Introduction
The Bcl-2 superfamily of proteins is classified
based on function and Bcl-2 homology (BH)-domain organization, such
as anti-apoptotic, pro-apoptotic and BH3-only proteins.
Anti-apoptotic Bcl-2 proteins maintain mitochondrial outer membrane
permeability by binding to Bax and Bak; thus, overexpression of
Bcl-2 results in apoptosis resistance in tumors (1). In contrast, pro-apoptotic Bcl-2
proteins increase mitochondrial outer membrane permeabilization
(MOMP), which is related to induction of apoptosis. BH3-only
proteins are classified into 2 groups: activators and sensitizers.
Activator BH3-only proteins, such as Bid, Bim, and Puma, bind to
anti- and pro-apoptotic proteins, whereas sensitizer BH3-only
proteins, such as Noxa, bind to anti-apoptotic proteins. When
anti-apoptotic proteins bind to BH3-only proteins, activator
BH3-only proteins and pro-apoptotic proteins induce MOMP, resulting
in the induction of apoptosis (2,3).
ABT-737 is a small-molecule BH3 mimetic, and directly binds to
Bcl-2, Bcl-xL and Bcl-w with a very high affinity, resulted in
induction of apoptosis by releasing Bax and Bak. Therefore, ABT-737
induces apoptosis in several types of cancer cells, such as
leukemia, myeloma and small-cell lung cancer cells (4–6). In
contrast, since ABT-737 has a very low affinity for Mcl-1 (7,8),
several types of cancer cells are resistant to ABT-737. Therefore,
to enhance the sensitivity of ABT-737, a strategy of combined
treatment is needed.
Aurora kinases are serine/threonine kinases, and are
important for cell proliferation, cell cycle regulation, mitotic
spindle formation, centrosome maturation, and cytokinesis (9,10).
Aurora kinase is divided into 3 groups: A, B, and C. Both Aurora
kinase A and B are expressed in most normal cells, while Aurora
kinase C is only expressed in testicular tissue (11). Recently, several studies found that
Aurora kinases are overexpressed in tumors, and the extent of
Aurora kinase overexpression is correlated with malignancy and poor
prognosis (12–14). Therefore, Aurora kinases are
important therapeutic targets. These molecular-targeted strategies
inhibit specific molecules, which vary between cancer and normal
cells. However, these strategies could be safer and more
effective.
In the present study, we investigated whether a
combined treatment with ABT-737 and an Aurora kinase inhibitor
induces apoptosis, and we aimed to identify the mechanism involved
in the combined treatment-mediated apoptosis in human breast
carcinoma MDA-MB-435S cells.
Materials and methods
Cells and materials
MDA-MB-435S, Caki, and A549 cells were purchased
from the American Type Culture Collection (ATCC; Manassas, VA,
USA). Human skin fibroblasts (HSFs) were a gift from Dr T.J. Lee
(Yeungnam University, Korea). The cells were cultured in Dulbecco’s
modified Eagle’s medium containing 10% fetal bovine serum, 20 μM
HEPES buffer, and 100 μg/ml gentamicin. ABT-737, VX-680 and MLN8237
were purchased from Sellek (Houston, TX, USA). Barasertib was
purchased from Tocris (Ellisville, MO, USA). z-VAD was purchased
from Sigma-Aldrich (St. Louis, MO, USA). Anti-death receptor 5
(DR5), anti-Bcl2, anti-Bcl-xL, anti-Mcl-1, anti-cIAP2 and anti-PARP
antibodies were purchased from Santa Cruz Biotechnology (Santa
Cruz, CA, USA). Anti-c-FLIP (L) antibody was obtained from Alexis
Corporation (San Diego, CA, USA). The anti-actin antibody was
obtained from Sigma-Aldrich.
Flow cytometric analysis
The cells were suspended in 100 μl of
phosphate-buffered saline (PBS), and 200 μl of 95% ethanol was
added during a vortex step. The cells were incubated at 4°C for 1
h, washed with PBS and resuspended in 250 μl of 1.12% sodium
citrate buffer (pH 8.4) together with 12.5 μg of RNase. The
incubation was continued at 37°C for 30 min. The cellular DNA was
then stained by applying 250 μl of propidium iodide (50 μg/ml) for
30 min at room temperature. The stained cells were analyzed by
fluorescent-activated cell sorting on a FACScan flow cytometer for
relative DNA content based on red fluorescence.
Western blot analysis
The cells were washed with cold PBS and lysed in ice
in a modified RIPA buffer (50 μM Tris-HCl (pH 7.4), 1% NP-40, 0.25%
Na-deoxycholate, 150 μM NaCl, 1 μM Na3VO4,
and 1 μM NaF) containing protease inhibitors (100 μM
phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml
pepstatin and 2 μM EDTA). The lysates were centrifuged at 10,000 ×
g for 10 min at 4°C, and the supernatant fractions were collected.
The proteins were separated by SDS-PAGE and transferred to an
Immobilon-P membrane. The specific proteins were detected using an
enhanced chemiluminescence (ECL) western blotting kit according to
the manufacturer’s instructions.
Cell death assessment by DNA
fragmentation assays
A Cell Death Detection ELISA Plus kit (Boehringer
Mannheim, USA) was used for assessing apoptotic activity by
detecting fragmented DNA within the nucleus in the ABT-737-,
VX-680-, and combined ABT-737 and VX-680-treated cells. Briefly,
each culture plate was centrifuged for 10 min at 200 × g, the
supernatant was removed, and the pellet was lysed for 30 min. After
centrifuging the plate again at 200 × g for 10 min, the supernatant
that contained the cytoplasmic histone-associated DNA fragments was
collected and incubated with an immobilized anti-histone antibody.
The reaction products were incubated with a peroxidase substrate
for 5 min and measured by spectrophotometry at 405 and 490 nm
(reference wavelength) with a microplate reader. The signals in the
wells containing the substrate alone were subtracted as the
background.
Determination of synergy
The possible synergistic effect of ABT-737 and
VX-680 was evaluated using the isobologram method. Briefly, the
cells were treated with different concentrations of ABT-737 and
VX680 alone or in combination. After 48 h, relative survival was
assessed and the concentration effect curves were used to determine
the IC50 (the half-maximal inhibitory concentration)
values for each drug alone and in combination with a fixed
concentration of the second agent (15).
Asp-Glu-Val-Asp-ase (DEVDase) activity
assay
To evaluate DEVDase activity, cell lysates were
prepared after their respective treatments with ABT-737 in the
presence or absence of VX-680. Assays were performed in 96-well
microtiter plates by incubating 20 μg of cell lysates in 100 μl of
reaction buffer (1% NP-40, 20 μM Tris-HCl, pH 7.5, 137 μM NaCl, 10%
glycerol) containing a caspase substrate
[Asp-Glu-Val-Asp-chromophore-p-nitroanilide (DVAD-pNA)] at 5 μM.
Lysates were incubated at 37°C for 2 h. Thereafter, the absorbance
at 405 nm was measured with a spectrophotometer.
Reverse transcription-polymerase chain
reaction
Total RNA was isolated using TRIzol reagent (Life
Technologies, Gaithersburg, MD, USA), and cDNA was prepared using
M-MLV reverse transcriptase (Gibco-BRL, Gaithersburg, MD, USA)
according to the manufacturer’s instructions. The following primers
were used for the amplification of human Mcl-1 and actin: Mcl-1
forward, 5′-GCGACTGGCAAAGCT TGGCCTCAA-3′ and reverse,
5′-GTTACAGCTTGGATCC CAACTGCA-3′; Bcl-2 forward, 5′-GTCCTCAGCCCTCGC
TCT-3′ and reverse, 5′-CACCTAATTGGGCTCCATCT-3′; c-FLIP forward,
5′-CCCAGTGGACAGCGAGC-3′ and reverse, 5′-ACTGCAGGCTTCCTGTGCGC-3′,
actin forward, 5′-GGCATCGTCACCAACTGGGAC-3′ and reverse, 5′-CGA
TTTCCCGCTCGGCCGTGG-3′. PCR amplification was carried out using the
following cycling conditions: 94°C for 3 min followed by 17 (actin)
or 23 cycles (Bcl-2, Mcl-1 and c-FLIP) of 94°C for 45 sec, 58°C for
45 sec, 72°C for 1 min, and a final extension at 72°C for 10 min.
The amplified products were separated by electrophoresis on a 1.5%
agarose gel and detected under UV light.
DNA transfection and the luciferase
assay
Transient transfection was performed in 6-well
plates. One day before the transfection, MDA-MB-435S cells were
plated at ~60–80% confluency. The Bcl-2/-3254 promoter-luciferase,
NF-κB-luciferase, and cAMP response element (CRE)-luciferase
plasmid were transfected into the cells using Lipofectamine™ 2000
(Invitrogen, Carlsbad, CA, USA). The NF-κB-luciferase (plasmid no.
26699) plasmid was purchased from Addgene (Cambridge, MA, USA)
(16). The CRE-luciferase plasmid
was purchased from Clontech (Palo Alto, CA, USA). To assess the
promoter-driven expression of the luciferase gene, the cells were
collected and disrupted by sonication in lysis buffer (25 μM
Tris-phosphate pH 7.8, 2 μM EDTA, 1% Triton X-100 and 10%
glycerol), and aliquots of the supernatants were used to analyze
the luciferase activity according to the manufacturer’s
instructions (Promega, Madison, WI, USA).
Statistical analysis
The data were analyzed using a one-way ANOVA
followed by post-hoc comparisons (Student-Newman-Keuls) using the
statistical package for Social Sciences version 22.0 (SPSS Inc.,
Chicago, IL, USA).
Results
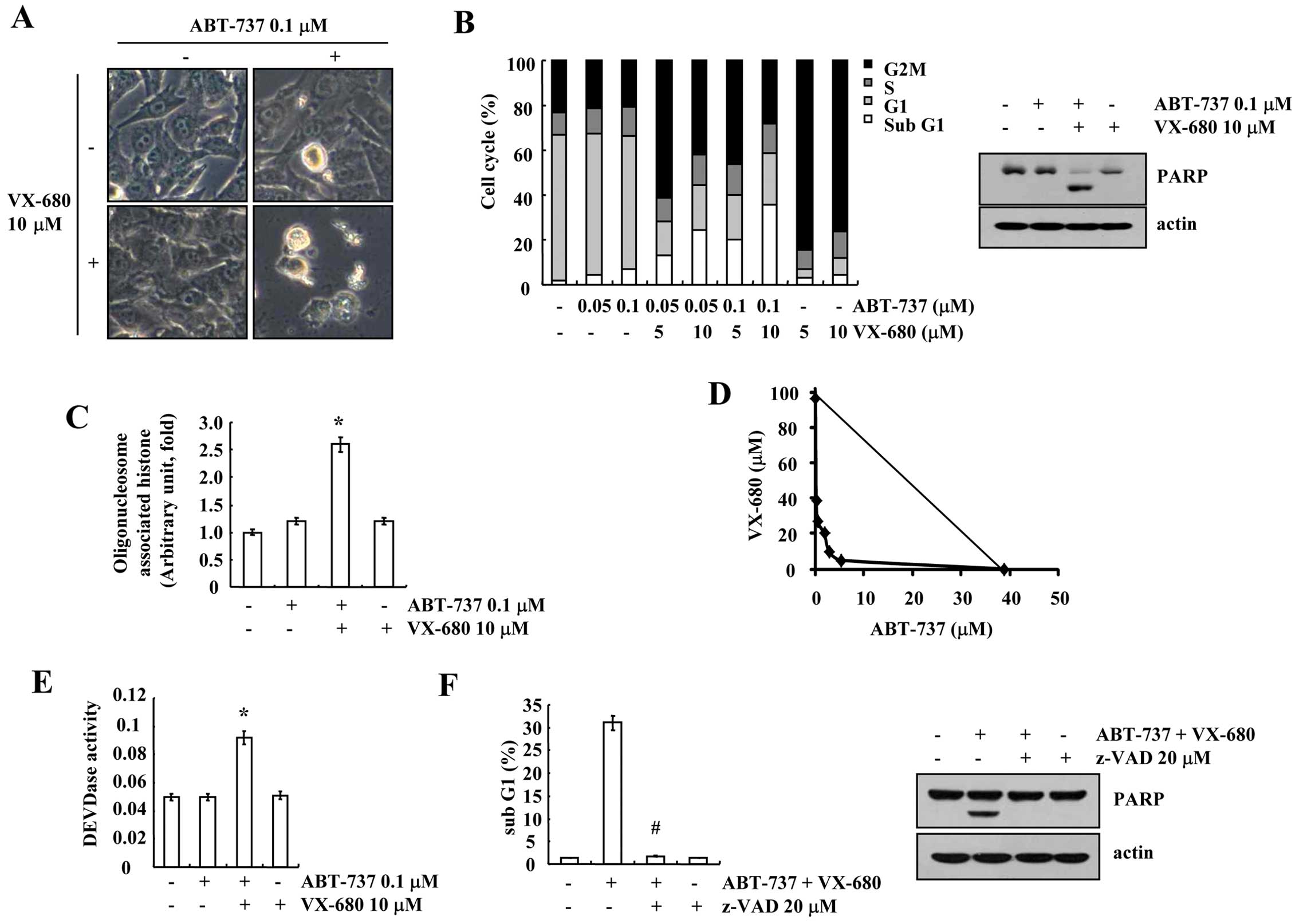
Combined treatment with ABT-737 and
VX-680 induces apoptosis in breast carcinoma MDA-MB-435S cells
Signaling molecule-target drugs inhibit specific
signaling, which is different between cancer and normal cells, and
a combination strategy reduces adverse effects, such as toxicity in
normal cells. Therefore, we examined whether a combined treatment
with sublethal dosages of ABT-737 and VX-680, which inhibits
ATP-binding of Aurora kinase A, B and C (17), induces apoptosis in breast carcinoma
MDA-MB-435S cells. ABT-737 plus VX-680 markedly induced cell
shrinkage and membrane blebbing (Fig.
1A). To determine whether ABT-737 plus VX-680 induces
apoptosis, FACS analysis to measure the DNA content and western
blotting to detect the cleavage of PARP, a substrate of caspase-3,
were performed. VX-680 induced G2/M arrest, but had no effect on
apoptosis. However, the combined treatment with ABT-737 and VX-680
increased the sub-G1 population and PARP cleavage (Fig. 1B). In addition, ABT-737 plus VX-680
markedly increased cytoplasmic histone-associated DNA fragmentation
(Fig. 1C). Next, to investigate
whether the combined treatment with ABT-737 and VX-680 has a
synergistic effect, MDA-MB-435S cells were treated with varied
concentrations of ABT-737 and VX-680. The isobologram analysis
revealed that the combined treatment with ABT-737 and VX-680 had
synergistic effects (Fig. 1D).
Next, to determine whether caspase is involved in the ABT-737 and
VX-680-induced apoptosis, we examined caspase activity. ABT-737 and
VX-680 increased caspase-3 activity, and a general caspase
inhibitor, z-VAD-fmk (benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl
ketone) markedly reduced apoptosis and PARP cleavage (Fig. 1E and F). These data revealed that
the combined treatment with ABT-737 and VX-680 induced
caspase-dependent apoptosis in the breast carcinoma MDA-MB-435S
cells.
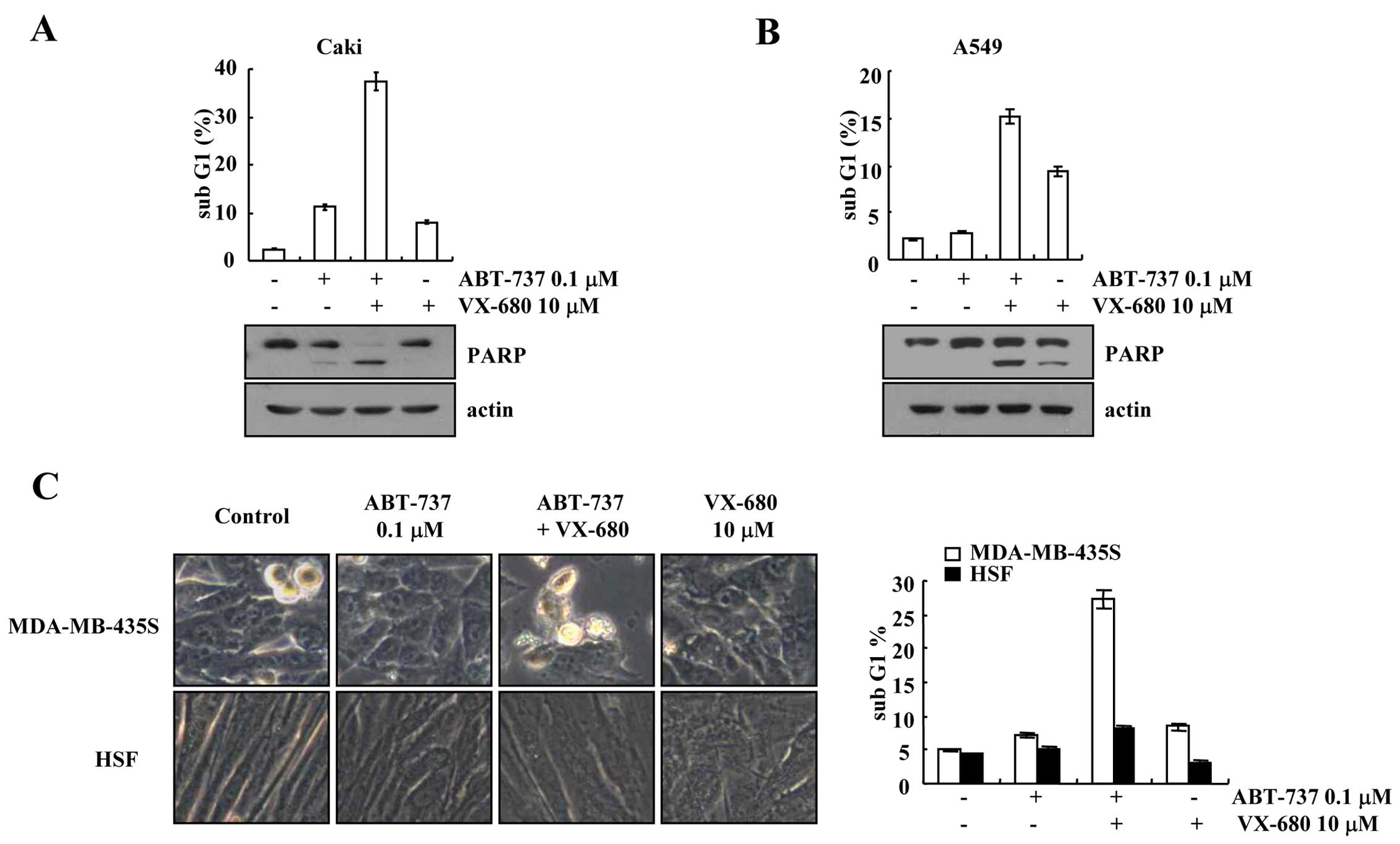
Effect of ABT-737 plus VX-680 on other
types of cancer cells and on normal cells
We investigated the effects of a combined treatment
with ABT-737 and VX-680 on other types of cancer cells [human renal
carcinoma (Caki cells), human lung carcinoma (A549 cells)] and
normal cells (HSFs)]. Combined treatment with ABT-737 and VX-680
markedly induced apoptosis and PARP cleavage in the Caki and A549
cells (Fig. 2A and B). When
MDA-MB-435S cells were treated with ABT-737 plus VX-680, the cell
morphology was altered including cellular shrinkage and blebbing,
while the morphology of the HSFs was not altered (Fig. 2C). Furthermore, combined treatment
with ABT-737 and VX-680 had no effect on the sub-G1 population in
the normal cells (Fig. 2C).
Therefore, our results indicate that the combined treatment of
ABT-737 and VX-680 induced apoptosis in cancer cells, but not in
normal cells.
Effect of ABT-737 plus VX-680 on
apoptosis-related proteins
Next, we investigated the expression levels of the
Bcl-2 family, the inhibitor of apoptosis (IAP) family and death
receptors in ABT-737 and VX-680-treated cells. As shown in Fig. 3A, ABT-737 and VX-680 alone had no
effect on apoptosis-related proteins, while the combined treatment
with ABT-737 and VX-680 reduced c-FLIP, Bcl-2 and Mcl-1 protein
expression. Furthermore, downregulation of Bcl-2 mRNA was detected,
while the mRNA expression levels of c-FLIP and Mcl-2 were not
altered in the ABT-737 plus VX-680-treated cells (Fig. 3B). To further confirm the mechanism
of Bcl-2 downregulation in ABT-737 plus VX-680-treated cells, we
examined whether the combined treatment with ABT-737 and VX-680
inhibits Bcl-2 promoter activity. As shown in Fig. 3C, ABT-737 plus VX-680 markedly
reduced Bcl-2 promoter (Bcl-2/-3254) activity. Furthermore, we
investigated whether the combined treatment with ABT-737 plus
VX-680 inhibits NF-κB and CRE-associated transcriptional
activities, which are important to Bcl-2 mRNA expression (18–20).
NF-κB and CRE transcriptional activities were markedly reduced in
the ABT-737 plus VX-680-treated cells (Fig. 3D). These results indicated that
NF-κB and CRE transcriptional activities are involved in ABT-737
plus VX-680-mediated downregulation of Bcl-2 expression.
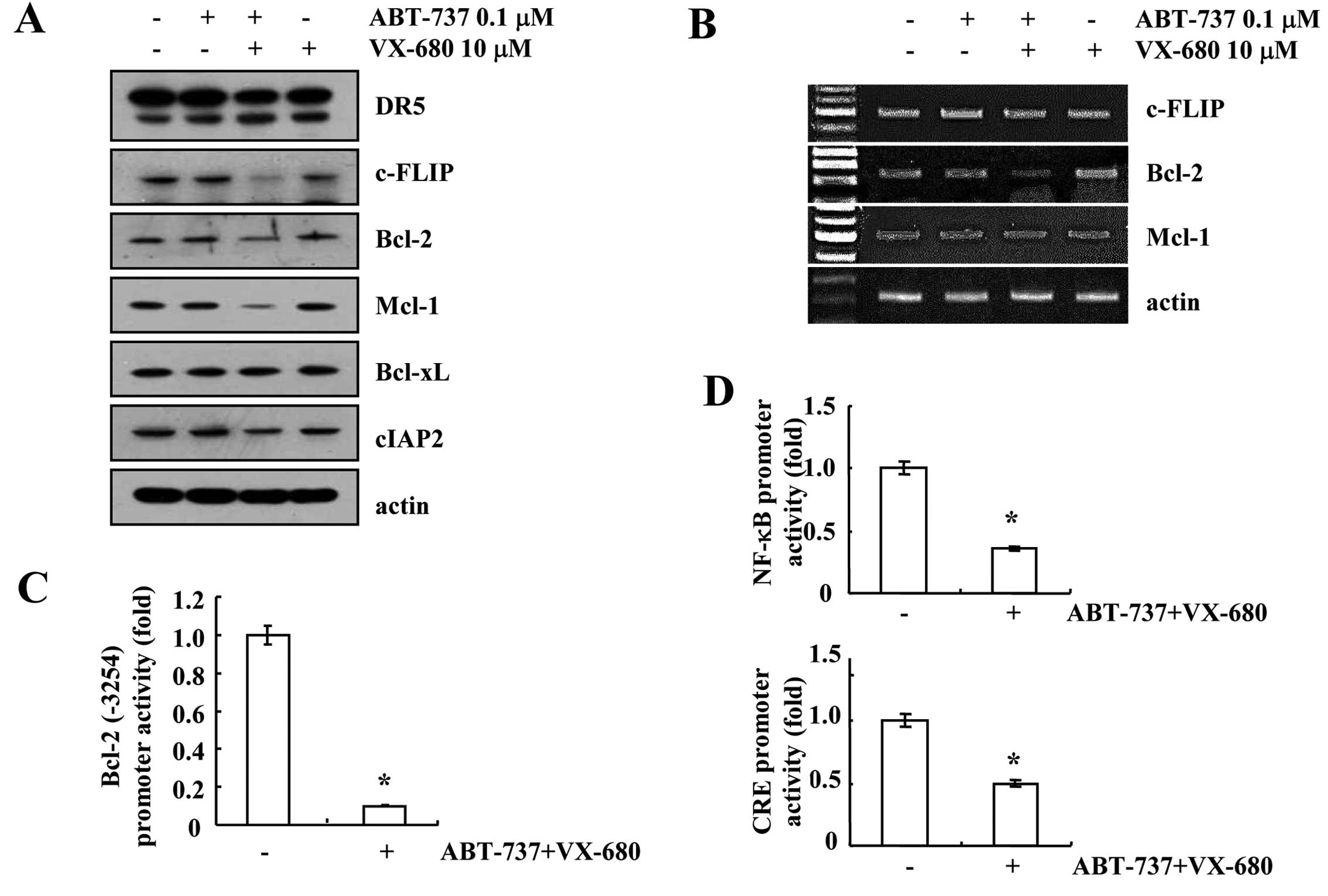 | Figure 3Combined treatment with ABT-737 and
VX-680 downregulates c-FLIP, Bcl-2 and Mcl-1 expression.
MDA-MB-435S cells were co-treated with 0.1 μM ABT-737 and 10 μM
VX-680 for 24 h. (A) The protein expression levels of DR5, c-FLIP,
Bcl-2, Mcl-1, Bcl-xL, cIAP2 and actin were determined by western
blotting. The level of actin was used as a loading control. (B)
c-FLIP, Bcl-2, Mcl-1 and actin mRNA were determined using RT-PCR.
The level of actin was used as a loading control. (C) MDA-MB-435S
cells were transiently transfected with a plasmid harboring the
luciferase gene under the control of the Bcl-2/-3254 promoter.
After transfection, MDA-MB-435S cells were treated with 0.1 μM
ABT-737 and 10 μM VX-680 for 24 h. After treatment, the cells were
lysed and then assayed for luciferase activity. (D) MDA-MB-435S
cells were transiently transfected with the NF-κB-luciferase
construct or the CRE-luciferase construct. After transfection, the
MDA-MB-435S cells were treated with 0.1 μM ABT-737 and 10 μM VX-680
for 18 h. After treatment, the cells were lysed, and the luciferase
activity was analyzed. *P<0.01 compared to the
control. DR5, death receptor 5; CRE, cAMP response element. |
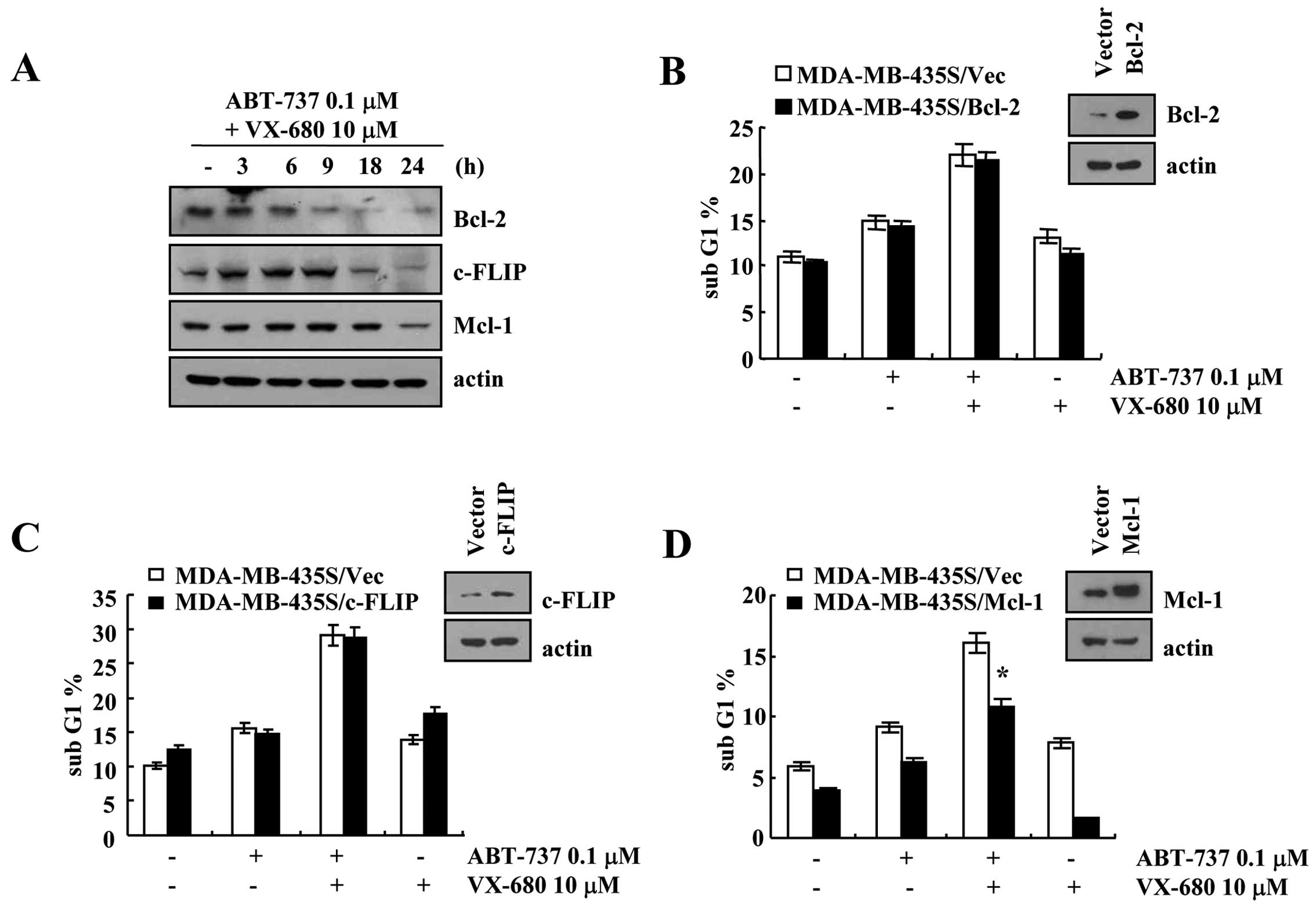
Effect of Bcl-2, c-FLIP and Mcl-1
overexpression on ABT-737 plus VX-680-mediated apoptosis
Combined treatment with ABT-737 and VX-680
downregulated Bcl-2, c-FLIP and Mcl-1 protein expression within 9,
18 and 24 h, respectively (Fig.
4A). To investigate whether downregulation of Bcl-2, c-FLIP and
Mcl-1 is associated with ABT-737 plus VX-680-induced apoptosis,
MDA-MB-435S cells were transiently transfected with Bcl-2, c-FLIP
and Mcl-1. As shown in Fig. 4,
overexpression of Bcl-2 or c-FLIP had no effect on the ABT-737 plus
VX-680-mediated apoptosis, but overexpression of Mcl-1 partially
blocked apoptosis in the ABT-737 plus VX-680-treated cells. These
data revealed that combined treatment with ABT-737 and VX-680
induced apoptosis in the Bcl-2- or c-FLIP-overexpressed cells.
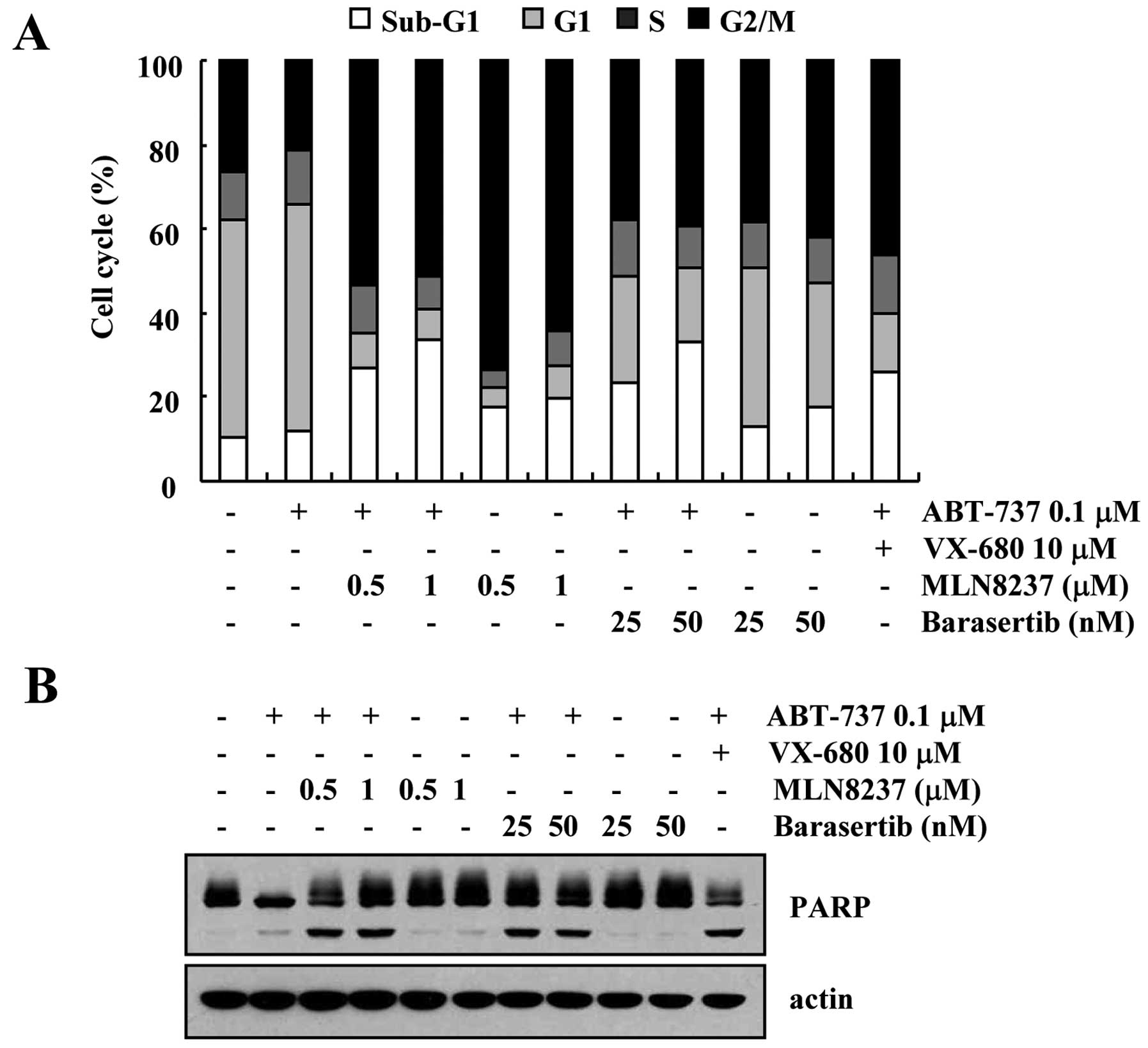
Effect of inhibition of Aurora kinase A
and B on apoptosis in VX-680-treated cells
In the present study, we used VX-680, which inhibits
both Aurora kinase A and B. To identify which Aurora kinase plays
an important role in ABT-737-induced apoptosis, we tested more
specific inhibitors of Aurora kinase A (MLN8237) and Aurora kinase
B (barasertib). As shown in Fig. 5,
both inhibitors of Aurora kinase A (MLN8237) and Aurora kinase B
(barasertib) induced an increase in the sub-G1 population and PARP
cleavage in the ABT-737-treated cells (Fig. 5A and B). Therefore, these results
revealed that inhibition of both Aurora kinase A and B could be
involved in the apoptosis in ABT-737-treated cells.
Discussion
In the present study, we demonstrated that combined
treatment with ABT-737 and VX-680 induced apoptosis in breast
carcinoma MDA-MB-435S cells through downregulation of Bcl-2, c-FLIP
and Mcl-1 expression. ABT-737 and VX-680 alone did not alter the
expression levels of Bcl-2, c-FLIP and Mcl-1. However, combined
treatment with ABT-737 and VX-680 induced the downregulation of
Bcl-2 expression at the transcriptional level and downregulation of
c-FLIP and Mcl-1 at the post-transcriptional level. Furthermore,
ABT-737 plus VX-680 had no effect on the apoptosis of normal cells.
Therefore, our results revealed that combined treatment with
ABT-737 and VX-680 is an effective strategy to induce apoptosis in
cancer cells.
In breast cancer cells, Aurora kinases are
overexpressed, thus targeting to Aurora kinases is a good strategy
by which to induce apoptosis. Pan-Aurora kinase inhibitor (VX-680)
alone did not induce apoptosis, whereas VX-680 induced G2/M arrest
(Fig. 1B). Combined treatment with
ABT-737 and VX-680 markedly induced apoptosis through caspase
activation (Fig. 1E). As shown in
Fig. 3A, ABT-737 plus VX-680
markedly reduced c-FLIP, Bcl-2, and Mcl-1 protein expression.
Firstly, downregulation of Bcl-2 is modulated at the
transcriptional levels. Several transcription factors regulate
Bcl-2 transcriptional level. Among them, p53 negatively modulates
Bcl-2 expression (21–23). We investigated whether combined
treatment with ABT-737 and VX-680 downregulates Bcl-2 mRNA by p53.
However, pifithrin-α (p53 inhibitor) had no effect on the
downregulation of ABT-737 plus VX-680-mediated Bcl-2 expression,
and combined treatment with ABT-737 and VX-680 reduced Bcl-2
expression in wild-type p53 HCT116 human colon carcinoma cells and
p53-null HCT116 cells (data not shown). Therefore, we ruled out the
possibility of p53-mediated modulation. NF-κB and CRE are
associated with Bcl-2 mRNA expression (18–20).
We found that combined treatment with ABT-737 and VX-680 markedly
decreased NF-κB and CRE transcriptional activities (Fig. 3D). These results indicate that the
ABT-737 plus VX-680-mediated inhibition of NF-κB and CRE
transcriptional activities is probably involved in the
downregulation of Bcl-2 expression. Next, to identify the
importance of Bcl-2 in ABT-737 plus VX-680-mediated apoptosis,
MDA-MB-435S cells were transiently transfected with the Bcl-2
expression plasmid. As shown in Fig.
4B, although Bcl-2 was overexpressed, the combined treatment
with ABT-737 plus VX-680 markedly induced apoptosis. ABT-737 binds
to Bcl-2, Bcl-xL and Bcl-w with a very high affinity, resulting in
induction of apoptosis. ABT-737 inhibited the anti-apoptotic
function of Bcl-2, thus the population of apoptotic cells was not
changed. Secondly, ABT-737 plus VX-680 reduced Mcl-1 and c-FLIP
expression at the post-transcriptional level. Ubiquitin-dependent
proteasomal degradation is a major mechanism of
post-transcriptional regulation (24), and Mcl-1 and c-FLIP are also mainly
degraded by the ubiquitin-proteasome pathway. Therefore, further
experiments are needed to identify the relationship of proteasome
activation in the downregulation of Mcl-1 and c-FLIP. As shown in
Fig. 4D, overexpression of Mcl-1
attenuated ABT-737 plus VX-680-induced apoptosis. Since ABT-737 has
a low affinity for Mcl-1, Mcl-1-overexpressing cells are resistant
to ABT-737-induced apoptosis. In contrast, when c-FLIP was
overexpressed, ABT-737 plus VX-680 induced apoptosis (Fig. 4C). Song et al, reported that
ABT-737 (>5 μM) induces DR5 expression in renal carcinoma cells
(25). Induction of DR5 expression
overcomes endogenous c-FLIP expression (25). However, in the present study,
ABT-737 did not induce DR5 expression (Fig. 3A), while ABT-737 plus VX-680 induced
apoptosis in the c-FLIP-overexpressing cells. Further experiments
are needed to identify the mechanism of ABT-737 plus
VX-680-mediated apoptosis in c-FLIP-overexpressing cells.
Taken together, our results revealed that the
combination treatment with ABT-737 and VX-680 induced apoptosis in
breast cancer cells, and overcame resistance by overexpression of
Bcl-2 and c-FLIP.
References
|
1
|
Yip KW and Reed JC: Bcl-2 family proteins
and cancer. Oncogene. 27:6398–6406. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Certo M, Del Gaizo Moore V, Nishino M, et
al: Mitochondria primed by death signals determine cellular
addiction to anti-apoptotic BCL-2 family members. Cancer Cell.
9:351–365. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim H, Rafiuddin-Shah M, Tu HC, et al:
Hierarchical regulation of mitochondrion-dependent apoptosis by
BCL-2 subfamilies. Nat Cell Biol. 8:1348–1358. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Oltersdorf T, Elmore SW, Shoemaker AR, et
al: An inhibitor of Bcl-2 family proteins induces regression of
solid tumours. Nature. 435:677–681. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Del Gaizo Moore V, Brown JR, Certo M, Love
TM, Novina CD and Letai A: Chronic lymphocytic leukemia requires
BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2
antagonist ABT-737. J Clin Invest. 117:112–121. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chauhan D, Velankar M, Brahmandam M, et
al: A novel Bcl-2/Bcl-X(L)/Bcl-w inhibitor ABT-737 as therapy in
multiple myeloma. Oncogene. 26:2374–2380. 2007. View Article : Google Scholar
|
|
7
|
Konopleva M, Contractor R, Tsao T, et al:
Mechanisms of apoptosis sensitivity and resistance to the BH3
mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 10:375–388.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
van Delft MF, Wei AH, Mason KD, et al: The
BH3 mimetic ABT-737 targets selective Bcl-2 proteins and
efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized.
Cancer Cell. 10:389–399. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Andrews PD, Knatko E, Moore WJ and Swedlow
JR: Mitotic mechanics: the auroras come into view. Curr Opin Cell
Biol. 15:672–683. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hirota T, Kunitoku N, Sasayama T, et al:
Aurora-A and an interacting activator, the LIM protein Ajuba, are
required for mitotic commitment in human cells. Cell. 114:585–598.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Farag SS: The potential role of Aurora
kinase inhibitors in haematological malignancies. Br J Haematol.
155:561–579. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gautschi O, Heighway J, Mack PC, Purnell
PR, Lara PN Jr and Gandara DR: Aurora kinases as anticancer drug
targets. Clin Cancer Res. 14:1639–1648. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gritsko TM, Coppola D, Paciga JE, et al:
Activation and overexpression of centrosome kinase BTAK/Aurora-A in
human ovarian cancer. Clin Cancer Res. 9:1420–1426. 2003.PubMed/NCBI
|
|
14
|
Katayama H, Brinkley WR and Sen S: The
Aurora kinases: role in cell transformation and tumorigenesis.
Cancer Metastasis Rev. 22:451–464. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tallarida RJ: Drug synergism: its
detection and applications. J Pharmacol Exp Ther. 298:865–872.
2001.PubMed/NCBI
|
|
16
|
Mitchell T and Sugden B: Stimulation of
NF-kappa B-mediated transcription by mutant derivatives of the
latent membrane protein of Epstein-Barr virus. J Virol.
69:2968–2976. 1995.PubMed/NCBI
|
|
17
|
Harrington EA, Bebbington D, Moore J, et
al: VX-680, a potent and selective small-molecule inhibitor of the
Aurora kinases, suppresses tumor growth in vivo. Nat Med.
10:262–267. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pugazhenthi S, Miller E, Sable C, et al:
Insulin-like growth factor-I induces bcl-2 promoter through the
transcription factor cAMP-response element-binding protein. J Biol
Chem. 274:27529–27535. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tamatani M, Che YH, Matsuzaki H, et al:
Tumor necrosis factor induces Bcl-2 and Bcl-x expression through
NFkappaB activation in primary hippocampal neurons. J Biol Chem.
274:8531–8538. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Catz SD and Johnson JL: Transcriptional
regulation of bcl-2 by nuclear factor kappa B and its significance
in prostate cancer. Oncogene. 20:7342–7351. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bourgarel-Rey V, Savry A, Hua G, et al:
Transcriptional down-regulation of Bcl-2 by vinorelbine:
identification of a novel binding site of p53 on Bcl-2 promoter.
Biochem Pharmacol. 78:1148–1156. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Haldar S, Negrini M, Monne M, Sabbioni S
and Croce CM: Down-regulation of bcl-2 by p53 in breast cancer
cells. Cancer Res. 54:2095–2097. 1994.PubMed/NCBI
|
|
23
|
Wu Y, Mehew JW, Heckman CA, Arcinas M and
Boxer LM: Negative regulation of bcl-2 expression by p53 in
hematopoietic cells. Oncogene. 20:240–251. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ciechanover A and Schwartz AL: The
ubiquitin system: pathogenesis of human diseases and drug
targeting. Biochim Biophys Acta. 1695:3–17. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Song JH, Kandasamy K and Kraft AS: ABT-737
induces expression of the death receptor 5 and sensitizes human
cancer cells to TRAIL-induced apoptosis. J Biol Chem.
283:25003–25013. 2008. View Article : Google Scholar : PubMed/NCBI
|