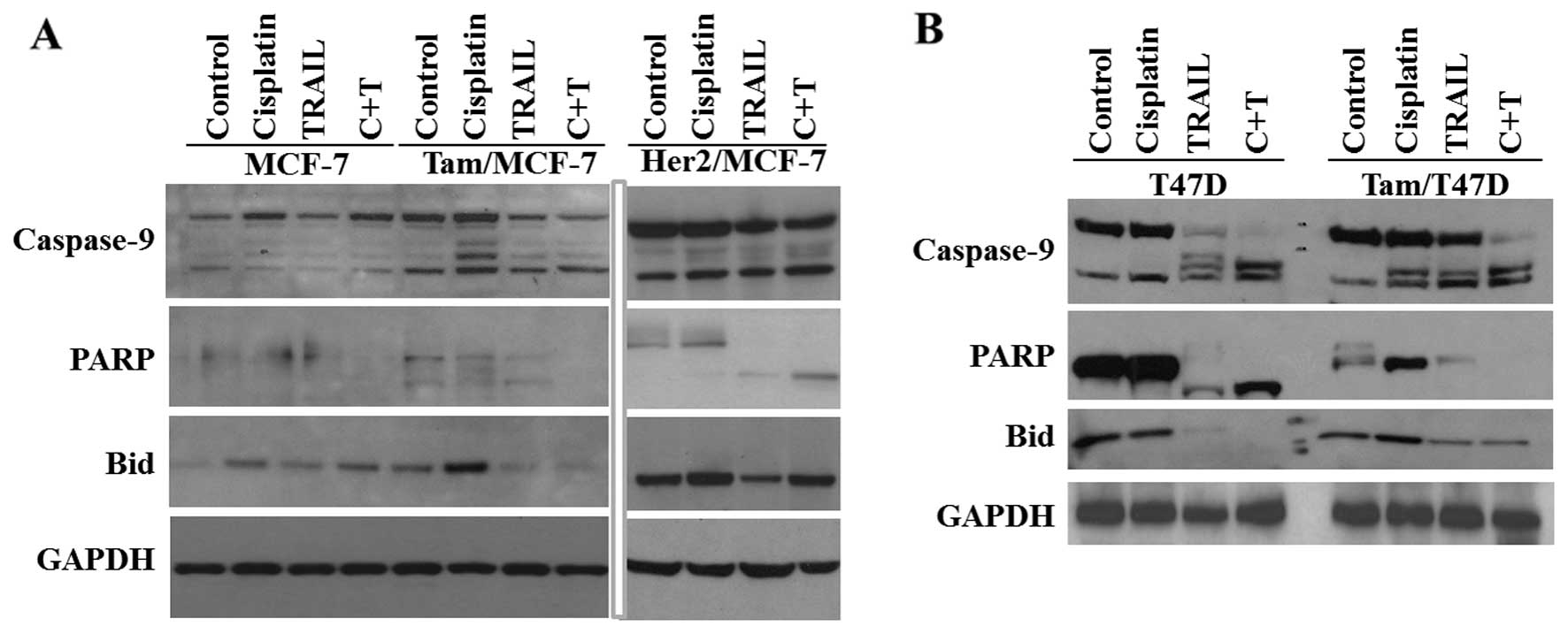
remains normal. We used MCF-7 and T47D anti-estrogen cells to
determine the possible mechanism by which cisplatin plus TRAIL
increase cell death. The results from the western blot analyses
revealed an increased caspase-9 activation in Tam/MCF-7 and
Her-2/MCF-7 anti-estrogen-resistant cells compared to -sensitive
cells and this led to increased PARP and Bid cleavage (Fig. 4A) and apoptosis (Fig. 3A). By contrast, T47D and Tam/T47D
(express caspase-3) showed a higher activation of caspase-3 and -9
in resistant and sensitive cells in the presence of cisplatin plus
TRAIL, leading to increased PARP and Bid cleavage (Fig. 4B).
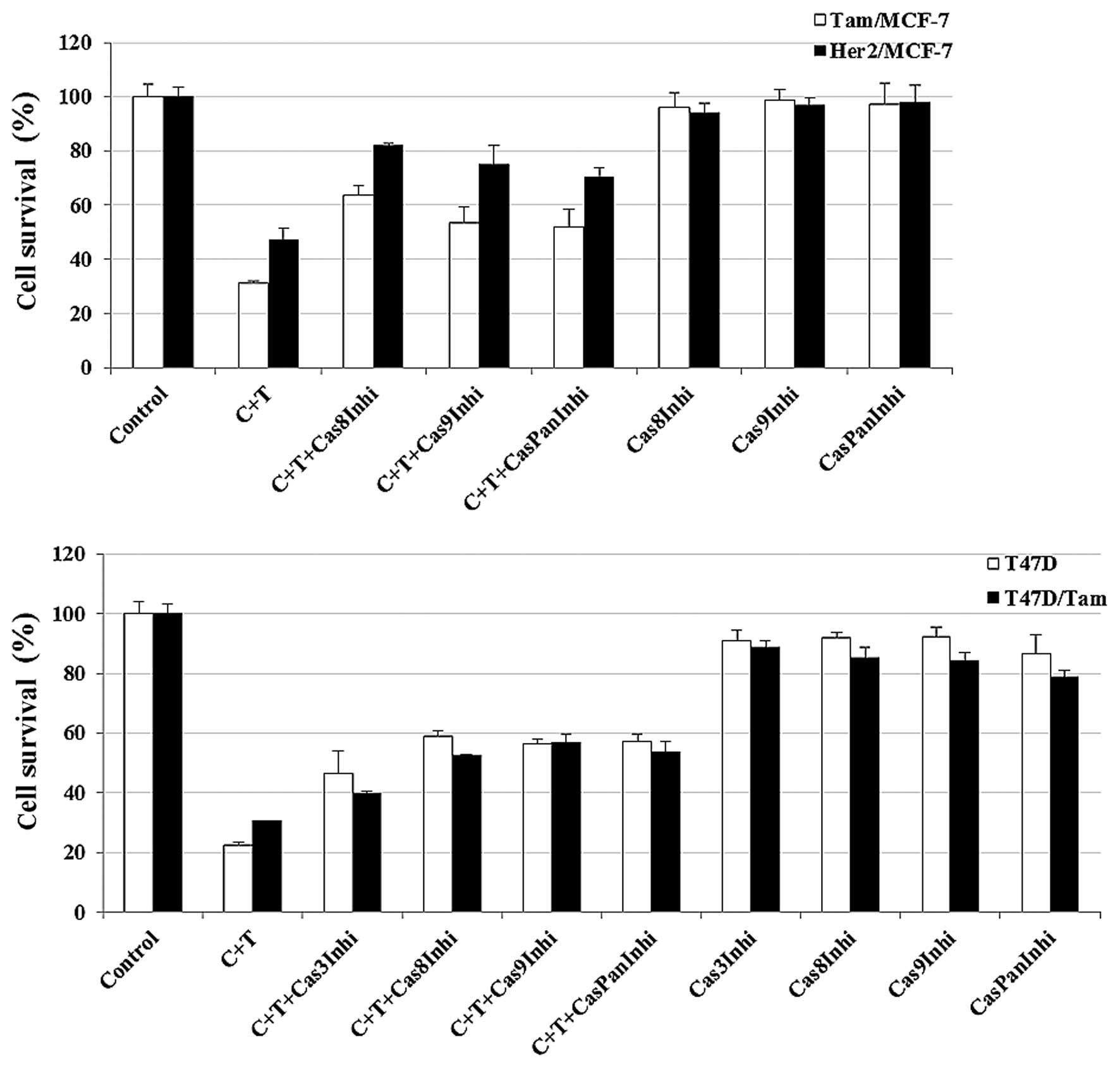
To determine whether caspase activation is essential
for cisplatin plus TRAIL-induced cell death, we blocked caspase-3,
-8, -9 or pan-caspase using relatively specific caspase
inhibitor(s). The effect of caspase inhibitors on cisplatin plus
TRAIL treatment in Tam/MCF-7 HER2/MCF-7 and Tam/T47D cells are
shown in Fig. 5. However, the
addition of caspase inhibitors alone to anti-estrogen-resistant
cells had a minimal or no effect on these cells. These data
suggested that cisplatin plus TRAIL-induced apoptosis is
predominantly mediated through the activation of caspases in
Tam-resistant cells.
Discussion
Anti-estrogenic drugs such as Tam provide an
effective therapy for women with hormone-dependent breast cancer in
the neo-adjuvant, adjuvant and advanced disease settings (25). However, a major clinical problem
with the use of anti-estrogens is that the majority of patients
with advanced disease eventually develop resistance to the
compounds and even stimulate tumor growth in some cases (26,27).
Thus, Tam resistance is a daunting challenge for the successful
treatment of ER-positive and hormone-dependent breast cancer.
Preclinical and clinical investigations conducted to understand
this resistance have led to the identification of multiple pathways
that contribute to anti-estrogen resistance, most of which arise
from alterations in ER− and/or growth factor receptor
signaling (28).
In this study, instead of managing or preventing
anti-estrogen resistance, we attempted the strategy of eliminating
endocrine-resistant cells by inducing enhanced cell death. The
results from the present investigations have shown that Tam/MCF-7
and Tam/T47D cells, when treated with cisplatin plus TRAIL,
exhibited significantly enhanced cell death as compared with the
untreated cells. Furthermore, a similar treatment had a minimal
effect on CRL8799 normal breast cells. In addition, results from
the present study provided evidence that cisplatin enhances
TRAIL-induced cell death by activation of caspases. Inhibition of
the activity of caspases decreased cell death in cisplatin alone
and cisplatin plus TRAIL-treated cells.
The results also showed that cisplatin plus TRAIL
significantly induces cell death in the de novo
Tam-resistant (HER2/MCF-7) and acquired Tam-resistant (Tam/MCF-7)
cells when compared to the MCF-7 parental cells. These observations
were similar to those reported by Yde et al (29): MCF-7-derived anti-estrogen-resistant
cells were more sensitive to cisplatin-induced cell death than the
parental cells. The results from this study suggest that treatment
with cisplatin or TRAIL alone induced cell death in 30–50% of
Tam-resistant cells, however, the maximum effect is evident when
these cells are treated with cisplatin plus TRAIL. There were
differences in the response between Tam-resistant and parental T47D
cells when compared to MCF-7 cells. Cisplatin plus TRAIL treatment
significantly inhibited cell survival in Tam-T47D and T47D cells.
However, the extent of cell death was different between Tam/MCF-7
and MCF-7 cells, the latter being relatively resistant to cisplatin
plus TRAIL treatment. The reason behind these discrepancies has yet
to be determined. One possibility is that MCF-7 cells lack
caspase-3 expression as a result of a functional deletion mutation
in caspase-3 gene, while expression of caspase-8 and -9 remains
normal (30). By contrast, T47D
expresses all caspases.
We have previously shown that re-introduction of
caspase-3 in MCF-7 cells increases TRAIL-induced apoptosis in
PKC-δ-induced anti-estrogen resistance (31). Caspase-3 mRNA expression levels in
breast cancer were shown to be ≥10- to 50-fold lower than those in
normal breast tissues (24) which
is a significant clinical observation. It is possible that
Tam/MCF-7 models can be used for more detailed mechanistic studies
to identify additional targets that can be used to treat
anti-estrogen-resistant breast tumors.
The results from the present investigations suggest
that treatment of Tam-resistant cells, i.e., Tam/MCF-7 and
Tam/T47D, with cisplatin can enhance TRAIL-induced apoptosis by
activating caspases while similar treatment of immortalized CRL8799
normal breast cells had a minimal effect. Individually, platinum
compounds, carboplatin or cisplatin and TRAIL have been approved by
the FDA to treat different forms of cancer. The results of the
present investigation suggest that cisplatin plus TRAIL treatment
can be used as a potential and novel treatment strategy for
anti-estrogen-resistant breast tumors.
Acknowledgements
This study is supported by the Department of
Pathology at WSU.
Abbreviations:
|
TRAIL
|
tumor necrosis factor-related
apoptosis-inducing ligand
|
|
PARP
|
poly(ADP-ribose) polymerase
|
|
ER
|
estrogen receptor
|
|
HER2
|
human epidermal growth factor receptor
2
|
|
Tam
|
tamoxifen
|
References
|
1
|
Anderson WF, Katki HA and Rosenberg PS:
Incidence of breast cancer in the United States: current and future
trends. J Natl Cancer Inst. 103:1397–1402. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Marshall E: Breast cancer. Dare to do
less. Science. 343:1454–1456. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Clarke R, Leonessa F, Welch JN and Skaar
TC: Cellular and molecular pharmacology of antiestrogen action and
resistance. Pharmacol Rev. 53:25–71. 2001.PubMed/NCBI
|
|
4
|
Lønning PE: Aromatase inhibitors in breast
cancer. Endocr Relat Cancer. 11:179–189. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nabha SM, Glaros S, Hong M, Lykkesfeldt
AE, Schiff R, Osborne K, et al: Upregulation of PKC-delta
contributes to antiestrogen resistance in mammary tumor cells.
Oncogene. 24:3166–3176. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shou J, Massarweh S, Osborne CK, Wakeling
AE, Ali S, Weiss H and Schiff R: Mechanisms of tamoxifen
resistance: increased estrogen receptor-HER2/neu cross-talk in
ER/HER2-positive breast cancer. J Natl Cancer Inst. 96:926–935.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Slamon DJ, Clark GM, Wong SG, Levin WJ,
Ullrich A and McGuire WL: Human breast cancer: correlation of
relapse and survival with amplification of the HER-2/neu oncogene.
Science. 235:177–182. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kaufmann M, Bajetta E, Dirix LY, Fein LE,
Jones SE, Zilembo N, et al: Exemestane is superior to megestrol
acetate after tamoxifen failure in postmenopausal women with
advanced breast cancer: results of a phase III randomized
double-blind trial. The Exemestane Study Group. J Clin Oncol.
18:1399–1411. 2000.PubMed/NCBI
|
|
9
|
Benz CC, Scott GK, Sarup JC, Johnson RM,
Tripathy D, Coronado E, et al: Estrogen-dependent,
tamoxifen-resistant tumorigenic growth of MCF-7 cells transfected
with HER2/neu. Breast Cancer Res Treat. 24:85–95. 1992. View Article : Google Scholar
|
|
10
|
Kurokawa H, Lenferink AE, Simpson JF,
Pisacane PI, Sliwkowski MX, Forbes JT, et al: Inhibition of
HER2/neu (erbB-2) and mitogen-activated protein kinases enhances
tamoxifen action against HER2-overexpressing, tamoxifen-resistant
breast cancer cells. Cancer Res. 60:5887–5894. 2000.PubMed/NCBI
|
|
11
|
Atanaskova N, Keshamouni VG, Krueger JS,
Schwartz JA, Miller F and Reddy KB: MAP kinase/estrogen receptor
crosstalk enhances estrogen-mediated signaling and tumor growth but
does not confer tamoxifen resistance. Oncogene. 21:4000–4008. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ashkenazi A, Pai RC, Fong S, Leung S,
Lawrence DA, Marsters SA, et al: Safety and antitumor activity of
recombinant soluble Apo2 ligand. J Clin Invest. 104:155–162. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Takeda K, Stagg J, Yagita H, Okumura K and
Smyth MJ: Targeting death-inducing receptors in cancer therapy.
Oncogene. 26:3745–3457. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lawrence D, Shahrokh Z, Marsters S,
Achilles K, Shih D, Mounho B, et al: Differential hepatocyte
toxicity of recombinant Apo2L/TRAIL versions. Nat Med. 7:383–385.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chinnaiyan AM, Prasad U, Shankar S,
Hamstra DA, Shanaiah M, Chenevert TL, et al: Combined effect of
tumor necrosis factor-related apoptosis-inducing ligand and
ionizing radiation in breast cancer therapy. Proc Natl Acad Sci
USA. 97:1754–1759. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Keane MM, Ettenberg SA, Nau MM, Russell EK
and Lipkowitz S: Chemotherapy augments TRAIL-induced apoptosis in
breast cell lines. Cancer Res. 59:734–741. 1999.PubMed/NCBI
|
|
17
|
Singh TR, Shankar S, Chen X, Asim M and
Srivastava RK: Synergistic interactions of chemotherapeutic drugs
and tumor necrosis factor-related apoptosis-inducing ligand/Apo-2
ligand on apoptosis and on regression of breast carcinoma in vivo.
Cancer Res. 63:5390–5400. 2003.PubMed/NCBI
|
|
18
|
Kourousis C, Kakolyris S, Androulakis N,
Heras P, Vlachonicolis J, Vamvakas L, et al: Salvage chemotherapy
with paclitaxel, vinorelbine, and cisplatin (PVC) in
anthracycline-resistant advanced breast cancer. Am J Clin Oncol.
21:226–232. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sledge GW Jr, Loehrer PJ Sr, Roth BJ and
Einhorn LH: Cisplatin as first-line therapy for metastatic breast
cancer. J Clin Oncol. 6:1811–1814. 1988.PubMed/NCBI
|
|
20
|
Lykkesfeldt AE and Briand P: Indirect
mechanism of oestradiol stimulation of cell proliferation of human
breast cancer cell lines. Br J Cancer. 53:29–35. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Miller RP, Tadagavadi RK, Ramesh G and
Reeves WB: Mechanisms of cisplatin nephrotoxicity. Toxins.
2:2490–2518. 2010. View Article : Google Scholar
|
|
22
|
Ashkenazi A and Dixit VM: Death receptors:
signaling and modulation. Science. 281:1305–1308. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lin J, Zhang Z, Zeng S, Zhou S, Liu BF,
Liu Q, et al: TRAIL-induced apoptosis proceeding from
caspase-3-dependent and -independent pathways in distinct HeLa
cells. Biochem Biophys Res Commun. 346:1136–1141. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Devarajan E, Sahin AA, Chen JS,
Krishnamurthy RR, Aggarwal N, Brun AM, et al: Down-regulation of
caspase 3 in breast cancer: a possible mechanism for
chemoresistance. Oncogene. 21:8843–8851. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fisher B, Dignam J, Bryant J, DeCillis A,
Wickerham DL, Wolmark N, et al: Five versus more than five years of
tamoxifen therapy for breast cancer patients with negative lymph
nodes and estrogen receptor-positive tumors. J Natl Cancer Inst.
88:1529–1542. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Berstein LM, Zheng H, Yue W, Wang JP,
Lykkesfeldt AE, Naftolin F, et al: New approaches to the
understanding of tamoxifen action and resistance. Endocr Relat
Cancer. 10:267–277. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Horwitz KB: When tamoxifen turns bad.
Endocrinology. 136:821–823. 1995.PubMed/NCBI
|
|
28
|
Clarke R, Liu MC, Bouker KB, Gu Z, Lee RY,
Zhu Y, et al: Antiestrogen resistance in breast cancer and the role
of estrogen receptor signaling. Oncogene. 22:7316–7339. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yde CW, Gyrd-Hansen M, Lykkesfeldt AE,
Issinger OG and Stenvang J: Breast cancer cells with acquired
antiestrogen resistance are sensitized to cisplatin-induced cell
death. Mol Cancer Ther. 6:1869–1876. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Janicke RU, Sprengart ML, Wati MR and
Porter AG: Caspase-3 is required for DNA fragmentation and
morphological changes associated with apoptosis. J Biol Chem.
273:9357–9360. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yin S, Sethi S and Reddy KB: Protein
kinase Cδ and caspase-3 modulate TRAIL-induced apoptosis in breast
tumor cells. J Cell Biochem. 111:979–987. 2010. View Article : Google Scholar : PubMed/NCBI
|