Introduction
The human ubiquitin-specific processing enzyme 22
(USP22) gene, which is located on chromosome 17 and consists of 14
exons, belongs to the deubiquitinating enzyme superfamily. Its
transcription product is a 525-amino acid polypeptide that is
ubiquitously expressed in human adult tissues, including the heart
and skeletal muscle, and is regularly expressed during early
embryogenesis in mice (1).
Recently, elevated levels of USP22 have been found in most types of
human cancers, including colorectal (2), gastric (3) and breast cancer (4), suggesting a crucial role in
tumorigenesis.
Structurally, USP22 contains a C-terminal peptidase
domain, which functions as an ubiquitin hydrolase and an N-terminal
zinc finger motif, mediating other protein associations. As the key
subunit of the human SAGA (hSAGA) transcriptional coactivator,
USP22 functions as a regulatory activator governing the expression
of several genes related to cancer and proliferation (5). Furthermore, USP22 has been shown to be
required for the transcription of target genes regulated by the Myc
oncoprotein, including cyclin D2, CAD, MTA1, with its
downregulation leading to cell-cycle arrest (5,6). USP22
has also been shown to deubiquitinate intracellular proteins,
including a shelterin protein (7),
a histone deacetylase (Sirt1) (8),
and the far upstream element-binding protein 1 (9). Murine studies have shown that Usp22
also regulates embryonic stem cell differentiation (10). Due to its extensive role in cancer
progression, USP22 is considered a putative cancer stem cell marker
and may serve as a novel cancer therapeutic target. However, the
mechanisms leading to USP22 transcriptional activation,
particularly in human cancer cells, are still unknown.
Previously, the USP22 promoter was cloned and
characterized in HeLa cells by generating a series of 5′ deletions,
to reveal that the −210 to +52 region contains the basal promoter
activity (11). The TFSEARCH
program was used to analyze this region and revealed the presence
of a putative CREB binding site. However, the expressional role of
this site in relation to USP22 promoter activity, or the role of
putative transcription factor(s) binding, has not been established.
Thus, the aim of the present study was to investigate the role of
the CREB binding site in relation to USP22 transcriptional activity
and identify any associated transcription factors. This study also
aimed to characterize any upstream pathways regulating USP22
transcription through this site. Overall, the present study
provides evidence to delineate the molecular mechanism governing
human USP22 gene expression.
Materials and methods
Cell culture
Human cervical carcinoma (HeLa) cells and
hepatocellular carcinoma (HepG2) cells were obtained from the
American Tissue Culture Collection (ATCC; Manassas, VA, USA) and
cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen,
Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS)
at 37°C in a 5% CO2 and 95% air incubator.
Plasmid constructs and cloning
Site-directed mutagenesis to inactivate the CREB
binding site was carried out within the p-210 construct containing
262 bp of the 5′-flanking sequence of the USP22 gene using the
MutanBEST kit (Takara, Shiga, Japan) according to the
manufacturer’s protocols. The following primers:
5′-GTagCGTAATCTCCGTCCGC-3′ (sense) and 5′-CCCCCCCCCCCCTCCCGGCC-3′
(antisense) were utilized and the mutation was confirmed by DNA
sequencing.
CREB-1 full-length cDNA was obtained by reverse
transcription PCR (RT-PCR). Briefly, total RNA was obtained from
HeLa cells and reverse-transcribed with SuperScript™ II primed by
oligo(dT)15 (Takara). cDNA products were amplified using
the following PCR primers: 5′-AGGATCCATGACCATGGAATCTGGAGCC-3′
(sense) and 5′-GGAATTCTTAATCTGATTTGTGGCAGTA-3′ (antisense),
digested with EcoRI and BamHI, and inserted into
pCDNA 3.1(+).
Transfection and the dual luciferase
reporter assay
Cells were plated in 24-well plates 24 h before
transfection, with each well treated with 0.5 μg of promoter
construct, 0.1 μg of pRL-TK (Promega, Madison, W, USA), and
Lipofectamine 2000 (Invitrogen). All transfection experiments were
repeated five times, with cells lysed 24 h after transfection and
lysates used for the dual luciferase assays (Promega) according to
the manufacturer’s protocol. The normalized luciferase activity was
expressed as a ratio of firefly luciferase activity to
Renilla luciferase for each sample.
Chromatin immunoprecipitation (ChIP)
assay
ChIP assays were performed using the EZ-ChIP kit
according to the manufacturer’s instructions (Pierce, Rockford, IL,
USA). Approximately 108 cells were fixed by adding
formaldehyde to a final concentration of 1% and incubated for 30
min at room temperature with modest shaking. Thereafter, the cells
were washed twice with cold phosphate-buffered saline, the pellet
was resuspended and lysed, and the nuclei were isolated and
sonicated until the chromatin had an average length of 400–500 bp.
Following centrifugation, the supernatant was incubated with 1 μg
of anti-CREB1 antibody or rabbit IgG overnight at 4°C for
immunoprecipitation. The following day, magnetic protein-G beads
were added and further incubated at 4°C for 1 h. After appropriate
washing, the antibody-transcription factor-DNA complex was eluted
from the beads, formaldehyde cross-links were reversed, and
proteins were digested with proteinase K at 67°C overnight. DNA was
purified and used for PCR with the primers,
5′-GGGCGGGCCCTCGTTAGCTT-3′ and 5′-GGAGGCTGCAAGGCAGGCAC-3′.
RNA interference
One day before transfection, HeLa cells were plated
in 24-well plates at a density of 5×104 cells/well, and
then transfected with 20 nM of siControl Non-Targeting siRNA duplex
(siControl) or human CREB1-specific siRNA duplex (sc-29281; Santa
Cruz Biotechnology, Santa Cruz, CA, USA), using an siRNA reagent
system (Santa Cruz Biotechnology) according to the manufacturer’s
instructions. After 24 h, the siRNA-transfected cells were
harvested for real-time PCR or transfected with p-210 for the
luciferase assay.
Quantitative real-time PCR (qRT-PCR)
Cells were grown to confluency and RNA was extracted
using TRIzol® reagent (Invitrogen). cDNA was synthesized
from 1 μg of total RNA using SuperScript II reverse transcriptase
(Invitrogen) and random hexamers in a total volume of 20 μl
according to the manufacturer’s instructions. The GAPDH gene was
used as an endogenous control to normalize for RNA loading. qRT-PCR
was performed using the SYBR-Green PCR Master Mix (Toyobo, Osaka,
Japan) on an ABI 7500 Real-Time PCR System (Applied Biosystems,
Foster City, CA, USA), with USP22 primers as previously described
and validated (11).
Western blot analysis
Cells were lysed with 1X SDS sample buffer, and the
protein was separated by 10% SDS-PAGE, and electroblotted onto a
nitrocellulose membrane. The membrane was then incubated with the
anti-USP22 antibody (Santa Cruz Biotechnology), the anti-CREB1
antibody (Santa Cruz Biotechnology), or the anti-GAPDH antibody
(Santa Cruz Biotechnology) and visualized using the Pierce ECL
system according to the manufacturer’s instructions.
Statistical analysis
Data are presented as mean ± SEM. Statistical
differences between sample means were determined using an unpaired,
two-tailed Student’s t-test, with P<0.05 deemed significant.
Results
CREB binding site mutation decreases
USP22 promoter activity
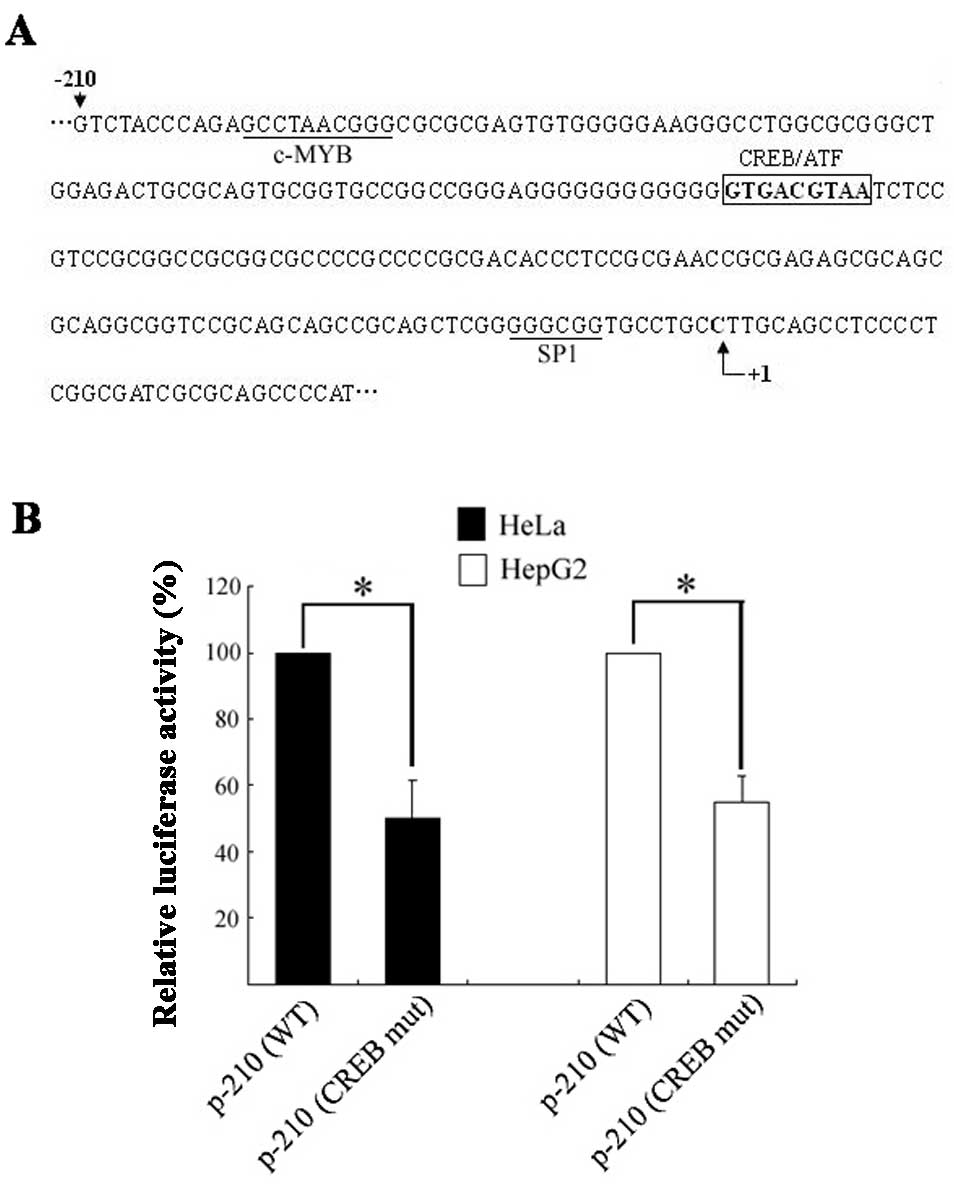
Analysis of the basic USP22 promoter region by the
TFSEARCH program revealed a putative CREB binding site (GTGAGCTAA)
at position −113 to −105 (Fig. 1A)
with a 100 point score, which is highly conserved among several
mammalian species, including the mouse and rat. To evaluate the
impact of this CREB motif on USP22 promoter activity, site-directed
mutagenesis was introduced within the basic promoter p-210, and the
resulting plasmids were transiently transfected into the HeLa and
HepG2 cells. The p-210/CREBmut construct showed an ~45% decrease in
luciferase activity in both the HeLa and HepG2 cells compared to
the wild-type p-210 (Fig. 1B),
suggesting that the CREB site is critical for constitutive USP22
promoter activity in the cells tested.
CREB interacts with the USP22
promoter
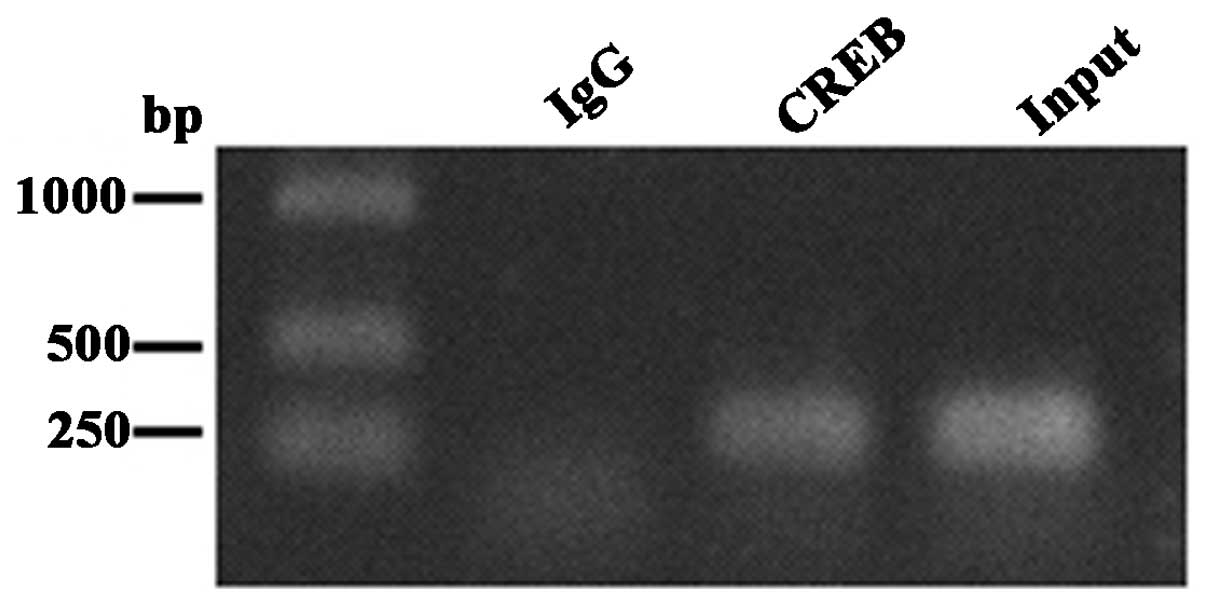
To determine whether CREB directly binds to the
USP22 promoter in vivo, chromatin immunoprecipitation (ChIP)
was performed using cultured HeLa cells. Chromatin was sonicated
into ~400–500-bp fragments and precipitated using the anti-CREB-1
antibody or rabbit IgG (negative control). The precipitated DNA was
subjected to PCR using primers designed to amplify a 272-bp
fragment of the USP22 promoter encompassing the CREB site.
Following immunoprecipitation, the samples treated with the
anti-CREB-1 antibody yielded a 272-bp PCR product, while no PCR
amplification product was detected following IgG incubation
(Fig. 2). These results indicate
that CREB-1 constitutively occupies this region of the USP22
promoter, and thus, is likely to contribute to basal
transcriptional activation.
Effect of CREB on USP22 promoter
activity
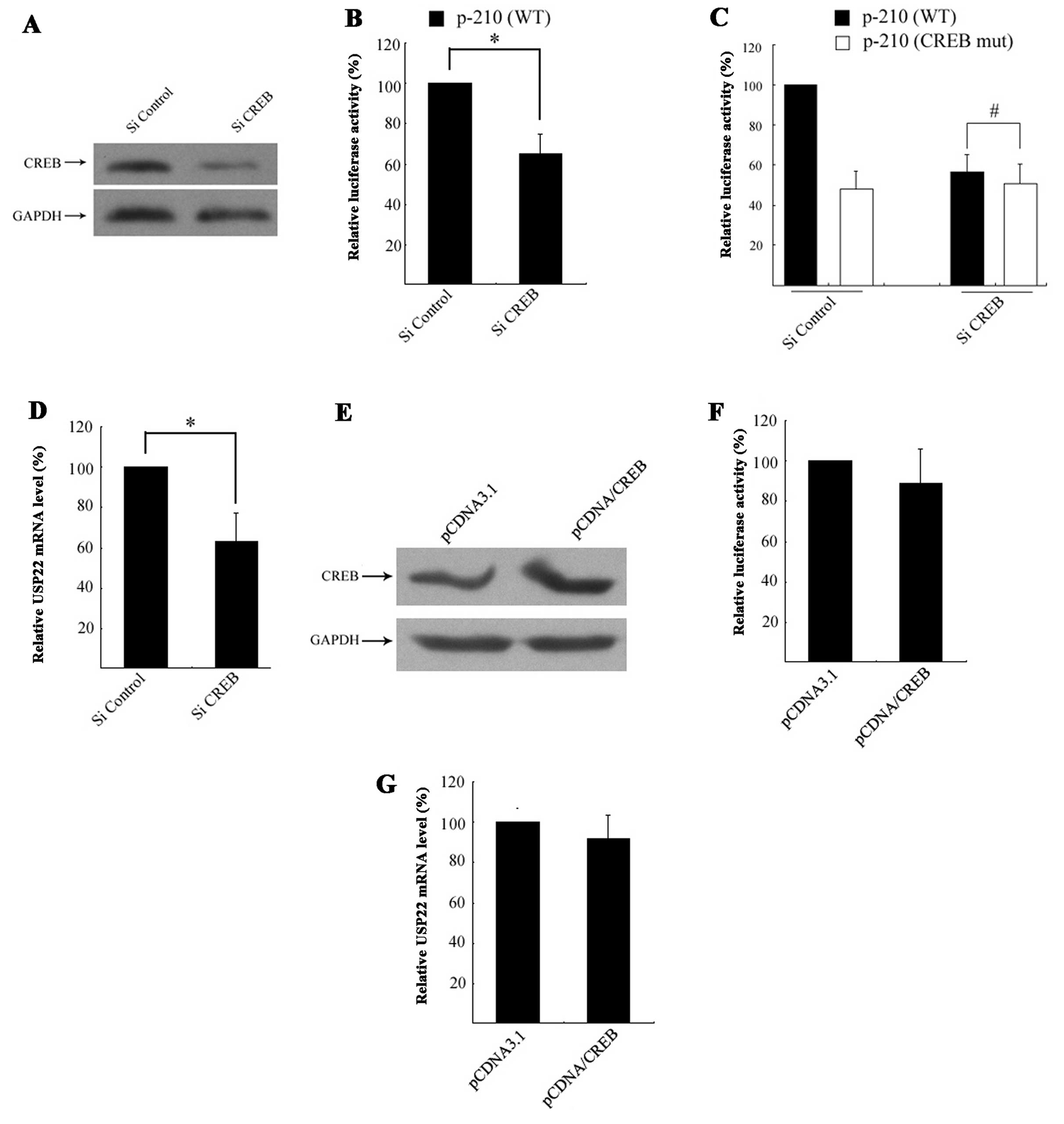
To determine whether CREB is necessary for basal
USP22 transcription, siRNA-mediated knockdown was employed.
CREB-specific siRNA resulted in an ~80% reduction in CREB protein
expression relative to the control (Fig. 3A), which resulted in a significant
decrease in p-210 (WT) luciferase activity, with no change in
luciferase activity observed in the control group (Fig. 3B). To confirm CREB regulation of the
USP22 promoter via the CREB site, HeLa cells were co-transfected
with CREB siRNA and the p-210/CREBmut plasmid. This showed that
siRNA-mediated knockdown had no significant impact on p-210/CREBmut
promoter activity (Fig. 3C).
Furthermore, the effect of CREB deletion on endogenous USP22
expression was determined by monitoring USP22 expression following
CREB siRNA transfection via qRT-PCR. The results showed that
siRNA-mediated knockdown of CREB led to a significant reduction in
USP22 mRNA expression levels (Fig.
3D).
Since it was now established that CREB is required
for USP22 transcription, the effect of exogenous CREB on USP22
transcriptional activity was further examined through the
transfection of HeLa cells with CMV-driven CREB expression plasmids
pCDNA/CREB (Fig. 3E). The results
showed that forced expression of exogenous CREB did not affect
USP22 promoter activity (Fig. 3F).
Accordingly, exogenous CREB did not promote endogenous USP22 mRNA
expression (Fig. 3G). These results
suggested that CREB behaves as a constitutive rather than an
inducible element for USP22 transcription.
H-89 decreases USP22 expression via the
CREB binding site
It has been reported that CREB transcriptional
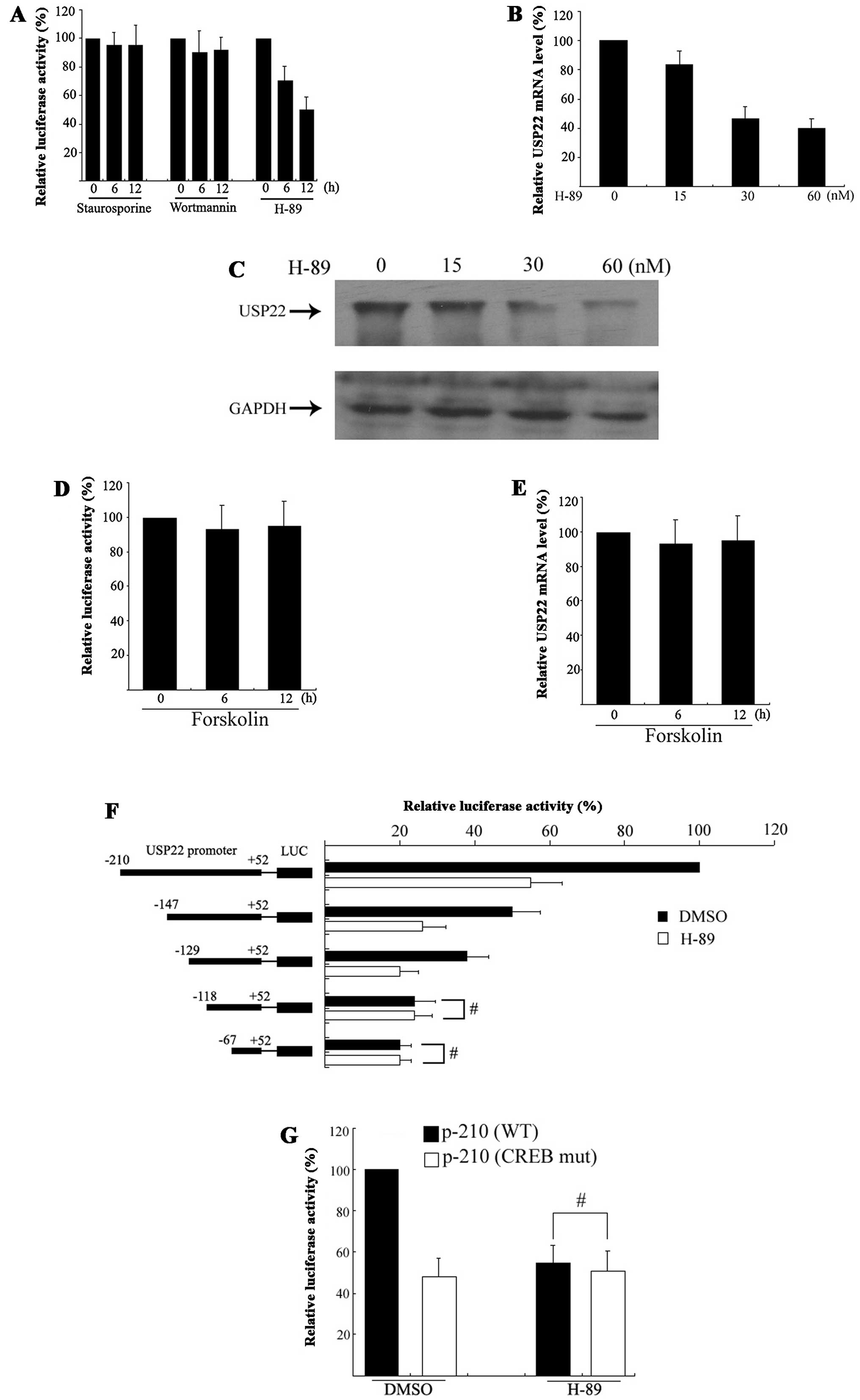
activity is controlled by several upstream signaling pathways.
Therefore, HeLa cell USP22 promoter activity was pharmacologically
targeted using H-89, staurosporine and wortmannin to selectively
inhibit PKA, PKC and PI3K, respectively. Based on published reagent
concentrations (12–14), HeLa cells were treated with 30 nM of
H-89, 10 nM of staurosporine, or 10 nM of wortmannin for 6 or 12 h
following a 24-h transfection with p-210. The dual luciferase assay
showed that H-89, but not staurosporine or wortmannin, decreased
promoter activity in a time-dependent manner (Fig. 4A), thus, indicating that the PKA
signaling pathway could play a positive role in the regulation of
USP22 transcription.
To determine whether H-89 suppresses endogenous
USP22 expression, HeLa cells were treated with 15, 30 or 60 nM of
H-89 for 12 h, with both USP22 mRNA and protein expression levels
analyzed. As shown in Fig. 4B and
C, 30 nM of H-89 induced a very significant decrease in USP22
mRNA and protein expression in the HeLa cells. Furthermore, HeLa
cells were treated with forskolin, which raises intracellular
levels of cAMP. Unexpectedly, incubation with 10 μM of forskolin
did not increase USP22 promoter activity or endogenous mRNA
expression (Fig. 4D and E). These
results indicate that the PKA pathway perhaps plays a constitutive
rather than an inducible role in USP22 promoter activity.
To delineate the region(s) that respond to H-89, a
series of 5′ truncated promoter fragments derived from p-210 were
developed and the HeLa cells were transfected. A dual luciferase
assay showed that USP22 promoter activity was serially decreased
when the 5′ terminal of P-120 was continuously cut. Furthermore,
when the 5′ terminal of P-120 was cut by 92 bp (P-118), a loss of
H-89 responsiveness was noted (Fig.
4F), suggesting that the H-89 response region is located
between −129 bp and −118 bp which contains the functional CREB
binding site. To determine whether H-89 decreases USP22 expression
via the CREB binding site, HeLa cells were transfected with
wild-type p-210 or p-210/CREBmut and incubated with H-89 for 12 h.
A dual luciferase assay showed that a mutation of the CREB binding
site abolished H-89 responsiveness (Fig. 4G), collectively suggesting that the
PKA pathway regulates USP22 expression and its effects may be
partly mediated by CREB.
Discussion
Recently, the deubiquitinating enzyme USP22 has
attracted attention due to the fact that its overexpression is
considered to be a hallmark of malignant cancer progression and is
linked to a poor prognosis (15,16).
However, the mechanisms leading to USP22 transcriptional activation
are still unclear. In the present study, we demonstrated that a
functional CREB-binding site is required for USP22 basic promoter
activity and the CREB transcription factor binding to this site
plays a positive role in constitutive transcription. Furthermore,
silencing of CREB expression by siRNA, as well as PKA inhibition by
H-89, decreased USP22 transcription, whereas overexpression of CREB
or PKA activation by forskolin did not present enhancing
effects.
Previously, we identified a 262-bp (−210 to +52)
region as the basic promoter for USP22 (11). Inspection of this region revealed
the existence of a putative CREB consensus site consisting of the
sequence GTGACGTAA at position −113 to −105. Usually, a classic
CREB site consists of the palindromic sequence, TGACGTCA, and
locates at 50–150 bp upstream from the start site of transcription
(17). Although the putative 9-bp
CREB binding site determined by TFSEARCH analysis is an atypical
CREB motif, prior reports have confirmed that it is recognized and
bound by CREB or other CREB/ATF members (18). In the present study, mutation of
this putative CREB site resulted in a significant reduction in
USP22 promoter activity, indicating that it plays a positive
constitutive role in USP22 promoter activity. Therefore,
delineating the transcription factor(s) binding to this site will
enable a better understanding of the mechanisms responsible for
USP22 expression.
In the present study, we did not characterize all
the transcription factors binding to this site, but confirmed that
CREB constitutively binds to this site in vitro. As a
ubiquitously expressed transcription factor, CREB is involved in
regulating various transcriptional genes. Many oncogenes have been
found to be activated by CREB, including cyclins (19), Bcl-2 family members (20) and Egr-1 (21), with elevated CREB levels noted in
several human cancers (22).
Therefore, CREB is considered a key factor in mediating the
malignant behavior of tumor cells. In addition to activating
transcription, CREB can also mediate transcriptional repression by
partnering with repressor proteins. For example, CREB acts as a
negative regulator of AP-2α by directly binding the AP-2α promoter
in melanoma cells (23). In the
present study, we demonstrated that CREB plays a positive role in
the regulation of USP22 transcription, with endogenous CREB
knockdown resulting in a reduction in USP22 promoter activity and
endogenous expression. These findings suggest that the regulation
of USP22 expression by CREB may have important consequences for
cancer progression.
It has been reported that CREB can stimulate both
basal transcription and inducible transcription through its
bipartite transcriptional activation domain (24). In this study, inhibition of CREB
decreased USP22 expression, but overexpression of exogenous CREB
did not enhance USP22 promoter activity, thus indicating that CREB
may not mediate inducible transactivation in USP22. This may be
explained by the assumption that activated CREB, rather than
elevated CREB expression, plays a crucial role in USP22
transcription. Previous studies have proven that activated CREB is
required for gene regulation, with this activation regulated
through the phosphorylation of serine 133 by kinases such as PKA
(25), PKC (26), calmodulin-dependent kinases
(27) or PI3K (28). To investigate the upstream signaling
pathways involved in USP22 transcriptional regulation via CREB,
several kinase inhibitors were employed. The results showed that
CREB functioning as a transcriptional element is controlled by PKA,
due to the H-89-mediated decrease in USP22 transcription via the
CREB binding site. In this study, the phosphorylation state of
serine 133 following H-89 treatment was not monitored. However,
previous studies have confirmed the ability of H-89 to
dephosphorylate CREB in various cell types (29). Unexpectedly, USP22 promoter activity
and endogenous USP22 mRNA levels were not altered in response to
the PKA activator forskolin, with a similar CREB role having been
previously reported. One study found that the CREB binding site in
the BRCA1 proximal promoter acts as a constitutive transcriptional
element (30). This constitutive
transactivation role was supported by the activation or
overexpression of CREB in cancers and the fact that the USP22
promoter lacks a canonical TATA box, making this type of promoter
unresponsive to cAMP (31).
In summary, the present study revealed that the CREB
binding site is crucial for basal USP22 promoter transcription.
Furthermore, the CREB transcription factor regulates USP22
transcription by directly binding to the CREB binding site, with
PKA regulation carried out through CREB. The understanding that
PKA/CREB is a key regulator of human USP22 transcription may be of
considerable value when selecting therapeutic targets for disease
treatment.
Acknowledgements
The present study was supported by the National
Nature Science Foundation of China (grant no. 31000581).
References
|
1
|
Lee HJ, Kim MS, Shin JM, Park TJ, Chung HM
and Baek KH: The expression patterns of deubiquitinating enzymes,
USP22 and Usp22. Gene Expr Patterns. 6:277–284. 2006. View Article : Google Scholar
|
|
2
|
Liu Y, Yang Y, Xu H and Dong X:
Implication of USP22 in the regulation of BMI-1, c-Myc, p16INK4a,
p14ARF, and cyclin D2 expression in primary colorectal carcinomas.
Diagn Mol Pathol. 19:194–200. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang DD, Cui BB, Sun LY, et al: The
co-expression of USP22 and BMI-1 may promote cancer progression and
predict therapy failure in gastric carcinoma. Cell Biochem Biophys.
61:703–710. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang Y, Yao L, Zhang X, et al: Elevated
expression of USP22 in correlation with poor prognosis in patients
with invasive breast cancer. J Cancer Res Clin Oncol.
137:1245–1253. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang XY, Varthi M, Sykes SM, et al: The
putative cancer stem cell marker USP22 is a subunit of the human
SAGA complex required for activated transcription and cell-cycle
progression. Mol Cell. 29:102–111. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhao Y, Lang G, Ito S, et al: A TFTC/STAGA
module mediates histone H2A and H2B deubiquitination, coactivates
nuclear receptors, and counteracts heterochromatin silencing. Mol
Cell. 29:92–101. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Atanassov BS, Evrard YA, Multani AS, et
al: Gcn5 and SAGA regulate shelterin protein turnover and telomere
maintenance. Mol Cell. 35:352–364. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lin Z, Yang H, Kong Q, et al: USP22
antagonizes p53 transcriptional activation by deubiquitinating
Sirt1 to suppress cell apoptosis and is required for mouse
embryonic development. Mol Cell. 46:484–494. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Atanassov BS and Dent SY: USP22 regulates
cell proliferation by deubiquitinating the transcriptional
regulator FBP1. EMBO Rep. 12:924–930. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sussman RT, Stanek TJ, Esteso P, Gearhart
JD, Knudsen KE and McMahon SB: The epigenetic modifier
ubiquitin-specific protease 22 (USP22) regulates embryonic stem
cell differentiation via transcriptional repression of
sex-determining region Y-box 2 (SOX2). J Biol Chem.
288:24234–24246. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xiong J, Che X, Li X, Yu H, Gong Z and Li
W: Cloning and characterization of the human USP22 gene promoter.
PLoS One. 7:e527162012. View Article : Google Scholar
|
|
12
|
Bubis M and Zisapel N: Modulation by
melatonin of protein secretion from melanoma cells: is cAMP
involved? Mol Cell Endocrinol. 112:169–175. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nakamura K, Bossy-Wetzel E, Burns K, et
al: Changes in endoplasmic reticulum luminal environment affect
cell sensitivity to apoptosis. J Cell Biol. 150:731–740. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zong Y, Sun L, Liu B, et al: Resveratrol
inhibits LPS-induced MAPK activation via activation of the
phosphatidylinositol 3-kinase pathway in murine RAW 264.7
macrophage cells. PLoS One. 7:e441072012. View Article : Google Scholar
|
|
15
|
Glinsky GV: Death-from-cancer signatures
and stem cell contribution to metastatic cancer. Cell Cycle.
4:1171–1175. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Glinsky GV: Genomic models of metastatic
cancer: functional analysis of death-from-cancer signature genes
reveals aneuploid, anoikis-resistant, metastasis-enabling phenotype
with altered cell cycle control and activated Polycomb Group (PcG)
protein chromatin silencing pathway. Cell Cycle. 5:1208–1216. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tinti C, Yang C, Seo H, et al:
Structure/function relationship of the cAMP response element in
tyrosine hydroxylase gene transcription. J Biol Chem.
272:19158–19164. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Euskirchen G and Snyder M: A plethora of
sites. Nat Genet. 36:325–326. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
White PC, Shore AM, Clement M, et al:
Regulation of cyclin D2 and the cyclin D2 promoter by protein
kinase A and CREB in lymphocytes. Oncogene. 25:2170–2180. 2006.
View Article : Google Scholar
|
|
20
|
Wilson BE, Mochon E and Boxer LM:
Induction of bcl-2 expression by phosphorylated CREB proteins
during B-cell activation and rescue from apoptosis. Mol Cell Biol.
16:5546–5556. 1996.PubMed/NCBI
|
|
21
|
Mayer SI, Willars GB, Nishida E and Thiel
G: Elk-1, CREB, and MKP-1 regulate Egr-1 expression in
gonadotropin-releasing hormone stimulated gonadotrophs. J Cell
Biochem. 105:1267–1278. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sakamoto KM and Frank DA: CREB in the
pathophysiology of cancer: implications for targeting transcription
factors for cancer therapy. Clin Cancer Res. 15:2583–2587. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Melnikova VO, Dobroff AS, Zigler M, et al:
CREB inhibits AP-2alpha expression to regulate the malignant
phenotype of melanoma. PLoS One. 5:e124522010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Quinn PG: Distinct activation domains
within cAMP response element-binding protein (CREB) mediate basal
and cAMP-stimulated transcription. J Biol Chem. 268:16999–17009.
1993.PubMed/NCBI
|
|
25
|
Rosenberg D, Groussin L, Jullian E,
Perlemoine K, Bertagna X and Bertherat J: Role of the PKA-regulated
transcription factor CREB in development and tumorigenesis of
endocrine tissues. Ann NY Acad Sci. 968:65–74. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Guo Y and Feng P: OX2R activation induces
PKC-mediated ERK and CREB phosphorylation. Exp Cell Res.
318:2004–2013. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Takeda H, Kitaoka Y, Hayashi Y, et al:
Calcium/calmodulin-dependent protein kinase II regulates the
phosphorylation of CREB in NMDA-induced retinal neurotoxicity.
Brain Res. 1184:306–315. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gibellini D, Bassini A, Pierpaoli S, et
al: Extracellular HIV-1 Tat protein induces the rapid Ser133
phosphorylation and activation of CREB transcription factor in both
Jurkat lymphoblastoid T cells and primary peripheral blood
mononuclear cells. J Immunol. 160:3891–3898. 1998.PubMed/NCBI
|
|
29
|
Delghandi MP, Johannessen M and Moens U:
The cAMP signalling pathway activates CREB through PKA, p38 and
MSK1 in NIH 3T3 cells. Cell Signal. 17:1343–1351. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Atlas E, Stramwasser M and Mueller CR: A
CREB site in the BRCA1 proximal promoter acts as a constitutive
transcriptional element. Oncogene. 20:7110–7114. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Conkright MD, Guzman E, Flechner L, Su AI,
Hogenesch JB and Montminy M: Genome-wide analysis of CREB target
genes reveals a core promoter requirement for cAMP responsiveness.
Mol Cell. 11:1101–1108. 2003. View Article : Google Scholar : PubMed/NCBI
|